

Abstract
The DEAD box RNA helicase, p68, has been implicated in various cellular processes and has been shown to possess transcriptional coactivator function. Here, we show that p68 potently synergises with the p53 tumour suppressor protein to stimulate transcription from p53-dependent promoters and that endogenous p68 and p53 co-immunoprecipitate from nuclear extracts. Strikingly, RNAi suppression of p68 inhibits p53 target gene expression in response to DNA damage, as well as p53-dependent apoptosis, but does not influence p53 stabilisation or expression of non-p53-responsive genes. We also show, by chromatin immunoprecipitation, that p68 is recruited to the p21 promoter in a p53-dependent manner, consistent with a role in promoting transcriptional initiation. Interestingly, p68 knock-down does not significantly affect NF-κB activation, suggesting that the stimulation of p53 transcriptional activity is not due to a general transcription effect. This study represents the first report of the involvement of an RNA helicase in the p53 response, and highlights a novel mechanism by which p68 may act as a tumour cosuppressor in governing p53 transcriptional activity.
Keywords: DEAD box protein, DNA damage response, p53, p68, transcriptional coactivator
Introduction
The DEAD box family of RNA helicases includes a large number of conserved proteins, which are found in all organisms from bacteria to humans and have been shown to be involved in virtually all cellular processes that require manipulation of RNA structure, including transcription, pre-mRNA processing, RNA degradation, RNA export, ribosome assembly and translation. Although the characteristic biochemical properties for this family are RNA-dependent ATPase and RNA helicase activities, relatively few members appear to be true processive helicases and it is clear that many are likely to be involved in unwinding of short base-paired regions of RNA or indeed in the disruption or rearrangement of RNA–protein interactions (Tanner and Linder, 2001).
p68 is a prototypic member of the DEAD box family (Ford et al, 1988) and is an established ATPase and RNA helicase (Hirling et al, 1989; Iggo and Lane, 1989). Previous reports have shown that p68 expression is growth and developmentally regulated, and that p68 is overexpressed and abnormally polyubiquitylated in colorectal tumours (Stevenson et al, 1998; Causevic et al, 2001). Recently, p68 has been shown to be essential for pre-mRNA splicing in vitro (Liu, 2002) and to play a role in the regulation of c-H-ras alternative splicing (Guil et al, 2003). Dbp2p, the yeast homologue of p68 (Iggo et al, 1991), was found to be important for both rRNA processing and nonsense-mediated mRNA decay (Bond et al, 2001), while, in an earlier study, overexpressed human p68 was found to stabilise T7 mRNAs in bacteria (Iost and Dreyfus, 1994). Although the roles of p68 in pre-mRNA/rRNA processing and mRNA decay/stability are consistent with its function as an RNA helicase, p68 has also been reported to act as a transcriptional coactivator for oestrogen receptor alpha (ERα), a function that appears to be independent of helicase activity (Endoh et al, 1999; Watanabe et al, 2001). Moreover, p68 has recently been shown to be recruited to the promoter of the ERα target gene pS2 (Metivier et al, 2003), consistent with it playing a role in ERα-dependent transcriptional initiation. p68 has also been reported to interact with the transcriptional coactivators CBP/p300 as well as RNA polymerase II and to stimulate transcriptional activation mediated by CBP/p300 although, in this case, p68 ATPase/RNA helicase activity appeared to be required (Rossow and Janknecht, 2003). These findings therefore suggest that, in addition to its role in RNA processing, p68 may also have an important function as a transcriptional regulator.
Given the implied role of p68 in growth regulation and tumour progression (Stevenson et al, 1998; Causevic et al, 2001), we investigated the ability of p68 to coactivate other transcription factors that are important in tumour development. One such protein is the critical tumour suppressor p53, a latent and labile transcription factor that is induced and activated in response to several stresses, including DNA damage (Vogelstein et al, 2000; Balint and Vousden, 2001). Activated p53 induces transcription of a host of downstream target genes, which are mainly involved in growth arrest, apoptosis and DNA repair. In addition, p53 also induces expression of its negative regulatory partner Mdm2 (Vogelstein et al, 2000; Balint and Vousden, 2001).
