

Abstract
Intramembrane proteases have the unusual property of cleaving peptide bonds within the lipid bilayer, an environment not obviously suited to a water-requiring hydrolysis reaction. These enzymes include site-2 protease, γ-secretase/presenilin, signal peptide peptidase and the rhomboids, and they have a wide range of cellular functions. All have multiple transmembrane domains and, because of their high hydrophobicity, have been difficult to purify. We have now developed an in vitro assay to monitor rhomboid activity in the detergent solubilised state. This has allowed us to isolate for the first time a highly pure rhomboid with catalytic activity. Our results suggest that detergent-solubilised rhomboid activity mimics its activity in biological membranes in many aspects. Analysis of purified mutant proteins suggests that rhomboids use a serine protease catalytic dyad instead of the previously proposed triad. This analysis also suggests that other conserved residues participate in subsidiary functions like ligand binding and water supply. We identify a motif shared between rhomboids and the recently discovered derlins, which participate in translocation of misfolded membrane proteins.
Keywords: catalytic dyad, derlin, proteolytic processing, rhomboid, serine protease
Introduction
The discovery over the last few years of proteases that cleave within transmembrane domains (TMDs) has posed fundamental questions about proteolytic mechanisms. These intramembrane proteases comprise three mechanistic classes. The first family, with site-2 protease (S2P) as the founding member, are metalloproteases (Rawson et al, 1997; Duncan et al, 1998). The second family is a group of aspartyl proteases including γ-secretase (De Strooper et al, 1998; Steiner et al, 1999; Wolfe et al, 1999) and signal peptide peptidase (SPP) (Ponting et al, 2002; Weihofen et al, 2002). The third family comprise the rhomboid serine proteases (Lee et al, 2001; Urban et al, 2001). Together they regulate many biological processes such as intercellular signalling, lipid metabolism and the unfolded protein response (reviewed in Brown et al, 2000; Weihofen and Martoglio, 2003; Wolfe and Kopan, 2004); γ-secretase also plays a key role in the pathogenesis of Alzheimer's disease (Selkoe and Schenk, 2003). Unlike soluble proteases, the catalytic heart of these enzymes appears buried within the lipid bilayer, an unexpected location for a hydrolytic reaction, which requires water. Addressing directly the mechanism of all these enzymes has been hampered by the fact that they are all very hydrophobic and therefore difficult to handle in biochemical assays. Only very recently has activity of γ-secretase and RseP, a bacterial S2P-like enzyme, been purified in their active forms (Akiyama et al, 2004; Fraering et al, 2004b); these represent the first direct biochemical analyses of intramembrane proteases.
Rhomboids were first discovered as activators of epidermal growth factor (EGF) receptor signalling in Drosophila (Sturtevant et al, 1993; Wasserman et al, 2000), where they release the soluble and active form of membrane tethered ligands, including Spitz, Gurken and Keren (Urban et al, 2002a). Rhomboids are very widely conserved (Wasserman et al, 2000; Koonin et al, 2003). For example, a rhomboid is essential for the production of a quorum sensing signal in the Gram-negative bacterium Providencia stuartii (Gallio et al, 2002; Urban et al, 2002b), and there is indication that rhomboids may participate in the invasive step of apicomplexan parasites such as Toxoplasma and Plasmodium (Urban and Freeman, 2003). A mitochondrial subclass of rhomboids has also been discovered (Esser et al, 2002; McQuibban et al, 2003). Beyond these examples, nothing is known about the biological function of most members of the family. Previous analysis of rhomboid proteolytic activity relied on the indirect read-out of the accumulation of a soluble form of a membrane-tethered substrate protein—typically an EGF-type ligand—in the medium or secretory pathway of cultured cells (Lee et al, 2001; Urban et al, 2001). The need to use whole cells as the basis for the assay introduced uncertainty about whether proteolysis was being influenced by other cellular events. This limitation prevented us from answering basic questions about the proteolytic mechanism.
The different families of intramembrane proteases appear structurally and evolutionarily unrelated. They share a polytopic membrane topology and have putative active sites within the plane of the lipid bilayer but, beyond this, there is no reason to think they use common mechanisms. Indeed, even before direct biochemical analysis has been possible, there are clear distinctions. For example, γ-secretase is a complex of proteins, including presenilin (for a review, see De Strooper, 2003), whereas there is no evidence for extra subunits required for the activity of the other intramembrane proteases. Each family has characteristic cleavage sites within TMDs: rhomboids cleave at or near the luminal/extracellular surface, S2P near the cytosolic surface, SPP in the centre of the TMD and γ-secretase at several sites. Rhomboids differ from all the other families by not requiring prior trimming of the substrate to generate only a short juxtamembrane domain. They are also the least characterised of these enzymes: no rhomboid activity has been reported, except after co-expression of substrate and protease in tissue culture cells; indeed, this has precluded a definitive, direct demonstration that they have proteolytic activity.
We have developed an in vitro assay to monitor rhomboid catalysed intramembrane proteolysis and have extended this approach to isolate for the first time a highly pure, detergent-solubilised rhomboid with activity. The availability of an efficient in vitro assay allows us to characterise biochemically the mechanism of catalysis by rhomboids. We conclude that they appear to use a catalytic dyad instead of the previously proposed, and more common, triad. We also present evidence that they might share a TMD interaction mechanism with the recently discovered derlins, which participate in the re-translocation of misfolded membrane proteins from the ER to the cytosol (Lilley and Ploegh, 2004; Ye et al, 2004).
Results
Activity of detergent-solubilised rhomboids
In order to investigate the mechanism of rhomboid catalysed proteolysis in detail, we have designed an in vitro activity assay. We started by expressing in Escherichia coli the Bacillus subtilis rhomboid YqgP, whose own substrate is unknown but which efficiently cleaves the Drosophila substrate Gurken in cell culture (Urban et al, 2002b). A membrane fraction from these cells was incubated with an in vitro translated [35S]methionine-labelled peptide spanning the whole predicted TMD of Gurken with an extension of 22 N-terminal juxtamembrane residues (plus an additional methionine required to initiate peptide translation). The rationale was that at least a proportion of the hydrophobic peptide would enter the translocon of the YqgP-expressing membranes and thereby be incorporated in the lipid bilayer; this has been shown to occur, at least with peptides that resemble signal sequences (Simon and Blobel, 1992). Such incorporation would allow the peptide to become a substrate for intramembrane proteolysis, as has been shown for SPP (Weihofen et al, 2000). After incubation at 30°C the peptide was cleaved (Figure 1A and B), although with low efficiency. Owing to the greater efficiency seen in the presence of detergent (see below), we presume the low efficiency was caused by limited incorporation of the substrate peptide into the membranes. The reaction was inhibited by 400 μM dichloroisocoumarin (DCI), one of only two serine protease inhibitors that has previously been shown to be effective against rhomboids, albeit with low potency (Urban et al, 2001). Cleavage did not occur in the absence of YqgP, or in a reaction containing a mutant enzyme, predicted to be catalytically inactive. We therefore conclude that the cleavage was catalysed by YqgP.
