

Abstract
The X-linked inhibitor of apoptosis protein (XIAP) uses its second baculovirus IAP repeat domain (BIR2) to inhibit the apoptotic executioner caspase-3 and -7. Structural studies have demonstrated that it is not the BIR2 domain itself but a segment N-terminal to it that directly targets the activity of these caspases. These studies failed to demonstrate a role of the BIR2 domain in inhibition. We used site-directed mutagenesis of BIR2 and its linker to determine the mechanism of executioner caspase inhibition by XIAP. We show that the BIR2 domain contributes substantially to inhibition of executioner caspases. A surface groove on BIR2, which also binds to Smac/DIABLO, interacts with a neoepitope generated at the N-terminus of the caspase small subunit following activation. Therefore, BIR2 uses a two-site interaction mechanism to achieve high specificity and potency for inhibition. Moreover, for caspase-7, the precise location of the activating cleavage is critical for subsequent inhibition. Since apical caspases utilize this cleavage site differently, we predict that the origin of the death stimulus should dictate the efficiency of inhibition by XIAP.
Keywords: apoptosis, BIR domain, caspase, IAP, protease
Introduction
The importance of the caspase family of proteases in apoptosis is well documented by biochemical, cell biologic and genetic studies (reviewed in Nicholson, 1999; Fesik and Shi, 2001; Denault and Salvesen, 2002). Mammals have developed regulatory proteins, members of the inhibitor of apoptosis (IAP) family, which target a subset of these enzymes. The prototype member of the family, X-linked IAP (XIAP), contains three distinct baculovirus IAP repeat (BIR) domains and a C-terminal RING finger. This protein inhibits caspases at both the initiation phase (caspase-9) and the execution phase (caspase-3 and -7) of apoptosis. The second BIR domain (BIR2) inhibits caspase-3 and -7, while the third BIR domain (BIR3) inhibits caspase-9 (Deveraux et al, 1999). The RING domain is an E3 ubiquitin ligase, promoting proteasomal degradation of XIAP, caspase-3 and second mitochondrial activator of caspases (Smac) also known as direct IAP binding protein with low pI (DIABLO; Du et al, 2000; Verhagen et al, 2000; Yang et al, 2000; Suzuki et al, 2001b; MacFarlane et al, 2002; Shin et al, 2003). No function has been attributed to the first BIR domain of XIAP.
Crystal structures of BIR–caspase complexes have elucidated much of what we understand about the mechanism of XIAP inhibition of caspases. Intriguingly, despite sharing 40% identity, the individual BIR domains seem to have evolved different strategies for inhibiting the same class of protease. BIR3 inhibits caspase-9 by taking advantage of the fact that active caspases are obligate dimers (Renatus et al, 2001; Shiozaki et al, 2003). The C-terminal helix of BIR3 enforces an interface with the newly revealed caspase-9 dimer interface, locking this initiator caspase in its inactive monomeric state (Shiozaki et al, 2003). This state is secured by binding a neoepitope (encompassing the N-terminus of the small subunit) to a surface groove on BIR3. This negatively charged surface groove is conserved in many BIR domains across species. We refer to this surface region as the ‘IBM interacting groove', because of its ability to bind to the extreme N-terminal IAP binding motif (IBM) of IBM containing proteins. These include the mammalian IAP antagonists Smac/DIABLO and HtrA2, and the Drosophila Hid, Grim and Reaper proteins (reviewed in Salvesen and Duckett, 2002; Vaux and Silke, 2003). The two essential units of BIR3 in this interaction are the IBM interacting groove and the C-terminal helix.
In contrast, the structures of BIR2 in complex with either caspase-3 or -7 reveal an inhibitory mechanism that seems to be unrelated to BIR3 and caspase-9 (Chai et al, 2001; Huang et al, 2001; Riedl et al, 2001b). Protease inhibition is achieved through binding the region immediately N-terminal to the BIR2 domain (linker) across the substrate groove in a reverse orientation with respect to substrate binding. This binding prevents caspase substrates from interacting with the catalytic machinery by steric occlusion. The essential unit in this interaction is the linker region preceding BIR2.
Whereas the interaction between caspase-9 and the IBM interacting groove on BIR3 is essential for binding and inhibition, there is surprisingly very little contact between the BIR2 domain and caspase-3 or -7. In fact, no electron density for the entire BIR2 domain was observed in the two structures with caspase-7 (Chai et al, 2001; Huang et al, 2001). Despite the lack of structure-based evidence, there are mounting indications that the BIR2 domain itself contributes to inhibition. Recombinant caspase-7 binds to a linker-deleted BIR2, although with lower affinity than to linker-BIR2 (Huang et al, 2001; Suzuki et al, 2001a). Indeed, the only structure of a caspase/BIR2 complex in which the BIR domain is visible reveals two other potential interactions. Within the asymmetric crystal unit, methionine 182 of caspase-3 docks into a hydrophobic pocket formed by Y154 and F228 of BIR2. An additional interaction is reminiscent of caspase-9/BIR3 complex, with the N-terminus of a caspase-3 small subunit docked into an equivalent surface groove on BIR2 (Riedl et al, 2001b). Although this binding was not observed within a biologically functional unit but as a crystal contact between symmetry mates, NMR studies also show significant chemical-shift changes in this part of the BIR2 domain when bound to caspase-3 (Sun et al, 1999). In our report on the structure of the caspase-3/BIR2 complex, we predicted that the putative IBM interacting groove of BIR2 may play a role in complex stabilization (Riedl et al, 2001b).
Based on conservation of mechanism, we hypothesize that the caspase-3 and -7 inhibitory mechanism of XIAP should include mechanistic components similar to those demonstrated in caspase-9 inhibition. If this is true, the IBM interacting groove of BIR2 should play a role in inhibition. This hypothesis would require a revision of the structure-based predictions for XIAP function, and will have consequences for therapeutic intervention in the caspase/IAP axis. To test this hypothesis, we dissected the inhibitory mechanism of caspase-3 and -7 with XIAP by focusing on the role of both the BIR2 domain and putative interaction sites on caspase-3 and -7.
Results
The BIR2 linker inhibits executioner caspases weakly
Mutagenesis and structural studies indicate that caspase-3 and -7 inhibition by XIAP is achieved through binding the linker region preceding BIR2 (linker-BIR2) across the substrate binding cleft of the protease (Sun et al, 1999; Chai et al, 2001; Huang et al, 2001; Riedl et al, 2001b). To determine whether the capacity of XIAP to inhibit caspase-3 and -7 is confined to this region, a linker peptide corresponding to residues 124–168 of XIAP was synthesized and kinetics of caspase inhibition analyzed. Previous studies showed that D148 is crucial for inhibition, so a control peptide containing asparagine at this position was also tested. A similar peptide (residues 134–154) has been investigated by others and it was concluded that it was insufficient for caspase inhibition (Sun et al, 1999). In contrast, our peptide inhibited with a Ki of 9–11 μM, demonstrating that it is sufficient for weak inhibition (Figure 1B and Table I). HPLC confirmed that it was not a substrate because it was not significantly cleaved after 3 h with 1 μM caspase-3 (data not shown). As expected, a control peptide (Asp148Asn) was even less efficient at caspase inhibition (Table I).
Figure 1.