In this report, we show that p68 is a potent transcriptional coactivator of p53, as shown by its ability to synergise with p53 to activate transcription from p53-responsive promoters. Additionally, endogenous p53 and p68 co-immunoprecipitate from nuclear protein extracts, suggesting that these proteins interact in the cell. Furthermore, by RNAi-mediated suppression of p68 expression in cells that express wild-type (WT) p53, we show that p68 is specifically required for the induction of expression of the cellular p53 target genes p21WAF-1, mdm2, Fas/APO1 and PIG3 in response to treatment with the DNA-damaging agent etoposide, while it has no effect on non-p53-responsive genes. This activity is specific to p68 since RNAi suppression of the highly related RNA helicase p72 (Lamm et al, 1996) has no effect on the induction of p53 transcriptional activity by DNA damage. We also show that p68 knock-down results in a reduction in apoptosis in response to p53 induction. Finally, we show by chromatin immunoprecipitation (ChIP) that p68 is recruited to the p21 promoter. These findings are therefore consistent with p68 being an important regulator of the p53 response and suggest a novel mechanism for regulating p53 transcriptional activity.
Results
p68 acts as a coactivator of p53 transcriptional activity
To determine initially whether p68 has the potential to modulate the transcriptional activity of p53, we transfected H1299 (p53-null) cells with p68 and p53 cytomegalovirus (CMV) expression plasmids together with the p53-responsive reporter plasmid PG13-luciferase and measured luciferase activity. p68 potently synergised with p53 to activate transcription from the PG13 promoter (Figure 1A), supporting the hypothesis that p68 might regulate p53 transactivation function, with the most dramatic effect being observed with 10 ng of the p53 expression plasmid. In addition, titration of the p68 expression plasmid (Figure 1B) confirmed that this was a concentration-dependent effect. Since the highly related RNA helicase p72 was also reported to coactivate ERα (Watanabe et al, 2001), we tested whether p72 could similarly coactivate p53 transcriptional activity by carrying out a similar titration of a p72 expression plasmid. Interestingly, although some stimulation of p53 transcriptional activity by p72 was observed (Figure 1B), this was considerably lower than that seen for p68, suggesting that p72 is not such a potent coactivator of p53. (Both p68 and p72 were expressed at similar levels in these cells; see below and Supplementary data 1.) We then tested whether p68 could stimulate p53 transcriptional activity from other p53-responsive promoters. These included the p21 and Bax promoters as well as the p53-responsive element from the c-Ha-Ras gene (pRasH-Adluc) together with the nonresponsive pAdluc as a control (Deguin-Chambon et al, 2000) (Figure 1C–E). As previously shown with PG13 (Figure 1A and B), p68 synergised strongly with p53 to transactivate the p21 (Figure 1C) and the pRasH-Adluc promoters (Figure 1E), while a weaker effect was seen with the Bax promoter (Figure 1D). Importantly, no cooperative activation was observed with the pAdluc promoter, which lacks p53-binding sites (Figure 1E). These findings thus demonstrate that p68 synergises with p53 to activate transcription from a variety of p53-responsive promoters. A low level of transcriptional activation was observed when p68 alone was transfected (Figure 1C–E), suggesting that p68 has a low level of basal transcriptional activity; however, it should be noted that the amounts of p68 plasmid DNA transfected were higher than those for p53. Since the PG13 reporter plasmid gave the strongest effect in these experiments, we decided to use this to further characterise p68 coactivation activity. To confirm that the observed coactivation of p53 by p68 was not due simply to the transfected p68 affecting p53 levels in the cell, we examined the levels of p53 protein in the presence and absence of transfected p68/p72 by Western blotting (see Supplementary data 1). Although there were some minor variations in the expression of p53 between different transfections, increasing the amounts of transfected p68/p72 had no significant effect on the levels of p53.
Figure 1.