Figure 1.
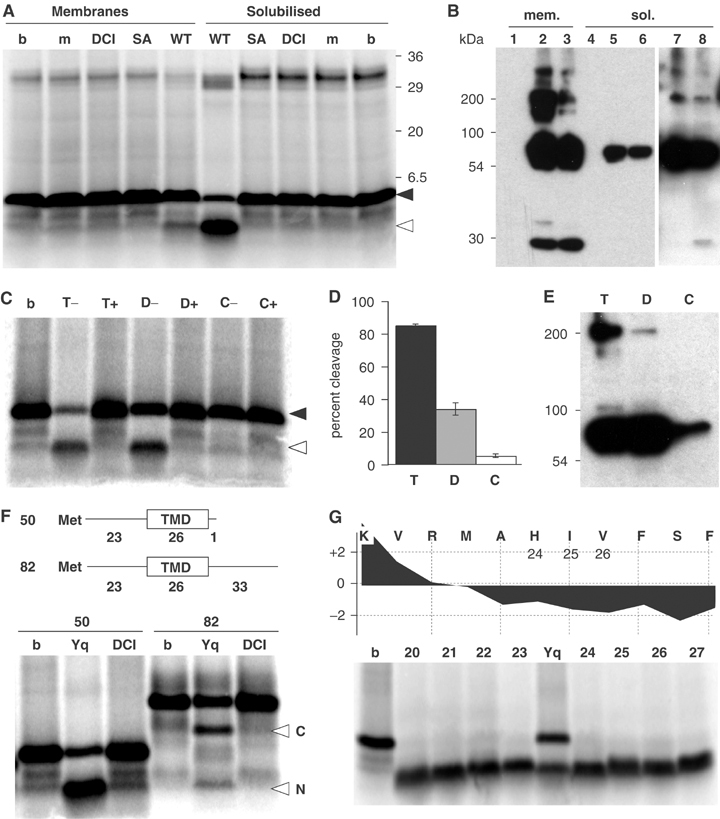
In vitro cleavage assay with recombinant B. subtilis rhomboid YqgP. (A) Cleavage of in vitro translated Gurken-TMD by a membrane fraction from E. coli expressing YqgP and TX100 solubilised YqgP (m, nonexpressing cells; WT, cells expressing YqgP; SA, cells expressing the catalytic mutant YqgP-S288A; DCI, assay of WT in presence of 400 μM DCI; b, buffer only). In both intact and solubilised membranes the substrate peptide was cleaved; this was dependent on serine-288 and was inhibited by DCI. The sizes of cleaved and uncleaved substrate peptide are indicated in this and subsequent panels by open and filled arrowheads, respectively. The band at ∼30 kDa corresponds to peptidyl-tRNA. (B) Rhomboid expression in intact membranes and after TX100 solubilisation analysed by Western blotting; YqgP (lanes 2 and 5), YqgP-S288A mutant (lanes 3 and 6) and mock expression (lanes 1 and 4). For TX100-solubilised WT and SA, a longer exposure is shown (lanes 7 and 8). Selective solubilisation of YqgP (∼60 kDa) and an SDS-stable multimer (∼200 kDa) was observed; however, degradation products and aggregates were only found in the detergent insoluble fraction (not shown). (C) TX100 and DDM retain rhomboid (YqgP) activity in the detergent-solubilised state (T, TX100; D, DDM; C, CHAPS; b, buffer only); 400 μM DCI was added as indicated (T+, D+ and C+). (D) Gurken-TMD cleavage by detergent-solubilised YqgP in three experiments; mean and range are indicated. (E) Western blot showing that TX100 and DDM but not CHAPS solubilised YqgP efficiently. (F) TX100-solubilised rhomboid (Yq) catalysed endoproteolysis of Gurken-TMD82 generated two detectable fragments (labelled with N and C); DCI, reaction in the presence of 400 μM DCI; b, buffer only. The reaction was run alongside cleavage of Gurken-TMD50. The two substrates are diagrammed, with the number of residues in each part of the peptide indicated. The two cleavage products of Gurken-TMD82 were seen consistently in multiple experiments. (G) N-terminal cleavage fragment of Gurken-TMD generated by TX100-solubilised YqgP (Yq) or buffer only (b) was compared with reference peptides corresponding to the indicated N-terminal cleaved fragment. The hydrophobicity plot of the relevant region is shown (using the scale of Kyte and Doolittle (1982), with a window size of 7) indicating the potential N-terminal TMD boundary.
An essential step towards obtaining pure, active rhomboid for biochemical analysis was to solubilise the protein while retaining its catalytic activity. Rhomboid activity was monitored after solubilisation by a number of different detergents; we discovered that YqgP retained activity after membranes were solubilised with the nonionic detergents Triton X-100 (TX100) (Figure 1A and C) or n-dodecyl-β-D-maltoside (DDM) (Figure 1C): in fact, cleavage was much more efficient than in intact membranes. The zwitterionic detergent CHAPS, which retains the activity of aspartyl intramembrane proteases γ-secretase/presenilin and SPP (Li et al, 2000; Weihofen et al, 2000), was ineffective at solubilising YqgP and little activity was detected (Figure 1C–E). Rhomboid activity depended on the presence of the catalytic serine and was inhibited by DCI, confirming that the reaction was specific (Figure 1A and C). The increased efficiency of substrate cleavage in vitro in the presence of detergent is presumably due to uncoupling from the rate-limiting step, the integration of the substrate TMD into the membrane. The cleaved products generated from either intact or solubilised membranes were identical in size, implying that detergent solubilisation did not alter the rhomboid cleavage specificity.
In order to identify the cleavage products, we generated a variant substrate peptide, extended at the C-terminus. By comparing the cleavage of the long and short substrates, we identified the product observed in the standard assay as the N-terminal fragment of the cleaved peptide (Figure 1F). Moreover, when the longer Gurken-TMD82 was used as a substrate, both the N- and C-terminal fragments of the proteolysis were detected (Figure 1F), formally demonstrating that rhomboid is an endopeptidase. Since we noticed that the shorter substrate was cleaved more efficiently than the extended one, the subsequent analysis in this study was performed with substrate TMDs with only short N-terminal extensions. Using this substrate, the cleavage site was approximately mapped to the region of residues alanine-24 to isoleucine-26 by comparing the N-terminal fragment with a series of reference peptides (Figure 1G). This site is very close to the predicted amino-terminus of the TMD, as also observed in intact cells (Urban et al, 2001; Lohi et al, 2004). This important result implies that cleavage in vitro mimics cleavage in intact cells, even though, until we can generate large enough quantities of substrate to analyse by mass spectrometry, the exact peptide bond that is cleaved by rhomboids remains uncertain.