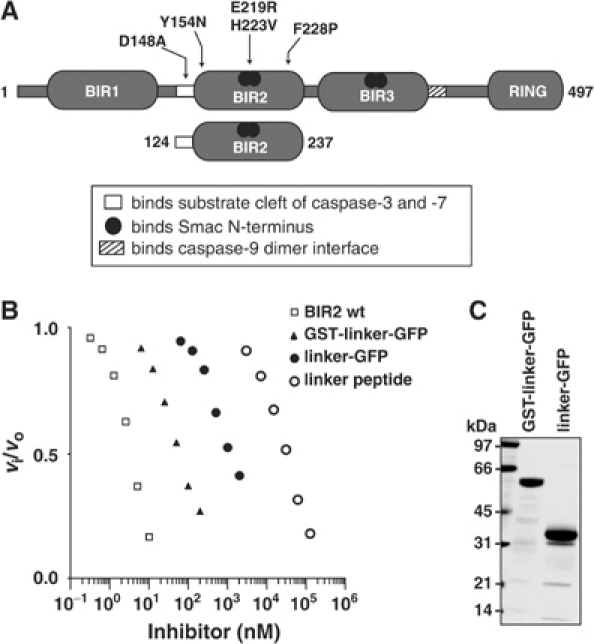
The BIR2 linker is a weak caspase-3 inhibitor. (A) Schematic diagram of XIAP and mutants used in this study. (B) Analysis of caspase-3 inhibition. A 100 pM portion of caspase-3 was preincubated with varying inhibitor concentrations at 37°C for 30 min and residual enzyme activity was analyzed by Ac-DEVD-afc hydrolysis. Relative activity is expressed as the ratio of inhibited to uninhibited enzyme activity (vi/vo). (C) Integrity of GST-linker-GFP and linker-GFP.
Table 1.
Inhibition constant (Ki) for linker proteins/peptides with caspases
| Caspase-3 (nM) | Caspase-7 (nM) | Oligomer status | ||
|---|---|---|---|---|
| BIR2 | <0.4 | <0.05 | Monomer | |
| Linker peptide | 9000 | 11 000 | ND | |
| Control peptide D148N | 319 000 | >100 000 | ND | |
| Linker-GFP | 276 | 632 | Monomer | |
| GST-linker-GFP | 12 | 51 | Trimer/oligomer | |
| ND: not determined. | ||||
We also produced GST-linker-GFP and linker-GFP (Figure 1C). We used these to test the hypothesis that, to be available for active site binding, the linker needs to be stabilized by a structured protein at both N- and C-termini (similar to its environment within full-length XIAP) or at the C-terminus only (like BIR2 alone). Replacing BIR2 with GFP did not restore full inhibitory function (Figure 1B and Table I). Including GST at the N-terminus improved inhibition 12- to 23-fold, yet it still fell short by 30- to 1000-fold compared to linker-BIR2. These data are in line with other reports using GST-linker chimeras: IC50 of 100 nM (Chai et al, 2001) and Kd of 35 nM (Huang et al, 2001) with caspase-7. GST-linker fusions have artificially high binding affinities, which may be due to oligomerization of the linker, driven by GST and confirmed by gel filtration analysis (Table I; Vargo et al, 2004). Importantly, our results demonstrate that the BIR2 domain itself, not just any carrier protein, endows the linker with tight binding inhibition of caspase-3 and -7. Our data also show that GST chimeras should be avoided in kinetic studies of caspase inhibition by IAPs because the oligomeric status leads to overestimation of inhibitory constants.
The cleavage position within the executioner caspase linker is important for efficient inhibition by BIR2 of XIAP
Cathepsin G (CatG) can activate procaspase-7 by cleavage at Q196 (Casp7-Q196), two residues upstream from the canonical activation site (D198) that is cleaved by Granzyme B (GraB) and initiator caspase-8 and -9 (Figure 2A; Zhou and Salvesen, 1997). The resulting small subunit N-terminus is ADSG—as opposed to SGPI—for GraB-activated caspase-7 (Casp7-D198). Kinetic characterization of Casp7-Q196 and Casp7-D198 shows that they are essentially identical enzymes (Supplementary Table I; Zhou and Salvesen, 1997). However, they differ dramatically in their interaction with XIAP and BIR2, which were unable to inhibit efficiently caspase-7 activated with CatG compared to GraB (Figure 2C and Table II). Loss of inhibitory activity was not due to degradation of XIAP or BIR2 by residual protease activity (Figure 2D). Therefore, like caspase-9, the position of cleavage within the executioner caspase linker is important for downstream regulation by XIAP (Srinivasula et al, 2001). We hypothesize that cleavage not only activates the caspase but also reveals a motif that interacts with the IBM interacting groove on BIR2, contributing to inhibition.
Figure 2.
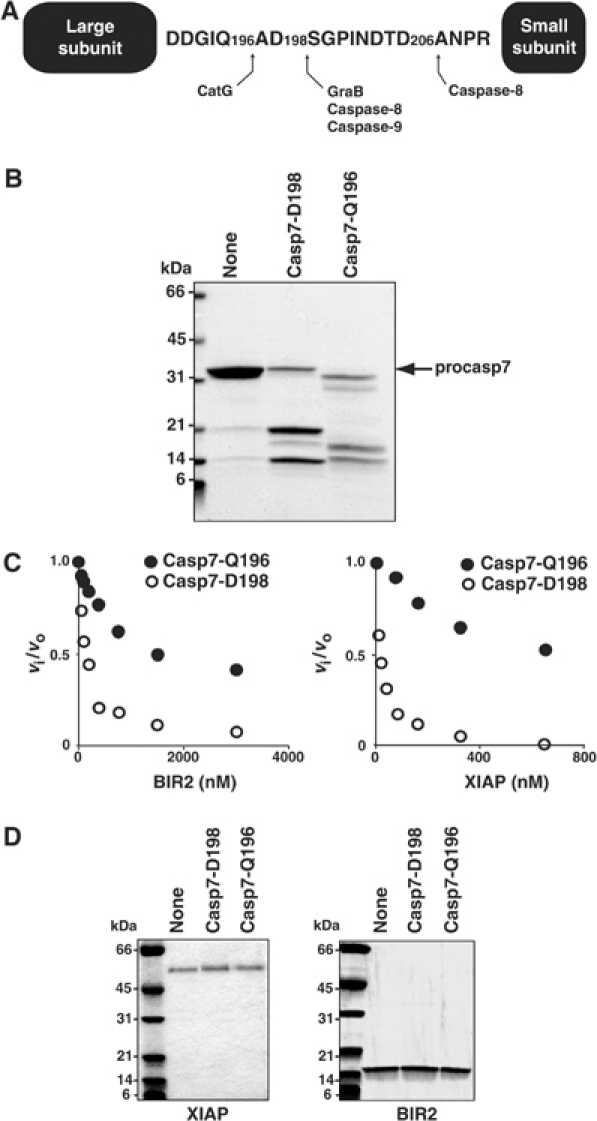
Caspase-7 must be activated by cleavage at the correct linker position for efficient inhibition by XIAP. (A) Schematic diagram of caspase-7 showing activating cleavage sites within the linker sequence (adapted from Zhou and Salvesen, 1997). (B) Procaspase-7 was activated by incubation with optimal amounts of the serine proteases GraB or CatG. A 20 μl portion of each reaction was analyzed by SDS–PAGE and stained with GELCODE Blue. (C) A 5 nM portion of active Casp7-D198 (○) or Casp7-D196 (•) was incubated with either BIR2 or XIAP for 15 min at 37°C in modified caspase buffer. Ac-DEVD-afc was added to each reaction and the relative activity expressed as the ratio of inhibited to uninhibited enzyme activity (vi/vo). (D) Samples from (C) containing the highest inhibitor concentration were analyzed by SDS–PAGE.
Table 2.
Inhibition constant (Ki, nM) for BIR2 with caspase-7 variants
| Casp7-Q196 | 287 |
| Casp7-D198 | 5.0 |
| Casp7-D206 | <0.3 |
Up to this point, our studies had been performed with recombinant caspase-7 that is processed at D198, with variable amounts of additional autocatalytic cleavage at D206 (Denault and Salvesen, 2003). The cleavage following D198 results in the liberation of SGPI—whereas the D206 cleavage results in ANPR—at the N-terminus of the small subunit (Figure 2A). We therefore asked whether processing at D198 or D206 differentially influences downstream regulation by BIR2 of XIAP.
We produced two different variants of caspase-7. Casp7-D198 was made by processing procaspase-7 with GraB. Casp7-D206 was generated by expression of a linker mutant where NDTD206 was replaced with IEPD206. This caspase-7 is cleaved at D198 followed rapidly by cleavage at D206 during expression in Escherichia coli, with removal of the S199-D206 peptide. Both enzymes contain an identical large subunit but differ in length and sequence of the small subunit N-terminus (Figure 3A). Characterization of these enzymes shows that they are catalytically equivalent (Supplementary Table I). However, BIR2 inhibits Casp7-D206 at least 10 times more efficiently than Casp7-D198 (Figure 3B and Table II). To determine whether this was due to an artifact of using a small synthetic substrate, we used a large protein substrate—the C2A mutant of baculovirus protein p35—which converts this caspase inhibitor to a substrate (Riedl et al, 2001c; Xu et al, 2001). Again, Casp7-D206 was inhibited more efficiently by BIR2 than Casp7-D198 (Figure 3C).