Activity of multiple rhomboids in vitro
Our in vitro assay was developed with a rhomboid from the Gram-positive bacterium B. subtilis. To explore the assay's utility for investigating rhomboids from different species, diverse rhomboids were similarly expressed in E. coli. These included rhomboids from the Gram-negative bacteria E. coli, Providencia stuartii and Pseudomonas aeruginosa; the thermophilic bacterium Aquifex aeolicus; and the eukaryotic Rhomboid-1 from Drosophila and RHBDL2 from humans. In each case, TX100 solubilised membranes were assayed for their ability to cleave three model substrate peptides based, respectively, on the TMDs of Gurken, Spitz and Delta (Figure 2A–C). Gurken-TMD was cleaved by YqgP (B. subtilis), GlpG (E. coli), predicted protein PA3086 (P. aeruginosa), AarA (P. stuartii), predicted protein AQ1327 (A. aeolicus), and also by Rhomboid-1 (Drosophila) and RHBDL2 (human). The only rhomboid tested not to cleave Gurken-TMD was YdcA from B. subtilis, which was previously found to be inactive in a cell culture assay (Urban et al, 2002b). All rhomboids were expressed at equivalent levels (Figure 2D). Spitz-TMD was significantly cleaved only by rhomboids from P. aeruginosa, P. stuartii and the thermophile A. aeolicus. The Drosophila and human rhomboids had little activity against Spitz-TMD, but these enzymes cleave Spitz at a lower efficiency than Gurken in the cell culture assay (Urban et al, 2002a). Delta-TMD was not a good substrate in vitro, consistent with the fact that Drosophila Delta protein is not a substrate for rhomboids in vivo (Urban and Freeman, 2003). The rhomboids from P. aeruginosa and A. aeolicus appeared to have slight activity against Delta-TMD; a similar activity in living cells was previously reported for this Pseudomonas rhomboid (Urban et al, 2002b).
Figure 2.
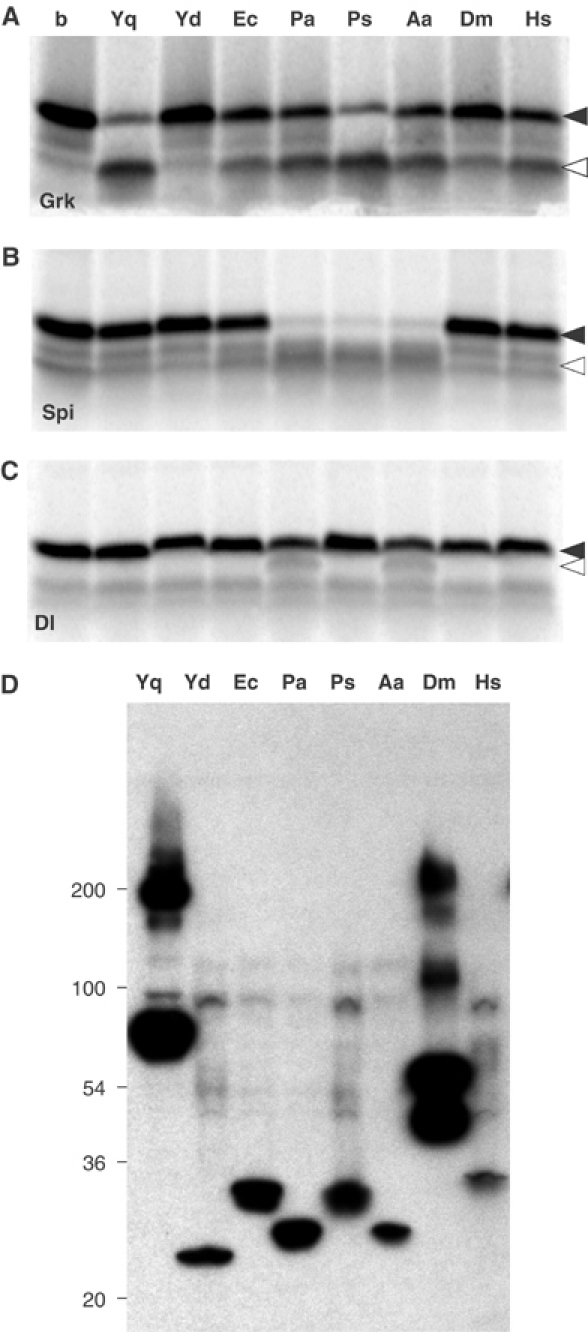
TX100-solubilised rhomboids were tested for the cleavage of three model substrates based on membrane-anchored Drosophila EGF-type ligands. (A) Cleavage assay using Gurken-TMD, (B) Spitz-TMD and (C) Delta-TMD with TX100-solubilised B. subtilis YqgP (Yq), B. subtilis YdcA (Yd), E. coli GlpG, (Ec), P. aeruginosa rhomboid (Pa), P. stuartii AarA (Ps), A. aeolicus rhomboid (Aa), Drosophila melanogaster Rhomboid-1 (Dm) and human RHBDL2 (Hs); b, buffer only. (D) Recombinant expression and solubilisation of mutant rhomboids was analysed by Western blotting. After solubilisation with TX100, similar amounts of monomeric form and, to varying extents, putative multimeric species were observed.
The only major distinction between these results and those in cell culture is the activity of A. aeolicus: its activity here implies that in some cases the detergent-solubilised assay can overcome limitations of the cell-based approach. In summary, the activity of rhomboids from across evolution can be reconstituted in the detergent-solubilised state and in most cases their activities reflect activity in whole cells. This is an important step towards determining the catalytic mechanism of these enzymes as well as providing an assay for identifying specific inhibitors.
Purification of active rhomboids
Detergent solubilisation of active rhomboids also allows purification, which is essential for biochemical and structural analysis. Our initial purification efforts focused on histidine-tagged B. subtilis YqgP and E. coli GlpG, two of the highest-expressing rhomboids. Both were purified using a combination of selective membrane solubilisation, metal-chelate chromatography (Ni2+-NTA), and anion exchange chromatography (Figure 3A; Materials and methods). Both proteins retained activity throughout the purification procedure (Figure 3B). Notably, E. coli GlpG was purified to a level where only a single band was visible on an overloaded Coomassie-stained gel (Figure 3A), making it the first active intramembrane protease available in homogeneous form. Until now, the most purified was RseP, a member of the S2P family, but in this case the degree of purity was considerably less (Akiyama et al, 2004). The fact that the highly pure rhomboid fraction retains activity is the first direct demonstration of proteolytic activity in a rhomboid protein and provides evidence that rhomboids do not function as part of a multiprotein complex, as is the case for presenilin (De Strooper, 2003).
Figure 3.
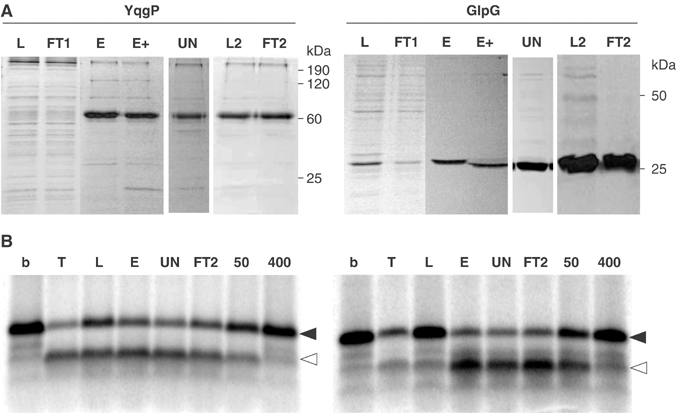
Rhomboid was purified and assayed for activity. (A) Aliquots of fractions from the purifications (as described in Materials and methods) were resolved by SDS–PAGE and stained with Coomassie blue. L, DDM-solubilised membrane protein load; FT1, Ni2+-NTA flow-through; E, Ni2+-NTA elute pool; E+, Ni2+-NTA elute pool after TEV cleavage; UN, representative fraction of UNO-Q eluate; L2, load 2 (UNO-Q eluate pool); FT2, second Ni2+-NTA flow-through (for GlpG, the gel was overloaded to demonstrate protein purity). The yield from a 2 L culture was 1 mg for GlpG, and 0.6 mg for YqgP. (B) YqgP and GlpG remained active against Gurken-TMD over the entire purification; b, buffer only; T, TX100-solubilised rhomboid; L, E, UN and FT2 as outlined in (A); DCI was added at 50 and 400 μM to assays with FT2 as indicated.
Rhomboid cleavage is ATP-independent
Our working hypothesis is that substrate presentation to rhomboids is topologically straightforward because the cleavage site of the substrate and the catalytic site of the enzyme reside at approximately the same level in the membrane. We could not, however, rule out a more complex model where energetically unfavourable distortions of the substrate and/or active site are required, as has been postulated for the S2P intramembrane protease (Ye et al, 2000). Indeed, the yeast mitochondrial rhomboid appears to cleave its substrate after the substrate has been pulled through the membrane by the ATP-dependent Tim23 translocation complex (Herlan et al, 2004). We therefore tested whether rhomboid cleavage itself was ATP dependent. By depleting or increasing ATP levels present in the reaction with purified YqgP and GlpG, we conclude that proteolysis is ATP independent (Figure 4A).
Figure 4.
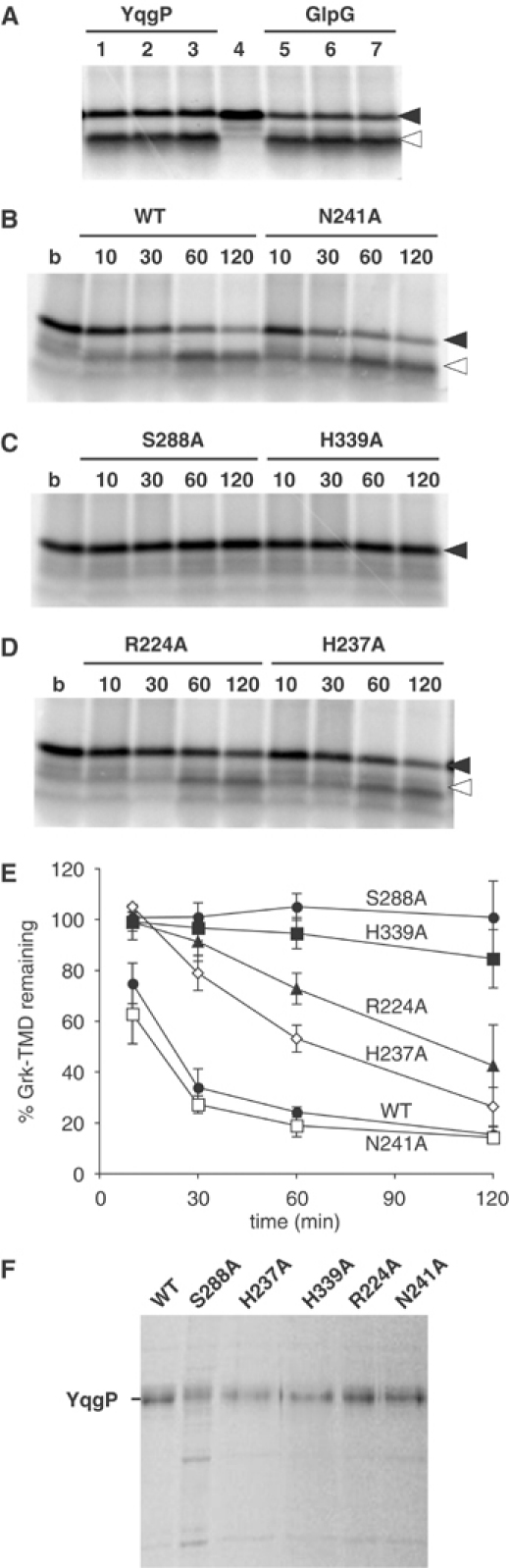
Enzymatic properties of purified rhomboid. (A) Rhomboid-catalysed cleavage is ATP independent. Cleavage assay with Ni2+-NTA purified YqgP (lane 1–3) and GlpG (lane 5–7); lanes 2 and 6, assay after ATP depletion; lanes 3 and 7, assay in the presence of an ATP regeneration system; lane 4, buffer only. (B–D) Comparative analysis of Ni2+-NTA purified YqgP mutants. Numbering refers to reaction time (min); b, control with buffer alone. (E) Progression curves of three independent cleavage assays with the indicated mutants; mean and range are indicated. Similar results were obtained with a second independent expression and purification (data not shown). (F) SDS–PAGE and Coomassie staining of Ni2+-NTA purified YqgP mutants.