Figure 3.
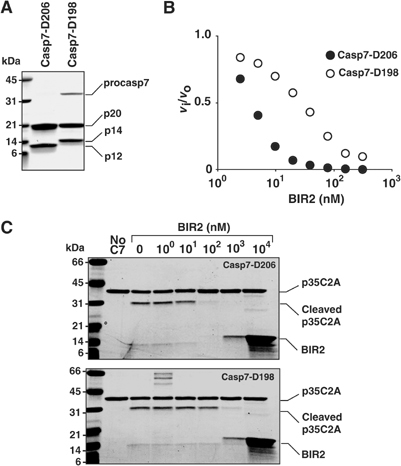
BIR2 is less efficient at inhibiting caspase-7 cleaved at D198 compared to D206. (A) Recombinant proteins were analyzed by SDS–PAGE. Casp7-D206: caspase-7 processed at both D198 and D206; Casp7-D198: caspase-7 cleaved at D198 with GraB. (B) A 5 nM portion of Casp7-D198 (○) or Casp7-D206 (•) was incubated with BIR2 in modified caspase buffer for 15 min at 37°C and 100 μM Ac-DEVD-afc was added to each reaction and the residual enzyme activity expressed as the ratio of inhibited to uninhibited enzyme activity (vi/vo). (C) A 5 nM portion of Casp7-D198 or Casp7-D206 was incubated with BIR2 for 15 min at 37°C. Recombinant p35 C2A (3 μM), a noninhibitory protein substrate, was added and incubated for a further 30 min at 37°C. Monitoring cleavage of p35 C2A by SDS–PAGE assessed residual enzyme activity. The data demonstrate that BIR2 is at least a 10-fold better inhibitor of Cas7-D206 than of Casp7-D198.
These data demonstrate that the position of cleavage within the caspase-7 interdomain linker is important for efficient inhibition by XIAP. In turn, this suggests that XIAP inhibition of executioner caspases involves a two-site binding mechanism. Within XIAP, the BIR2 linker binds directly across the active site of the caspase but with low affinity. We hypothesize that the BIR2 domain provides a second binding site that stabilizes the inhibitory interaction. A crystal structure of the caspase-3/BIR2 complex reveals two other potential interactions involving the BIR2 domain: (a) M182 of caspase-3 docks into a hydrophobic pocket formed by Y154 and F228 of BIR2 and (b) the caspase-3 small subunit N-terminus and the IBM interacting groove of BIR2 (Riedl et al, 2001b). We produced XIAP and BIR2 variants to test biochemically the contribution these interactions make to inhibition of caspase-3 and -7 (Figure 1A).
Caspase-3 M182 binding to BIR2 hydrophobic pocket does not contribute to inhibition
Methionine 182 of capase-3 forms interactions with Y154 and F228 of BIR2 (Riedl et al, 2001b). Considering there is no electron density for this region of BIR2 when it is bound to caspase-7, the significance of this interaction for executioner caspase inhibition remains unclear. Mutating either the caspase-3 interacting residue (M182A) or the BIR2 interacting residues (Y154N and F228P, converting them to the residues found in the BIR2 domain of cIAP1 to conserve structure) had no significant effect on caspase-3 and -7 inhibition, ruling out this interaction as significant in generating tight inhibition (Table III and Supplementary Figure 1).
Table 3.
Inhibition constant (Ki, nM) for XIAP mutants with caspases
| BIR2 (124–237) |
XIAP (1–497) |
|||
|---|---|---|---|---|
| Casp3 | Casp7 | Casp3 | Casp7 | |
| Wild-type | <0.4 | <0.05 | <0.8 | <0.07 |
| D148A | >1000 | 1.7 | >1000 | 1.1 |
| E219R H223V | 2.3 | 2.2 | 3.1 | 1.1 |
| D148A E219R H223V | >1000 | >1000 | >1000 | >1000 |
| Y154N | <0.4 | <0.05 | ND | ND |
| Y154N E219R H223V F228P | 2.8 | 3.5 | ND | ND |
| ND: not determined. | ||||
The IBM interacting groove of BIR2 is important for efficient inhibition of caspase-3 and -7
Based on the crystal contact interactions, single point mutations were introduced to ablate the putative IBM interacting groove on BIR2 (Figure 4A). To maintain structural integrity within the BIR domain, residues were mutated to corresponding residues found in other BIR domains (BIR1 of XIAP, or BIR2 of cIAP1). Unfolding studies with guanidinium chloride confirmed that BIR2 (124–237) mutants were folded and had conformational stabilities comparable to wild-type BIR2 (data not shown). Against caspase-3 and -7, all proteins containing wild-type BIR2 domain showed comparable inhibition constants: Ki<1 nM for caspase-3 and <0.1 nM for caspase-7 (Table III). These inhibition constants are comparable to previous reports (Deveraux et al, 1997; Takahashi et al, 1998; Sun et al, 1999; Riedl et al, 2001b; Silke et al, 2001; Suzuki et al, 2001a). It was not possible to measure the Ki more accurately because the interaction is too tight for the sensitivity of our detection system. XIAP and BIR2 possessing mutations in the IBM interacting groove were consistently weaker inhibitors of caspase-3 and -7 (Table III). In agreement with previous reports, mutation of D148A within the N-terminal BIR2 linker results in complete loss of caspase-3 inhibition (Sun et al, 1999; Suzuki et al, 2001a). By comparison, mutation of D148 had less impact on caspase-7 inhibition. This is consistent with GST pull-down experiments where caspase-7 co-precipitates with GST-XIAP D148A and GST-BIR2 D148A (Suzuki et al, 2001a). Strikingly, when D148A is combined with the E219R H223V surface groove mutations, complete loss of caspase-7 inhibition occurs. These data show that the IBM interacting groove on BIR2 is a secondary binding site contributing to the overall efficiency of inhibition, and may be more important for caspase-7 than caspase-3.
Figure 4.
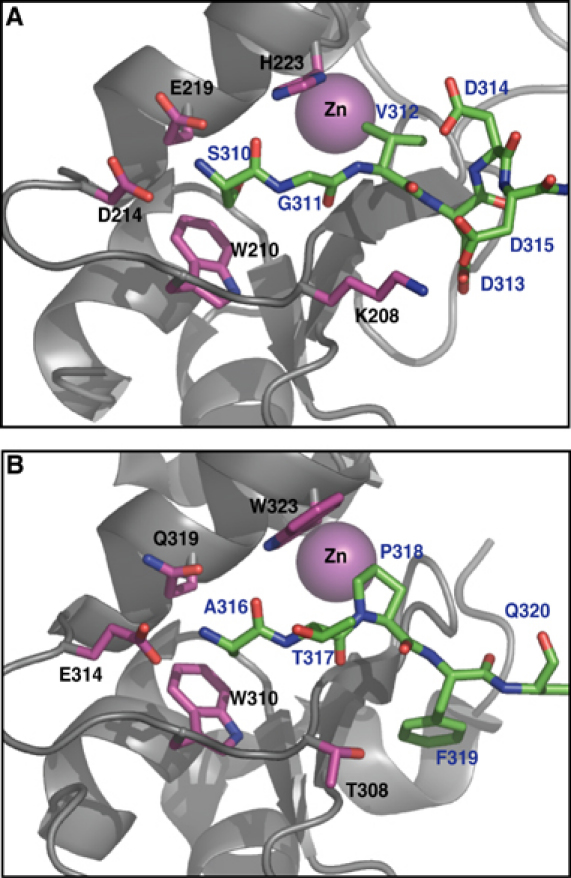
(A) As a crystal contact interaction between asymmetric units, the caspase-3 small subunit N-termini (SGVDDD315; green sticks) docks into a conserved surface groove on the BIR2 domain (taken from PDB 1I3O; Riedl et al, 2001b). This binding mode is almost identical to (B) caspase-9 small subunit N-termini (green sticks) binding to the analogous surface groove on BIR3 of XIAP (taken from PDB 1NW9; Shiozaki et al, 2003). BIR domains are in gray trace with interacting residues in magenta. The BIR-coordinated zinc atom does not directly contact the ligands, but is included as reference.