Requirement for key residues
A number of residues that appear important for rhomboid activity have been identified by their widespread conservation and, in some cases, their necessity, as determined by site-directed mutagenesis in the cell culture assay (Urban et al, 2001, 2002b). This led to the proposal of a serine protease catalytic mechanism, centred around a catalytic triad. We have now assayed and quantified the requirement for many of these key residues in vitro, by testing the activity of purified mutant proteins (Figure 4B–E). The expression in E. coli of all these mutants was equivalent (Figure 4F). The serine-288 and histidine-339 proposed to participate in a catalytic triad were essential (Figure 4C and E); this is also true for glycine-286 two residues N-terminal to the proposed catalytic serine that, by analogy to classical serine proteases, is predicted to contribute to an oxyanion-binding pocket that stabilises the reaction intermediate (data not shown). However, the third member of the suggested triad, asparagine-241, was not required for catalytic activity: the N241A mutant has activity indistinguishable from wild type in vitro (Figure 4B and E). Combined with the previous observation that this mutation retained some activity in the cellular assay (Urban et al, 2002b), this result demonstrates that asparagine-241 is not an obligate member of a catalytic triad. We conclude that rhomboids instead function with a serine–histidine dyad, which is a less common serine protease mechanism (Paetzel and Dalbey, 1997). The observed effect of mutation of this asparagine in the cellular assay, which for most rhomboids tested blocked cleavage completely, implies that its requirement must be specific to the reaction that occurs in a lipid bilayer. It could, for example, be involved in providing the water molecule required for the deacylation step of the proteolysis (Hedstrom, 2002). In the detergent-solubilised state, water would be expected to be more freely accessible than in a lipid bilayer.
We have also mutated a well-conserved histidine (H237 in YqgP) in the second TMD. Previous in vivo assays with Drosophila Rhomboid-1 suggested that, despite its conservation, this residue did not participate substantially in the cleavage reaction. In contrast, YqgP activity was significantly compromised in the H237A mutant although, unlike the proposed dyad residues, this mutant did not destroy all activity (Figure 4D and E). We speculate that this residue, which contributes to the amphipathic nature of the second TMD, plays a structural role or contributes to binding of the substrate backbone. This partial requirement illustrates the ability of the new assay to detect quite subtle differences in activity.
A highly conserved tryptophan–arginine (WR) pair of residues in the large loop near the luminal face of TMD2 has previously been shown to be required for rhomboid activity in cells (Urban et al, 2001). There is, however, no obvious link with the proposed serine protease mechanism and its function remains elusive. Unexpectedly, the corresponding R224A purified mutant protein of YqgP retained cleavage activity in the detergent-solubilised state, albeit with a reduced level compared to wild type, and the N241A and the H237A mutants (Figure 4D and E). We conclude that the WR motif is only essential when in a lipid bilayer and is therefore not part of the core proteolytic mechanism. Again, the differences between the activity of detergent-solubilised rhomboids and those expressed in intact membranes allow us to distinguish residues involved in different aspects of the cleavage mechanism. Interestingly, we noticed that the same WR motif, in an identical position, is widely conserved in the derlins, a recently discovered family of polytopic membrane proteins that are involved in the extraction of misfolded membrane proteins from the ER (Figure 5A) (Lilley and Ploegh, 2004; Ye et al, 2004). These observations raise the possibility of some shared mechanism between rhomboids and derlins.
Figure 5.
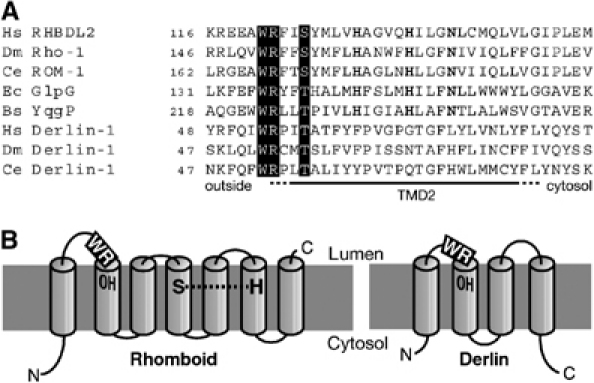
Possible functional relationship between rhomboids and derlins. (A) Alignment of TMD2 of a variety of rhomboids and derlins; the conserved WR motif and hydroxyl residue are highlighted and residues conserved in rhomboids are bold. The species and GenBank accession numbers are: human (Hs) RHBDL2 (AAM95697) and Derlin-1 (NP_077271); D. melanogaster (Dm) Rho-1 (AAF47496) and Derlin-1 (NP_608632); C. elegans (Ce) ROM-1 (AAA91218) and Derlin-1 (NP_492721); E. coli (Ec) GlpG (BAB37690); B. subtilis (Bs) YqgP (NP_390367). (B) Topology model of human RHBDL2 compared with Derlin-1. The conserved WR and the hydroxyl residue (OH) in TMD2 are highlighted; the proposed rhomboid catalytic dyad residues (S and H) are indicated.
Discussion
It is clear that intramembrane proteolysis regulates many biological processes. As with other intramembrane proteases such as γ-secretase/presenilin and SPP (Martoglio and Golde, 2003; Selkoe and Schenk, 2003), rhomboids may turn out to be useful therapeutic targets (Opitz et al, 2002; Urban and Freeman, 2003). The lack of an in vitro assay for rhomboid activity has been a limiting factor in the biochemical analysis of their proteolytic mechanisms. We have now demonstrated rhomboid activity both in detergent-solubilised membrane extracts and in a highly purified fraction. These two versions of the in vitro assay have different utilities. The activity of multiple rhomboids can be rapidly determined in TX100 membrane extracts; and in purified fractions, precise quantification of relative activities can be measured. Here we have exploited both approaches to show that diverse rhomboids have differential activities on three model substrates, and to compare the activities of a number of purified mutant proteins.
There are several strands of evidence that the rhomboid assay in vitro reflects the proteolytic activity in cellular membranes. The cleaved products of substrate incubated with intact or solubilised membranes are indistinguishable; the cleavage site in vitro also appears to correspond to that seen in a cell culture assay (Urban et al, 2001; Lohi et al, 2004); the activities of different rhomboids against three different substrates is very similar in vitro and in cell culture (Urban et al, 2001, 2002b); and finally, the key catalytic residues essential in tissue culture are also essential when the enzyme is detergent solubilised. Nevertheless, there are also significant differences between the results from solubilised rhomboids and whole-cell assays. Although some of these may be caused by taking the rhomboids out of their natural lipid bilayer environment, many may be instructive. We suspect that the reduced efficiency of cleavage of the extended substrate TMD is a limitation of the detergent-based assay. There is no prior evidence to suggest that rhomboid-catalysed cleavage depends on a prior juxtamembrane cleavage and a similar low turnover is observed in in vitro γ-secretase cleavage assays using substrates of comparable length (Li et al, 2000). This may be a general property of these types of assay, possibly reflecting an effect of longer hydrophilic tails increasing substrate peptide solubility and thereby influencing its distribution between protease-containing detergent micelles and the aqueous phase.
In contrast to this technical restriction, comparison of cleavage efficiency of different rhomboid mutants between in vitro and cell-based assays is quite informative. Residues necessary for the core proteolytic activity will be essential in any assay, whereas those involved in subsidiary functions such as substrate binding and water supply are likely to be affected by the difference between the membrane and detergent-solubilised state. In this way, we have significantly modified our view of the catalytic mechanism of rhomboids, now proposing that they rely on a catalytic dyad instead of a triad. We have also been able to exploit comparison of in vitro and cellular assays to shed some light on the likely roles of arginine-224 and histidine-237 of YqgP.
The choice of the detergent that allows retention of the native structure and the biological function of membrane proteins is critical; in this aspect, the rhomboids differ from other intramembrane proteases. The zwitterionic detergents CHAPSO and CHAPS have been successfully used to solubilise the γ-secretase complex and SPP, respectively (Li et al, 2000; Weihofen et al, 2000). Recently, the nonionic detergent DDM was used for the purification of the S2P homologue RseP (Akiyama et al, 2004). We have shown that rhomboids were not solubilised efficiently by CHAPS, but are active in DDM and TX100. In contrast to the similarity of rhomboid cleavage site in both detergents and intact membranes, γ-secretase specificity is sensitive to solubilisation conditions (Fraering et al, 2004b) and TX100 and DDM disrupt the γ-secretase activity (Li et al, 2000; Fraering et al, 2004a). We also note that, even in detergents compatible with activity, γ-secretase and SPP are inactive when the detergent concentration is above the critical micelle concentration (CMC) (MK Lemberg and Bruno Martoglio, unpublished; Fraering et al, 2004b). Combined with the lipid requirement observed after purification of the γ-secretase complex (Fraering et al, 2004b), this suggests that the proteolytic activity in these assays might require mixed lipid–detergent–protein structures or partially reconstituted proteoliposomes, which form spontaneously on dilution to detergent concentrations below the CMC. Rhomboids, on the other hand, are active even in presence of 0.5% TX100 and DDM (data not shown), both of which have a CMC below 0.1%. This suggests that rhomboids may differ from γ-secretase and SPP in their ability to cleave peptide bonds in detergent micelles.
The shared WR motif between rhomboids and derlins is intriguing (Figure 5A). Both are polytopic membrane proteins with the motif residing in the luminal loop, just N-terminal to the second predicted TMD (Figure 5B). They also share a highly conserved residue with an hydroxyl side-group, also in the second TMD. Further support for a possible functional relationship between rhomboids and derlins is our observation that reiterative homology searching with the PSI-Blast algorithm (Altschul et al, 1997) reveals a consistent (though weak) sequence relationship between the derlins and rhomboids, suggesting that they may in fact be distantly related. Since the derlins function in a process that involves translocation of transmembrane proteins across the ER membrane prior to proteasomal degradation (Lilley and Ploegh, 2004; Ye et al, 2004), it is difficult to guess what mechanistic features they might share with rhomboids. A possible common theme between the two protein families might be the destabilisation or unfolding of transmembrane helices to allow, in one case, translocation to the cytosol, or in the other, cleavage in the plane of the membrane. We emphasise, however, that this possibility remains speculative until the mechanism of both rhomboids and derlins are better understood.
Materials and methods
Plasmid constructs
The rhomboid coding sequences (Lee et al, 2001; Urban et al, 2001, 2002b) were subcloned into a pET25b(+)-derived E. coli expression vector (Novagen). When cloned into this vector, the constructs encode a C-terminal six-histidine tag preceded by a linker containing a TEV protease cleavage site (…ASENLYFQ^G…). YqgP mutants were generated with the QuickChange Site-Directed Mutagenesis Kit (Stratagene). All constructs were confirmed by sequencing.
Preparation of membrane fractions and solubilisation
To produce recombinant rhomboid, 2 l of LB medium were inoculated with E. coli BL21-Gold(DE3) cells harbouring the expression vector and the extra plasmid pRARE2 (Novagen) encoding supplementary tRNAs. Culture was grown at 30°C until reaching an OD600 of 0.3–0.4, and induction of expression was initiated by the addition of 0.6 mM IPTG. Incubation was continued for 12 h at 16°C, at which point cells where harvested and resuspended in 40 ml buffer A (20 mM HEPES/NaOH, pH 7.4, 100 mM NaCl, 5 mM MgCl2, 10% glycerol), containing EDTA-free Complete Protease Inhibitor cocktail (1 × PI, Roche). All subsequent steps were performed at 4°C. Cells were lysed by two passes through an Avestin EmulsiFlex-C5 and centrifuged at 50 000 g for 30 min. After washing with buffer A and recentrifugation, membranes were resuspended in 3 ml buffer B (50 mM HEPES/NaOH pH 7.4, 100 mM NaCl, 15% glycerol, 1 × PI) using a Dounce homogeniser. The membrane suspension was either directly used for cleavage assay or solubilised by the addition of 2% detergent from a 4% solution in buffer B (Triton-X 100, protein grade, Calbiochem; DDM and CHAPS, solubilisation grade, Anatrace). The resulting solution was incubated for 1 h on ice, and non-solubilised proteins were removed by centrifugation at 100 000 g for 30 min. Detergent-solubilised rhomboids were flash frozen in liquid nitrogen and stored at −80°C.
Nickel affinity chromatography
For purification of DDM-solubilised rhomboid, E. coli membrane fraction (obtained as described above) was resuspended in buffer D (50 mM HEPES/NaOH, pH 7.4, 200 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 10% glycerol, 5 mM imidazole, 1% DDM, 1 × PI) containing 0.05% cholesteryl hemisuccinate (CHS, Anatrace), stirred gently for 1 h at 4°C, and then centrifuged at 100 000 g for 30 min. The 100 000g-cleared supernatant was loaded onto a 1 × 2.5 cm Ni2+-NTA Superflow gravity column (Qiagen), which was washed with 10 volumes of buffer E (50 mM HEPES/NaOH, pH 7.4, 1 mM MgCl2, 1 mM CaCl2, 10% glycerol, 30 mM imidazole, 0.1% DDM, 0.02% CHS) containing 500 mM NaCl, followed by 10 column volumes of buffer E containing 200 mM NaCl. Bound protein was eluted with buffer F (50 mM HEPES/NaOH, pH 7.4, 200 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 10% glycerol, 400 mM imidazole, 0.1% DDM, 0.02% CHS). The elute pool was flash frozen in liquid nitrogen, and stored at −80°C. Protein concentrations were determined using a detergent-compatible EZQ Protein Quantitation Kit (Molecular Probes) with chicken ovalbumin as standard.