Biological significance of the two proposed interaction sites
We tested the significance of the IBM interacting groove in cells triggered to undergo apoptosis via the extrinsic apoptosis pathway or by a direct executioner caspase route. A caspase activity assay (DEVDase) and morphology assay (Annexin V binding) were used to assess apoptosis, and equivalent expression of each XIAP mutant was confirmed by Western blot (Figure 5A). The XIAP IBM interacting groove mutant E219R H223V is less effective than wild-type XIAP at inhibiting endogenously activated executioner caspases (primarily, caspase-3) as assessed by hydrolysis of Ac-DEVD-afc (Figure 5A). However, there was no significant difference in cell death protection between wild-type and E219R H223V mutant in response to exogenous TRAIL or Fas/CD95 transfection (Figure 5B and C). In contrast, the D148A linker mutation completely abrogates caspase inhibition and reduces protection from apoptosis. Significantly, when both binding sites are abrogated (D148A E219R H223V), there is an enhancement of caspase activity and cell death in response to TRAIL (Figure 5A and B). To address specifically caspase-7 regulation, we transfected caspase-7 minus its regulatory N-peptide into 293A cells. This enzyme induces apoptosis without activating any other caspases (Denault and Salvesen, 2003). In response to ectopic expression of active caspase-7, compared to wild-type XIAP the E219R H223V mutant was as defective as the D148A mutant in protecting from cell death (P>0.05; Figure 5D). In summary, the importance of both the linker and IBM interacting groove of BIR2 for executioner caspase inhibition was confirmed in the context of dying cells.
Figure 5.
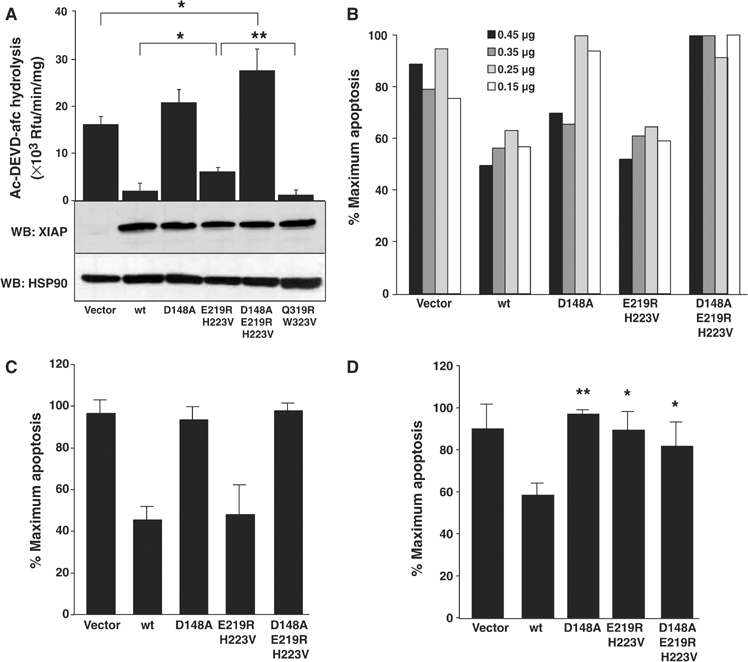
Ablation of the IBM interacting groove of BIR2 reduces XIAP's potency as an executioner caspase inhibitor. (A) 293A cells were transfected with 0.5 μg of myc-XIAP wild-type or mutant constructs, treated with 100 ng/ml TRAIL for 2 h and lysed in mRIPA buffer. One-tenth of the lysate was added to 100 μM Ac-DEVD-afc. Initial rates were analyzed and normalized for protein amount. Lysates from duplicate, untreated transfectants were balanced for equal protein, electrophoresed by reducing SDS–PAGE and immunoblotted with mouse anti-XIAP or mouse anti-HSP90 antibody as a loading control. 293A cells were (B) transfected with 0.45, 0.35, 0.25 or 0.15 μg myc-XIAP variant plasmid and treated with 100 ng/ml TRAIL for 2 h; (C) cotransfected with 0.5 μg Fas and 0.5 μg myc-XIAP mutants; (D) cotransfected with 0.25 μg ΔN-caspase-7 and 2.75 μg myc-XIAP mutants. (B–D) FACS analysis of Annexin V-PE-stained cells was performed. (A, C, D) Data were analyzed with a two-tailed paired Student's t-test (n=3; *P<0.05; **P<0.01). For (D), P-values are derived from comparison with wild-type XIAP.
The IBM interacting grooves on BIR2 and BIR3 are similar in surface charge and topography. Consequently, there is a formal possibility that the BIR3 groove may also contribute to caspase-3 or -7 inhibition in the context of full-length XIAP. Mutation of this groove (Q319R W323V) had no effect on protection from TRAIL-induced apoptosis (Figure 5A) or on caspase-3 and -7 inhibition in kinetic assays with recombinant protein, while losing all affinity for caspase-9 (data not shown). This is in agreement with the finding that caspase-3 and -9 can bind XIAP simultaneously (Bratton et al, 2002).
BIR2 binds ANPR-Smac with higher affinity than SGPI-Smac
We propose that the N-terminus of the inhibited caspase small subunit occupies the IBM interacting groove of BIR2 and that this interaction stabilizes binding of the BIR2 linker across the catalytic binding site. We used wild-type Smac, and mutants containing various N-termini, to assess the binding specificity of BIR2. To confirm that Smac binds at this site, we performed a co-precipitation assay with GST-BIR2 mutated at its IBM interacting groove (E219R H223V) and demonstrated that Smac no longer binds (Figure 6A). We then asked whether the IBM interacting groove in BIR2 could bind sequences corresponding to the N-terminus of the two alternatively cleaved caspase-7 small subunits (ANPR and SGPI). MVPI-Smac was used as a negative control (Chai et al, 2000). ANPR-Smac binds to GST-BIR2 as efficiently as wild-type Smac under these conditions (AVPI; Figure 6C). SGPI-Smac bound less efficiently, which agrees with the observation that Ala and Ser are tolerated at the N-terminus of BIR2 binding peptides from combinatorial libraries, but Ser is less represented than Ala (BE Turk and LM Martins, personal communication). These results also explain why Casp7-D198 is more poorly inhibited by BIR2 compared to Casp7-D206 (Figure 3). Finally, we asked whether a Smac IBM peptide could antagonize caspase-3 inhibition by BIR2 E219R H223V. Smac 7-mer efficiently interferes with enzyme inhibition (Figure 6D). This indicates that Smac peptide and caspase-3 compete for a shared binding site on BIR2. Strikingly, caspase inhibition by BIR2 E219R H223V was unaffected. We conclude that Smac/DIABLO directly competes with the executioner caspase small subunit for binding to the IBM interacting groove on BIR2, again demonstrating the significance of this second binding component for caspase-3 and -7 inhibition.
Figure 6.
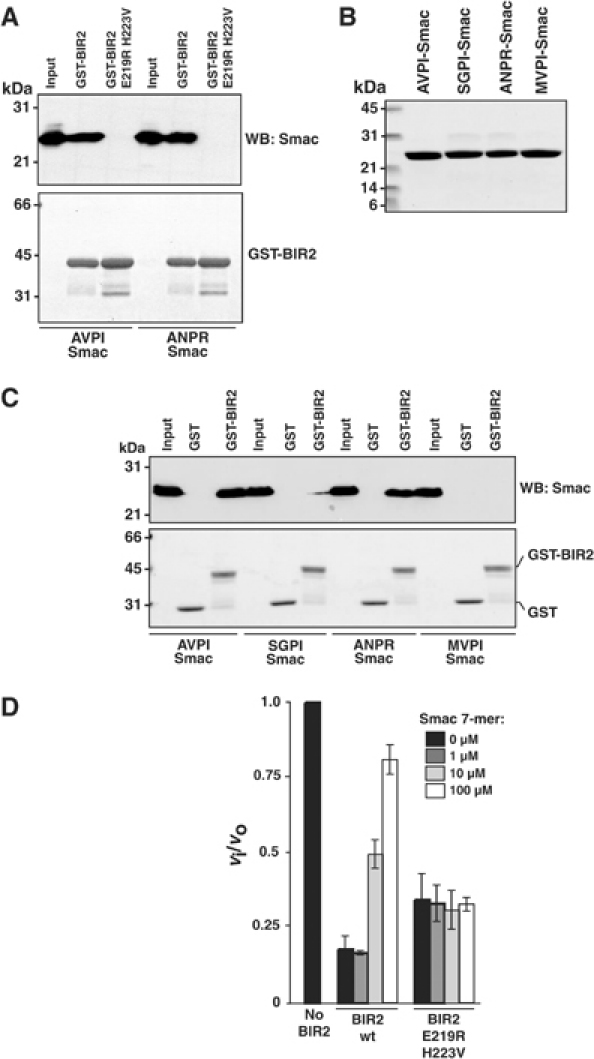
Specificity of the IBM interacting groove of BIR2. (A) A 5 μl portion of GST-BIR2 beads or GST-BIR2 E219R H223V beads was incubated with 100 nM AVPI-Smac or ANPR-Smac in binding buffer for 30 min at 4°C and bound proteins eluted by boiling in SDS sample buffer. Samples were analyzed by SDS–PAGE. ‘Input' corresponds to 20% of the total input prior to addition of beads. Top panel: immunoblot with rabbit anti-Smac antibody; bottom panel: GELCODE Blue stain of corresponding gel. (B) SDS–PAGE analysis of recombinant Smac mutants. (C) A 5 μl portion of GST or GST-BIR2 Sepharose beads was incubated with 100 nM AVPI-Smac, SGPI-Smac, ANPR-Smac or MVPI-Smac in binding buffer for 30 min at 4°C. Samples were processed as in (A). (D) A 300 pM portion of caspase-3 was incubated for 30 min with 5 nM wild-type BIR2 or 30 nM BIR2 E219R H223V in the presence of 0, 1, 10 or 100 μM Smac 7-mer (AVPIAQK). Ac-DEVD-afc (100 μM) was added and the residual enzyme activity expressed as the ratio of inhibited to uninhibited enzyme activity (vi/vo).
Discussion
The fundamental mechanism of specific protein interactions is usually conserved during protein evolution. According to conservation of mechanism, the two units of XIAP that inhibit caspases should preserve a fundamental interaction strategy. On the basis of structural studies, this concept could be questioned. The key elements of caspase-9 inhibition by BIR3 are the IBM interacting groove and the C-terminal helix (Shiozaki et al, 2003). In contrast, the key element of caspase-3 and -7 inhibition by BIR2 seems to be the completely nonconserved N-terminal linker region (Chai et al, 2001; Huang et al, 2001; Riedl et al, 2001b). The most conserved surface structure of BIR domains is the IBM interacting groove. It is found on many BIR domains including the BIR2 and BIR3 of XIAP, and the BIR1 and BIR2 of an ortholog in Drosophila melanogaster, DIAP1. The surface groove of DIAP1 BIR2 is involved in binding and ubiquitination of Dronc, the initiator caspase in flies (Wilson et al, 2002; Chai et al, 2003). In addition, the IBM interacting groove of DIAP1 BIR1 is absolutely required for inhibition of the executioner caspase DrICE by an unknown mechanism (Yan et al, 2004). We suggest the IBM interacting groove is a conserved interaction element of BIR domains and that for XIAP BIR2 it confers tight inhibition of caspase-3 and -7 by providing a second binding site.
A clue that the conserved IBM interacting groove may be important for executioner caspase inhibition is seen in the structure of caspase-3 bound to XIAP BIR2. The small subunit N-terminus is bound to the IBM interacting groove (Riedl et al, 2001b). This interaction does not occur within a biologically functional unit (i.e. one caspase-3 dimer and two BIR2 molecules) but across two, symmetry-related, biologically functional units throughout the crystal (Figure 7). This ‘domain swapping' may be necessary to stabilize crystal packing. We propose that the interaction seen in this crystal structure is not what occurs in solution. Rather the caspase small subunit N-terminus binds the IBM interacting groove of the BIR domain, inhibiting the adjacent catalytic domain within the caspase-3 dimer. Only minimal rearrangements of the caspase are required to achieve this interaction (Figure 7). It is important to understand why this interaction was also not seen in the structures of the BIR2/caspase-7 complex (Chai et al, 2001; Huang et al, 2001). Interestingly, the entire BIR2 domain was not observed in the crystal structures, despite being present during crystallization. Both structures lose resolution beyond residue 152. This means that the entire BIR domain is in motion in the crystals, with a flexible axis around residue 153. In fact, the BIR2 domain is within a solvent channel in these crystals and does not participate in crystal packing. However, an overlay of the BIR2 domain from the caspase-3/BIR2 complex reveals that it is possible for the caspase-7 N-terminus to proceed into the surface groove of BIR2 without causing steric clashes. The reason this interaction is missing in the crystal may be due to the preferred crystallization of molecules that lack this interaction, perhaps forced by crystal packing. It is a rarely publicized facet of protein crystallography that molecules that crystallize do not necessarily represent the most prevalent conformation in solution. In this context, neither the BIR2/caspase-7 nor BIR2/caspase-3 structures reveal the solution binding mechanism. Maybe crystals can only form when the BIR2 IBM interacting groove is occupied in a domain swapped orientation.
Figure 7.
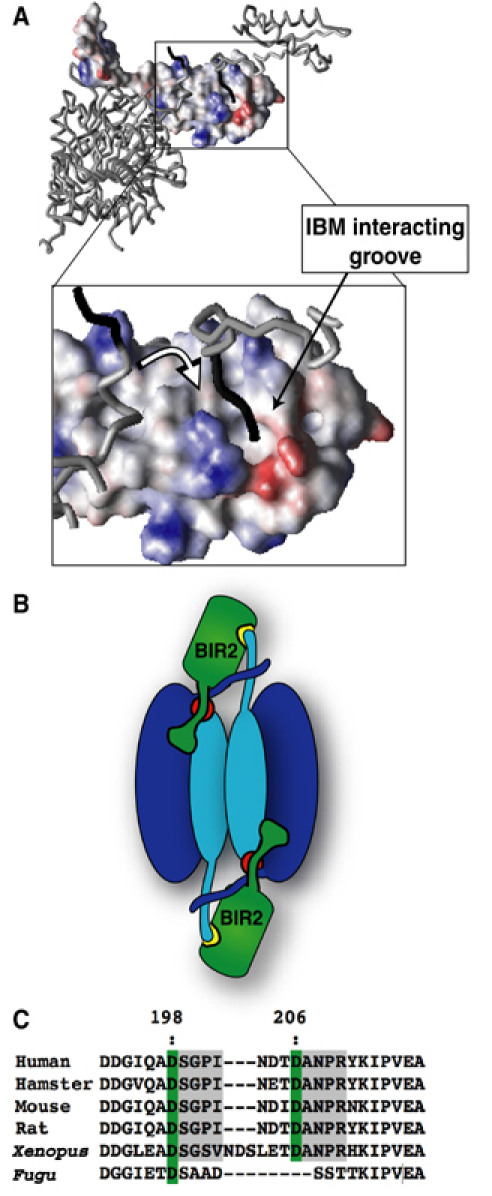
Proposed two-site binding model for caspase-3 and -7 inhibition by XIAP. The BIR2 domain is shown in surface representation (A), with the N-terminus of caspase-3 small subunit lining the IBM interacting groove in a ‘domain swapping' mode, as seen in the crystal structure of caspase-3/BIR2 (Riedl et al, 2001b). We propose that in solution the contact is made by the small subunit N-terminus of the domain that forms a partner to the inhibited domain (see white arrow in the inset). The model is more clearly seen in a cartoon (B), where the red circles represent the catalytic sites of a caspase dimer, and the yellow patch represents the IBM interacting groove on BIR2. Panel (C) highlights the degree of species conservation across the interdomain linker of caspase-7. The distant Fugu protein is probably close to the ancestor of both caspase-3 and -7.