Removal of the fusion tail and anion-exchange chromatography
Removal of the C-terminal fusion tag was achieved by cleavage with histidine-tagged TEV protease (Science Reagents), which was typically performed after supplementation of 0.5 mM Tris(2-carboxyethyl)phoshine hydrochloride (Bioshop) for 12 h at 4°C in the ratio 1:20 (w/w, TEV protease to fusion protein). Following cleavage, the protein solutions were diluted with four volumes of buffer G (10 mM HEPES pH7.4, 0.05% DDM) and immediately applied at 1.0 ml/min to an UNO Q-1 ion exchange column (Biorad), pre-equilibrated in buffer H (15 mM HEPES/NaOH pH 7.4, 0.05% DDM). Protein was resolved with a 60 ml linear gradient to buffer I (15 mM HEPES/NaOH, pH 7.4, 1 M NaCl, 0.05% DDM), at 1.0 ml/min. GlpG eluted between 25 and 30% Buffer I, while YqgP eluted between 55 and 60% Buffer I. Pooled fractions from the UNO Q-1 ion exchange were loaded onto a second 1 × 2.5 cm Ni2+-NTA Superflow gravity column and purified rhomboid was collected in the flow-through, flash frozen in liquid nitrogen, and stored at −80°C.
Rhomboid cleavage assay
Radiolabelled substrate consisting of the substrate TMD was generated by cell-free in vitro translation using wheat germ extract (Promega) and [35S]methionine (Amersham-Pharmacia) as described (Lemberg and Martoglio, 2003). The peptide substrate Gurken-TMD corresponds to an N-terminal methionine plus residues 224–272 of Gurken; the substrate Spitz-TMD to residues 113–161 of Spitz; and the substrate Delta-TMD to an N-terminal methionine plus residues 573–621 of Delta. Gurken-TMD82 corresponds to an N-terminal methionine plus residues 224–295 of Gurken plus nine additional residues at the C-terminus (YPYDVPDYA). To obtain a template for in vitro transcription, the coding region of interest was amplified by PCR from the pcDNA3.1 plasmids described previously (Lee et al, 2001; Urban et al, 2002a; Urban and Freeman, 2003). As forward primer, we use an oligonucleotide coding the SP6 promoter, Kozak initiation sequence, ATG for initiation, followed by the respective coding region. As reverse primer, we use an oligonucleotide 5′-N6CTAN20-3′ (where the N20 region is complementary to the cDNA) in order to introduce a TAG stop codon at the desired position. For the cleavage assay, 2 μl of translation mixture was added to a 38 μl reaction containing the rhomboid in 50 mM HEPES/NaOH, pH 7.4, 10% glycerol and 50 mM EDTA (the amount used was 1 μl of E. coli membrane suspension; 2 μl crude detergent-solubilised proteins; 5 μg protein for Figures 3B and 4A). For Figure 4B–E, 40 μl Ni2+-NTA purified rhomboid (at 0.125 mg/ml in buffer F) was added to 14 μl 4 × assay buffer (200 mM HEPES/NaOH, pH 7.4, 40% glycerol and 200 mM EDTA) and subsequently supplemented with 1 μl substrate mixture. Where indicated, 3,4-DCI (Calbiochem) from a 50 × stock in DMSO was added. ATP was depleted by the addition of 0.3 U of hexokinase and 1 mM glucose; for ATP supplementation, 2 μl of ATP mix was added (12.5 mM ATP, 3.5 U/μl creatine kinase, and 100 mM creatine phosphate, 50 mM HEPES-KOH, pH 7.4). Samples were incubated at 30°C as outlined: 2 h for E. coli membrane fractions (Figure 1A); 1 h for detergent-solubilised proteins (Figures 1 and 2); 30 min and 1 h for samples from the YqgP and GlpG purification, respectively (Figure 3B); 2 h for Figure 4A; as indicated for Figure 4B and C. Proteins were next precipitated with 10% trichloroacetic acid and samples were prepared for SDS–PAGE as described below.
SDS–PAGE and Western blotting
Recombinant rhomboids were analysed by SDS–PAGE using Tris/Glycine polyacrylamide gels (4–20% for Figures 1B, E and 2D; 12% for Figures 3B and 4F), followed either by Western blot analysis with an anti-polyhistidine antibody (1/3000) (Sigma) or by Coomassie Blue staining. Protein samples were mixed with SDS-sample buffer (containing 2% β-mercaptoethanol) and loaded on the gel without prior heating. Radiolabelled peptides were analysed by 10% BisTris/MES SDS–PAGE (Invitrogen). The trichloroacetic acid-precipitated peptides were washed with acetone and dissolved in SDS-sample buffer by incubation at 65°C for 15 min. Following electrophoresis, gels were incubated in fixation buffer (0.4 M borate/phosphate, pH 6.2, 0.25% glutaraldehyde) for 2 × 15 min, followed by incubation with wash buffer (40% methanol, 10% acetic acid) for 10 min. Gels were dried and labelled proteins were visualised by a Typhoon 8600 Phosphorimager (Molecular Dynamics). Quantification of substrate turnover was performed using the software ImageQuant 5.2. [35S]labelled reference peptides corresponding to the N-terminal portion of the substrate peptide Gurken-TMD (Figure 1G) were generated by cell-free in vitro translation using wheat germ extract (Promega) as described (Lemberg and Martoglio, 2003).
Acknowledgments
We thank Aled Edwards, Chris Tate and Markus Zettl for much advice; and Angus McQuibban and Sean Munro for their helpful comments on the manuscript. MKL is a recipient of an EMBO Long Term Fellowship. Work in CK's lab is supported by Genome Canada and the Ontario Genomics Institute.
References
- Akiyama Y, Kanehara K, Ito K (2004) RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J 23: 4434–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100: 391–398 [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-secretase complex. Neuron 38: 9–12 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387–390 [DOI] [PubMed] [Google Scholar]
- Duncan EA, Dave UP, Sakai J, Goldstein JL, Brown MS (1998) Second-site cleavage in sterol regulatory element-binding protein occurs at transmembrane junction as determined by cysteine panning. J Biol Chem 273: 17801–17809 [DOI] [PubMed] [Google Scholar]
- Esser K, Tursun B, Ingenhoven M, Michaelis G, Pratje E (2002) A novel two-step mechanism for removal of a mitochondrial signal sequence involves the mAAA complex and the putative rhomboid protease Pcp1. J Mol Biol 323: 835–843 [DOI] [PubMed] [Google Scholar]
- Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, Wolfe MS (2004a) Detergent-dependent dissociation of active gamma-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry 43: 323–333 [DOI] [PubMed] [Google Scholar]
- Fraering PC, Ye W, Strub JM, Dolios G, LaVoie MJ, Ostaszewski BL, van Dorsselaer A, Wang R, Selkoe DJ, Wolfe MS (2004b) Purification and characterization of the human gamma-secretase complex. Biochemistry 43: 9774–9789 [DOI] [PubMed] [Google Scholar]
- Gallio M, Sturgill G, Rather P, Kylsten P (2002) A conserved mechanism for extracellular signaling in eukaryotes and prokaryotes. Proc Natl Acad Sci USA 99: 12208–12213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedstrom L (2002) Serine protease mechanism and specificity. Chem Rev 102: 4501–4524 [DOI] [PubMed] [Google Scholar]
- Herlan M, Bornhovd C, Hell K, Neupert W, Reichert AS (2004) Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol 165: 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Rogozin IB, Davidovic L, Letellier MC, Pellegrini L (2003) The rhomboids: a nearly ubiquitous family of intramembrane serine proteases that probably evolved by multiple ancient horizontal gene transfers. Genome Biol 4: R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157: 105–132 [DOI] [PubMed] [Google Scholar]
- Lee JR, Urban S, Garvey CF, Freeman M (2001) Regulated intracellular ligand transport and proteolysis controls EGF signal activation in Drosophila. Cell 107: 161–171 [DOI] [PubMed] [Google Scholar]
- Lemberg MK, Martoglio B (2003) Analysis of polypeptides by sodium dodecyl sulfate–polyacrylamide gel electrophoresis alongside in vitro-generated reference peptides. Anal Biochem 319: 327–331 [DOI] [PubMed] [Google Scholar]
- Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, Register RB, Sardana MK, Shearman MS, Smith AL, Shi XP, Yin KC, Shafer JA, Gardell SJ (2000) Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: 689–694 [DOI] [PubMed] [Google Scholar]
- Lilley BN, Ploegh HL (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429: 834–840 [DOI] [PubMed] [Google Scholar]
- Lohi O, Urban S, Freeman M (2004) Diverse substrate recognition mechanisms for rhomboids; thrombomodulin is cleaved by mammalian rhomboids. Curr Biol 14: 236–241 [DOI] [PubMed] [Google Scholar]
- Martoglio B, Golde TE (2003) Intramembrane-cleaving aspartic proteases and disease: presenilins, signal peptide peptidase and their homologs. Hum Mol Genet 12 (Suppl 2): R201–R206 [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Saurya S, Freeman M (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423: 537–541 [DOI] [PubMed] [Google Scholar]
- Opitz C, Di Cristina M, Reiss M, Ruppert T, Crisanti A, Soldati D (2002) Intramembrane cleavage of microneme proteins at the surface of the apicomplexan parasite Toxoplasma gondii. EMBO J 21: 1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paetzel M, Dalbey RE (1997) Catalytic hydroxyl/amine dyads within serine proteases. Trends Biochem Sci 22: 28–31 [DOI] [PubMed] [Google Scholar]
- Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, Golde TE (2002) Identification of a novel family of presenilin homologues. Hum Mol Genet 11: 1037–1044 [DOI] [PubMed] [Google Scholar]
- Rawson RB, Zelenski NG, Nijhawan D, Ye J, Sakai J, Hasan MT, Chang TY, Brown MS, Goldstein JL (1997) Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol Cell 1: 47–57 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D (2003) Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43: 545–584 [DOI] [PubMed] [Google Scholar]
- Simon SM, Blobel G (1992) Signal peptides open protein-conducting channels in E. coli. Cell 69: 677–684 [DOI] [PubMed] [Google Scholar]
- Steiner H, Romig H, Pesold B, Philipp U, Baader M, Citron M, Loetscher H, Jacobsen H, Haass C (1999) Amyloidogenic function of the Alzheimer's disease-associated presenilin 1 in the absence of endoproteolysis. Biochemistry 38: 14600–14605 [DOI] [PubMed] [Google Scholar]
- Sturtevant MA, Roark M, Bier E (1993) The Drosophila rhomboid gene mediates the localized formation of wing veins and interacts genetically with components of the EGF-R signaling pathway. Genes Dev 7: 961–973 [DOI] [PubMed] [Google Scholar]
- Urban S, Freeman M (2003) Substrate specificity of rhomboid intramembrane proteases is governed by helix-breaking residues in the substrate transmembrane domain. Mol Cell 11: 1425–1434 [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M (2001) Drosophila Rhomboid-1 defines a family of putative intramembrane serine proteases. Cell 107: 173–182 [DOI] [PubMed] [Google Scholar]
- Urban S, Lee JR, Freeman M (2002a) A family of Rhomboid intramembrane proteases activates all Drosophila membrane-tethered EGF ligands. EMBO J 21: 4277–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Schlieper D, Freeman M (2002b) Conservation of intramembrane proteolytic activity and substrate specificity in prokaryotic and eukaryotic rhomboids. Curr Biol 12: 1507–1512 [DOI] [PubMed] [Google Scholar]
- Wasserman JD, Urban S, Freeman M (2000) A family of rhomboid-like genes: Drosophila rhomboid-1 and roughoid/rhomboid-3 cooperate to activate EGF receptor signalling. Genes Dev 14: 1651–1663 [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B (2002) Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296: 2215–2218 [DOI] [PubMed] [Google Scholar]
- Weihofen A, Lemberg MK, Ploegh HL, Bogyo M, Martoglio B (2000) Release of signal peptide fragments into the cytosol requires cleavage in the transmembrane region by a protease activity that is specifically blocked by a novel cysteine protease inhibitor. J Biol Chem 275: 30951–30956 [DOI] [PubMed] [Google Scholar]
- Weihofen A, Martoglio B (2003) Intramembrane-cleaving proteases: controlled liberation of proteins and bioactive peptides. Trends Cell Biol 13: 71–78 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Kopan R (2004) Intramembrane proteolysis: theme and variations. Science 305: 1119–1123 [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398: 513–517 [DOI] [PubMed] [Google Scholar]
- Ye J, Dave UP, Grishin NV, Goldstein JL, Brown MS (2000) Asparagine–proline sequence within membrane-spanning segment of SREBP triggers intramembrane cleavage by site-2 protease. Proc Natl Acad Sci USA 97: 5123–5128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA (2004) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429: 841–847 [DOI] [PubMed] [Google Scholar]