We propose that BIR2 and caspase-3 or -7 binding complies with a two-site interaction model where each site contributes to the overall binding affinity, and therefore inhibitory strength. The first site is the BIR2 linker, which binds weakly across the active site with a Kd of ∼10 μM (Figure 1 and Table I). This interaction inhibits enzyme activity. The second site at least partly comprises the IBM interacting groove, which binds the N-terminus of the caspase small subunit. This is supported by a significant loss of inhibitory strength when the IBM interacting groove is mutated or occupied by Smac peptide (Table III and Figure 6). In such two-site interactions, the overall Kd will be a product of the individual values for each site. Entropy is decreased as a consequence of converting a bimolecular reaction to a unimolecular one. This would place the overall Kd (or Ki) in the high picomolar to low nanomolar range, in line with our experimental observations (Tables II and III).
The two-site model also explains how compounds identified from a chemical library screen can disrupt caspase-3 inhibition and antagonize XIAP in cells (Wu et al, 2003). Modeling of the most potent antagonist suggests that it anchors into the IBM interacting groove and also occupies a hydrophobic pocket comprising Y154 and F228. The authors suggest that the latter interaction disrupts caspase-3 inhibition. However, our data showed that these residues do not contribute to caspase inhibition (Table III and Supplementary Figure 1). We suggest that this compound antagonizes caspase inhibition by directly competing with the small subunit N-terminus for the IBM interacting groove, and the hydrophobic moiety increases binding affinity of the overall molecule for BIR2.
The two-site binding mechanism would also account for the finding that Smac/DIABLO, which is dimeric, requires both the BIR2 and BIR3 domains for efficient antagonism of apoptosis (Chai et al, 2000; Huang et al, 2003). One model for the mechanism of Smac/DIABLO function is that the IBM of each monomer within the dimer binds to the BIR2 and BIR3 domains within one XIAP molecule, and that the BIR2 N-terminal linker may be ‘hidden' underneath the Smac/DIABLO dimer interface, making it inaccessible to the caspase-3 or -7 active site (Vaux and Silke, 2003). Our data show that the Smac IBM binds directly to the surface groove on BIR2 and that Smac peptide is able to antagonize inhibition in the absence of a full-length Smac dimer (Figure 6). Although high concentration of Smac 7-mer was required to antagonize linker-BIR2 (Kd 9.4±0.6 μM), full-length Smac dimer binds to XIAP BIR2+BIR3 (124–356) with a Kd of 316 pM (Liu et al, 2000; Huang et al, 2003). We propose that in vivo, Smac/DIABLO antagonizes XIAP inhibition of caspase-3 and -7 by directly competing for the small subunit N-terminus in a similar manner to caspase-9 antagonism.
Our evidence that the caspase small subunit N-terminus is the other component of the second binding site comes in part from the drastic weakening of inhibition when caspase-7 displays a different small subunit N-terminus. This is achieved by activating the zymogen by cleavage upstream of the normal processing sites (Figure 2). It is noteworthy that inhibitory strength of BIR2 increases as the site of interdomain processing is shifted from residues 196 to 198 to 206 in the caspase-7 interdomain linker. This could be due to differences in the affinity of the resultant N-terminal neoepitopes for the IBM interacting groove or the length of the resulting small subunit N-terminal strand, or a combination of both. Pull-down data using Smac mutated to simulate caspase-7 cleaved at each of the interdomain processing sites reveal that this interaction is specific, with each terminus having different binding affinity (Figure 6).
The role of the BIR2 IBM interacting groove in apoptosis regulation may be multifaceted and depend on which apoptotic pathway is engaged, which executioner caspase is activated and whether antagonists such as Smac/DIABLO or HtrA2 are involved. When the extrinsic death pathway is triggered (Fas or TRAIL), mutation of the BIR2 IBM interacting groove leads to increased executioner caspase activity. This does not necessarily result in increased apoptosis (Figure 5). These stimuli primarily activate caspase-3, the predominant executioner caspase in most cells. In contrast, when apoptosis was triggered by caspase-7 overexpression, complete loss in apoptosis protection occurred when the BIR2 IBM interacting groove of XIAP was ablated. This may suggest that the second site contributes more to caspase-7 versus caspase-3 inhibition and is supported by the kinetic data (Table III). It should also be noted that mutation of the BIR2 IBM interacting groove of XIAP results in decreased affinity for proapoptotic antagonists Smac/DIABLO and HtrA2 (Figure 6; Silke et al, 2002; Verhagen et al, 2002). In apoptotic pathways involving these proteins, it is difficult to predict whether the BIR2 IBM groove mutations would be pro- or antiapoptotic, since this would depend on the relative concentration of activated caspase and the amount of antagonists released from mitochondria.
Endogenous procaspase-7 can be activated by an initial cleavage at D198 (Denault and Salvesen, 2003). This can be accomplished by GraB during cytotoxic T-cell killing of target cells, caspase-9 during activation of the intrinsic (mitochondrial) cell death pathway or by caspase-8 during activation of the extrinsic (death receptor) pathway. Caspase-8 can directly cleave both D198 and D206 (Denault and Salvesen, 2003). Our finding that caspase-7 activated by cleavage at D206 is more efficiently inhibited by XIAP (Figure 3) may have important implications for downstream inhibition by XIAP. Significantly, caspase-8 is not only involved in cell death but may also be involved in specific cell proliferation (reviewed in Barnhart and Peter, 2002). Cleavage at the second site (D206) might provide an additional level of regulation by ensuring that caspase-7 activity is tightly controlled. A thorough study delineating the cellular situations in which the second activation site is used should provide further insight into the need for dual cleavage sites. Interestingly, the two possible N-terminal epitopes exposed during activation are conserved throughout mammals, and even amphibians, suggesting evolutionary pressure to retain these sequences (Figure 7C). In contrast, caspase-3 activation has only been reported to occur at Asp175, and this protease is less efficiently inhibited than caspase-7. Although they share overlapping substrate specificity, the exact role caspase-3 and -7 play both in apoptosis and other physiological processes is not understood. XIAP may have evolved to regulate caspase-3 and -7 with distinct potencies to provide exquisite control of cell proliferation and cell death.
In conclusion, both BIR2 and BIR3 inhibit their target caspases by a two-site interaction mechanism. They have conserved a functional IBM interacting groove that participates in inhibition by binding neoepitopes revealed following activation of their target enzymes. This interaction, primarily a docking contact, represents the conserved mechanism and also provides a platform for regulation by antagonists Smac/DIABLO and HtrA2. The primary inhibition site, however, is mechanistically different for each domain: blocking the active site in caspase-3 and -7, or dissociating the dimer of caspase-9. It is far from clear how the inhibitory mechanism diverged. It is much clearer that these distinct mechanisms direct the exquisite specificity that allows XIAP BIR domains to target selectively individual caspases in a way that other inhibitory strategies, both natural (Stennicke et al, 2002) and artificial (Nicholson, 2000; Ullman et al, 2003), have yet to achieve.
Materials and methods
Materials
Benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (Z-VAD-fmk), acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (Ac-DEVD-afc), acetyl-Leu-Glu-His-Asp-7-amino-4-trifluoromethylcoumarin (Ac-LEHD-afc) and acetyl-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-pNA) were from Enzyme System Products. IPTG was from Bio Vectra dcl. Purified native GraB was a kind gift of Dr Chris Froelich (Northwestern University Medical School, IL). Purified native CatG was a kind gift of Dr Jan Potempa (Jagellonian University, Krakow, Poland). Linker and control peptides were from the Eastern Quebec Proteomics Center (Laval University, Sainte-Foy, Canada). Smac 7-mer (AVPIAQK) was a kind gift of Dr John Reed (The Burnham Institute, CA). Annexin V-PE was from Caltag. Monoclonal XIAP/hILP and HSP90 antibody were from BD Transduction Laboratories. Polyclonal Smac antibody was from Cell Sciences.
Plasmids and recombinant proteins
The XIAP fragment BIR2 (124–237) with C202A and C213G (Sun et al, 1999) was cloned into a modified pET15b (Novagen) vector containing 8xHis residues at the N-terminus and expressed in BL21 (DE3) E. coli by induction with 0.2 mM IPTG at 30°C for 4 h. Full-length XIAP was cloned into pET15b and expressed at 22°C for 18 h without IPTG. All mutants were generated by site-directed mutagenesis using Quickchange (Stratagene). Caspase-3, caspase-7 and procaspase-7 were as described (Stennicke and Salvesen, 1999). To generate caspase-7 cleaved at D198 and D206 (Casp7-D206), NDTD206 was mutated to IEPD206. Expression in E. coli results in autocatalytic processing at D198 and D206. Caspase-7 cleaved at D198 (Casp7-D198) was generated by cleaving procaspase-7 zymogen with GraB, as previously described (Riedl et al, 2001a). Wild-type Smac lacking the mitochondrial targeting sequence (residues 56–239) was amplified from human thymus cDNA library and cloned into pET23b with a C-terminal 6xHis tag. SGPI-Smac, ANPR-Smac and MVPI-Smac were generated by Quickchange and proteins were N-terminally sequenced using Edman degradation. pGEX-4T-1/GST-GFP was generated by cloning a BamHI (Klenow-treated)–NotI fragment of pEGFP-N2 (Clontech) into pGEX-4T-1 digested with SmaI–NotI. pGEX-4T-1/GST-linker(124–168)-GFP was generated by amplifying residues 124–168 from GST-BIR2 and cloning into the EcoRI site of pGEX-4T-1/GST-GFP. Linker(124–168)-GFP was generated by cleaving recombinant GST-linker-GFP with 5 U of thrombin (Sigma) overnight at 4°C on glutathione Sepharose beads. Recombinant GST, GST-BIR2, GST-BIR2 E219R H223V and GST-linker-GFP were expressed and purified as previously described (Takahashi et al, 1998). p35 C2A was expressed and purified as previously described (Riedl et al, 2001c). The pcDNA3/myc-XIAP plasmid was from Dr John Reed. The pcDNA3/ΔN-caspase-7 was previously described (Denault and Salvesen, 2003). All constructs were sequenced across the entire cDNA. All recombinant proteins with a poly-His tag were purified by Ni+-affinity chromatography as described (Stennicke and Salvesen, 1999).
Enzyme kinetics
Caspases were active-site titrated with Z-VAD-fmk as previously described (Stennicke and Salvesen, 2000). Determination of KM and kcat for Ac-DEVD-afc was as previously described (Zhou et al, 1997). Inhibition constants (Ki) were determined as previously described with modifications (Zhou et al, 1997). Briefly, in a 100 μl assay, caspase-3 (50 or 100 pM) or caspase-7 (100 or 200 pM) was incubated with varying inhibitor concentrations in modified caspase buffer (50 mM HEPES, 100 mM NaCl, 10% (w/v) sucrose, 0.1% (w/v) CHAPS, 20 mM β-mercaptoethanol, pH 7.4) for 30 min at 37°C. Residual enzyme activity was determined by hydrolysis of 100 μM Ac-DEVD-afc for caspase-3 and -7, and Ac-LEHD-afc for caspase-9, at 37°C with an fMAX Fluorescence Plate Reader (Molecular Devices), excitation wavelength of 405 nm and emission wavelength of 510 nm. The Ki for each enzyme–inhibitor pair was determined from the uninhibited rate (vo) and inhibited rates (vi), such that a plot of (vo/vi)−1 versus [I] gives a slope of 1/Ki(apparent). From the KM and substrate concentration [S], the true Ki=Ki(apparent)/(1+[S]/KM) was determined.
Activation of procaspase-7 by serine proteases
Procaspase-7 was activated by cleavage with GraB or CatG as described with some modifications (Zhou and Salvesen, 1997). Procaspase-7 was initially incubated with 500 nM Z-VAD-fmk for 30 min at 37°C in caspase buffer to inhibit irreversibly any active caspase-7. The protein was then purified over a PD-10 column to remove any unassociated Z-VAD-fmk. Procaspase-7 was activated with GraB or CatG for 30 min at 37°C in PBS. The extent of procaspase-7 activation was monitored by hydrolysis of Ac-DEVD-pNA in caspase buffer. Once maximal activation was achieved, samples were incubated with 1 mM PMSF for 5 min at 37°C to inactivate GraB and CatG. Caspase-7 is not inhibited by PMSF at this concentration. GraB-activated caspase-7 (Casp7-D198) and CatG-activated caspase-7 (Casp7-D196) were used in inhibition studies with XIAP and BIR2.
Size-exclusion chromatography
A Superdex 200HR 10/30 column (Pharmacia) with 20 mM Tris, 150 mM NaCl, 5 mM EDTA, pH 8 buffer was used to determine the oligomeric state of BIR2, GST-linker-GFP and linker-GFP. The column was calibrated with standards (Bio-Rad).
Electrophoresis and immunoblotting
Samples were electrophoresed on 8–18% linear gradient acrylamide SDS–PAGE under reducing conditions as described (Denault and Salvesen, 2003). Gels were stained with GELCODE Blue Stain Reagent (Pierce). Immunoblotting was as described (Denault and Salvesen, 2003).
Transfections and induction of cell death
Adherent QBI-HEK 293A cells (293A) were maintained as described and are sensitive to TRAIL (Wu et al, 2000; Denault and Salvesen, 2003). Cells were transfected with FuGENE 6 Transfection Reagent (Roche). Cells were stained 24 h post-transfection with Annexin V-PE and analyzed by FACS on a Becton Dickinson FACSort. For some experiments, transfectants were treated with 100 ng/ml rhsKillerTRAIL (Alexis Biochemicals, CA) and either stained with Annexin V-PE and analyzed by FACS or lysed on ice with modified radioimmunoprecipitation buffer (mRIPA; 10 mM Tris, 150 mM NaCl, 1% (v/v) NP-40, 0.5% (v/v) deoxycholate, 0.1% (w/v) SDS, 5 mM EDTA, pH 7.4). Lysates were clarified by centrifugation and assayed for executioner caspase activity in caspase buffer (20 mM PIPES, 100 mM NaCl, 10% (w/v) sucrose, 0.1% (w/v) CHAPS, 10 mM DTT, 1 mM EDTA, pH 7.2) with 100 μM Ac-DEVD-afc. Protein concentrations were determined (Bio-Rad DC Protein Assay) and caspase activity was normalized for protein content. Untreated duplicate samples were processed for immunoblotting. Individual experiments were normalized by dividing each sample by the highest value (by Annexin V-PE staining) and multiplying by 100 to give ‘% Maximum Apoptosis'. Statistical analysis was performed using the Student's paired t-test with two-tailed distribution.
GST co-precipitations
GST, GST-BIR2 or GST-BIR2 E219R H223V from E. coli lysates was bound to glutathione Sepharose beads for 30 min at room temperature in PBS. Beads were washed three times in binding buffer (20 mM Na-phosphate buffer pH 7, 100 mM NaCl, 0.5 mM EDTA, 1 mM DTT, 0.05% (v/v) Tween 20) and resuspended at 50% (w/v). A 5 μl portion of beads was incubated with 100 nM AVPI-Smac, SGPI-Smac, ANPR-Smac or MVPI-Smac in a total of 50 μl at 4°C for 30 min. Beads were washed three times in binding buffer and proteins eluted by boiling in SDS sample buffer containing 20 mM DTT prior to electrophoresis on an 8–18% linear gradient acrylamide SDS–PAGE. Samples were either transferred to PVDF and immunoblotted with polyclonal Smac antibody or the gel was stained with GELCODE Blue to demonstrate integrity of GST proteins.
Supplementary Material
Supplementary Figure 1
Supplementary Table 1
Acknowledgments
We thank Scott Snipas and Annamarie Price for expert technical assistance, Drs Chris Froelich and Jan Potempa for providing proteases and Dr Phil Bird for the use of lab space. This work supported by NIH grant AG15402, and FLS was supported by a CJ Martin Training Fellowship from NHMRC (Australia). FLS is a PI on NHMRC Program Grant 284233.
References
- Barnhart BC, Peter ME (2002) Two faces of caspase-8. Nat Immunol 3: 896–898 [DOI] [PubMed] [Google Scholar]
- Bratton SB, Lewis J, Butterworth M, Duckett CS, Cohen GM (2002) XIAP inhibition of caspase-3 preserves its association with the Apaf-1 apoptosome and prevents CD95- and Bax-induced apoptosis. Cell Death Differ 9: 881–892 [DOI] [PubMed] [Google Scholar]
- Chai J, Du C, Wu JW, Kyin S, Wang X, Shi Y (2000) Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406: 855–862 [DOI] [PubMed] [Google Scholar]
- Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y, Dataa P (2001) Structural basis of caspase-7 inhibition by XIAP. Cell 104: 769–780 [DOI] [PubMed] [Google Scholar]
- Chai J, Yan N, Huh JR, Wu JW, Li W, Hay BA, Shi Y (2003) Molecular mechanism of Reaper–Grim–Hid-mediated suppression of DIAP1-dependent Dronc ubiquitination. Nat Struct Biol 10: 892–898 [DOI] [PubMed] [Google Scholar]
- Denault JB, Salvesen GS (2002) Caspases: keys in the ignition of cell death. Chem Rev 102: 4489–4500 [DOI] [PubMed] [Google Scholar]
- Denault JB, Salvesen GS (2003) Human caspase-7 activity and regulation by its N-terminal peptide. J Biol Chem 278: 34042–34050 [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS, Reed JC (1999) Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J 18: 5242–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304 [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42 [DOI] [PubMed] [Google Scholar]
- Fesik SW, Shi Y (2001) Structural biology: controlling the caspases. Science 294: 1477–1478 [DOI] [PubMed] [Google Scholar]
- Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H (2001) Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell 104: 781–790 [PubMed] [Google Scholar]
- Huang Y, Rich RL, Myszka DG, Wu H (2003) Requirement of both the BIR2 and BIR3 domains for the relief of XIAP-mediated caspase inhibition by Smac. J Biol Chem 278: 49517–49522 [DOI] [PubMed] [Google Scholar]
- Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW (2000) Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 408: 1004–1008 [DOI] [PubMed] [Google Scholar]
- MacFarlane M, Merrison W, Bratton SB, Cohen GM (2002) Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem 277: 36611–36616 [DOI] [PubMed] [Google Scholar]
- Nicholson DW (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ 6: 1028–1042 [DOI] [PubMed] [Google Scholar]
- Nicholson DW (2000) From bench to clinic with apoptosis-based therapeutic agents. Nature 407: 810–816 [DOI] [PubMed] [Google Scholar]
- Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci USA 98: 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W (2001a) Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci USA 98: 14790–14795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS (2001b) Structural basis for the inhibition of caspase-3 by XIAP. Cell 104: 791–800 [DOI] [PubMed] [Google Scholar]
- Riedl SJ, Renatus M, Snipas SJ, Salvesen GS (2001c) Mechanism-based inactivation of caspases by the apoptotic suppressor p35. Biochemistry 40: 13274–13280 [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol 3: 401–410 [DOI] [PubMed] [Google Scholar]
- Shin H, Okada K, Wilkinson JC, Solomon KM, Duckett CS, Reed JC, Salvesen GS (2003) Identification of ubiquitination sites on the X-linked inhibitor of apoptosis protein. Biochem J 373: 965–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y (2003) Mechanism of XIAP-mediated inhibition of caspase-9. Mol Cell 11: 519–527 [DOI] [PubMed] [Google Scholar]
- Silke J, Ekert PG, Day CL, Hawkins CJ, Baca M, Chew J, Pakusch M, Verhagen AM, Vaux DL (2001) Direct inhibition of caspase 3 is dispensable for the anti-apoptotic activity of XIAP. EMBO J 20: 3114–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Hawkins CJ, Ekert PG, Chew J, Day CL, Pakusch M, Verhagen AM, Vaux DL (2002) The anti-apoptotic activity of XIAP is retained upon mutation of both the caspase 3- and caspase 9-interacting sites. J Cell Biol 157: 115–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES (2001) A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 410: 112–116 [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Ryan CA, Salvesen GS (2002) Reprieval from execution: the molecular basis of caspase inhibition. Trends Biochem Sci 27: 94–101 [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Salvesen GS (1999) Caspases: preparation and characterization. Methods 17: 313–319 [DOI] [PubMed] [Google Scholar]
- Stennicke HR, Salvesen GS (2000) Caspase assays. Methods Enzymol 322: 91–100 [DOI] [PubMed] [Google Scholar]
- Sun C, Cai M, Gunasekera AH, Meadows RP, Wang H, Chen J, Zhang H, Wu W, Xu N, Ng SC, Fesik SW (1999) NMR structure and mutagenesis of the inhibitor-of-apoptosis protein XIAP. Nature 401: 818–822 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Nakata K, Reed JC, Takahashi R (2001a) X-linked inhibitor of apoptosis protein (XIAP) inhibits caspase-3 and -7 in distinct modes. J Biol Chem 276: 27058–27063 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Nakabayashi Y, Takahashi R (2001b) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci USA 98: 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R, Deveraux Q, Tamm I, Welsh K, Assa-Munt N, Salvesen GS, Reed JC (1998) A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem 273: 7787–7790 [DOI] [PubMed] [Google Scholar]
- Ullman BR, Aja T, Deckwerth TL, Diaz JL, Herrmann J, Kalish VJ, Karanewsky DS, Meduna SP, Nalley K, Robinson ED, Roggo SP, Sayers RO, Schmitz A, Ternansky RJ, Tomaselli KJ, Wu JC (2003) Structure–activity relationships within a series of caspase inhibitors: effect of leaving group modifications. Bioorg Med Chem Lett 13: 3623–3626 [DOI] [PubMed] [Google Scholar]
- Vargo MA, Nguyen L, Colman RF (2004) Subunit interface residues of glutathione S-transferase A1-1 that are important in the monomer–dimer equilibrium. Biochemistry 43: 3327–3335 [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J (2003) Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun 304: 499–504 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritx RL, Simpson RJ (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ, Vaux DL (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem 277: 445–454 [DOI] [PubMed] [Google Scholar]
- Wilson R, Goyal L, Ditzel M, Zachariou A, Baker DA, Agapite J, Steller H, Meier P (2002) The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol 4: 445–450 [DOI] [PubMed] [Google Scholar]
- Wu M, Das A, Tan Y, Zhu C, Cui T, Wong MC (2000) Induction of apoptosis in glioma cell lines by TRAIL/Apo-2l. J Neurosci Res 61: 464–470 [DOI] [PubMed] [Google Scholar]
- Wu TY, Wagner KW, Bursulaya B, Schultz PG, Deveraux QL (2003) Development and characterization of nonpeptidic small molecule inhibitors of the XIAP/caspase-3 interaction. Chem Biol 10: 759–767 [DOI] [PubMed] [Google Scholar]
- Xu G, Cirilli M, Huang Y, Rich RL, Myszka DG, Wu H (2001) Covalent inhibition revealed by the crystal structure of the caspase-8/p35 complex. Nature 410: 494–497 [DOI] [PubMed] [Google Scholar]
- Yan N, Wu JW, Chai J, Li W, Shi Y (2004) Molecular mechanisms of DrICE inhibition by DIAP1 and removal of inhibition by Reaper, Hid and Grim. Nat Struct Mol Biol 11: 420–428 [DOI] [PubMed] [Google Scholar]
- Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288: 874–877 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Salvesen GS (1997) Activation of pro-caspase-7 by serine proteases includes a non-canonical specificity. Biochem J 324: 361–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Snipas S, Orth K, Muzio M, Dixit VM, Salvesen GS (1997) Target protease specificity of the viral serpin CrmA. Analysis of five caspases. J Biol Chem 272: 7797–7800 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Table 1