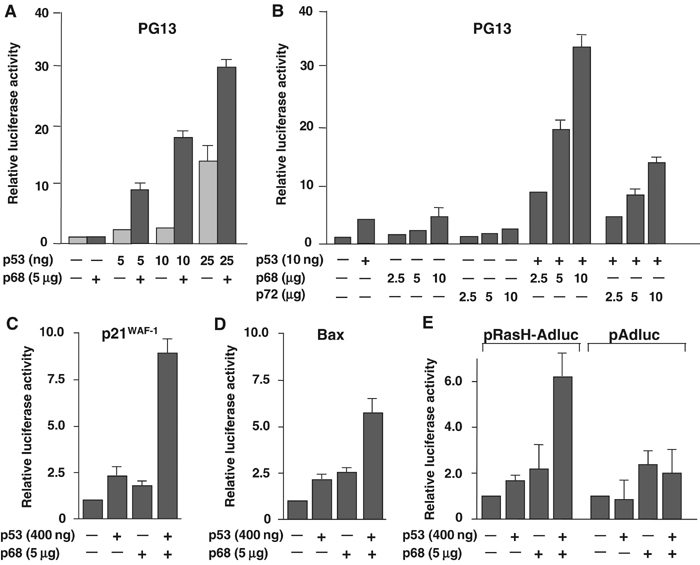
p68 stimulates p53 transcriptional activity from p53-responsive promoters. Effect of p68 on transactivation of the p53-responsive promoters PG13 (A, B), p21 (C), Bax (D) and pRasH-Adluc (E), fused to the luciferase reporter (pAdluc was used as a non-p53-responsive control (E)). In each case, the relative luciferase activity is shown with the basal activity of the promoter being taken as 1. Panels A and B show titres of the p53 and p68 plasmid DNAs, respectively, and the amounts used per ml of transfection mix are indicated. The amounts of reporter plasmid DNA used per ml of transfection mix were as follows: PG13, 2.5 μg; p21, 3 μg; Bax, 3 μg; pRasH-Adluc/pAdluc, 2.5 μg. Unless otherwise stated, the amounts of p53 plasmid transfected in these experiments had been optimised previously for the different promoters and were as follows: PG13, 10 ng; p21, Bax and pRasH-Adluc/pAdluc, 400 ng. Similarly, unless otherwise stated, 5 μg of p68 plasmid DNA was used. Graphs A and B represent the average results from two independent transfections, while graphs C–E represent average results from three independent experiments.
We next tested whether p68 coactivation was dependent on transcriptionally active p53 or whether similar effects could be seen with a transcriptionally inactive (L22Q/W23S) mutant (Venot et al, 1999). As shown in Figure 2A, p68 did not coactivate the L22Q/W23S p53 mutant. (Similar results were obtained with a His 175 p53 mutant; data not shown.) Since previous reports had suggested that coactivation of ERα was not dependent on p68 helicase activity (Endoh et al, 1999), we also tested an ATPase/helicase-inactive mutant of p68 (NEAD p68). Interestingly, NEAD p68, which is expressed at similar levels to WT p68 (data not shown), was equally capable of coactivating p53 (Figure 2B). This suggests that p68 helicase activity is not required for p53 coactivation, although we cannot rule out the possibility that, in this system, the transfected (helicase-inactive) p68 interacts with endogenous WT p68 (Ogilvie et al, 2003) to form a complex that may have sufficient residual helicase activity.
Figure 2.
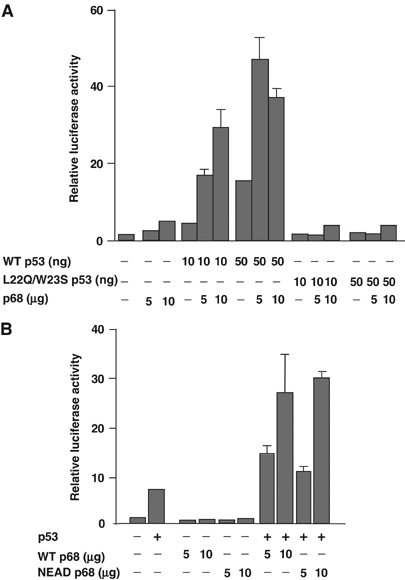
Coactivation requires transcriptionally active p53 but not helicase-active p68. (A) Coactivation of WT and L22Q/W23S (transcriptionally inactive) p53 transcriptional activity by p68. (B) Coactivation of WT p53 transcriptional activity by WT and NEAD (helicase-inactive) p68. Activity is determined by measurement of transactivation of the PG13 promoter fused to the luciferase reporter. The relative luciferase activity is shown, with the basal activity of the promoter being taken as 1. The amounts of p68 plasmid DNA used per ml of transfection mix are indicated and in all cases 10 ng of p53 plasmid DNA and 2.5 μg of PG13 reporter plasmid DNA were used per ml of transfection mix. Graphs represent the average results from two independent transfections.
p68 interacts with p53 in vitro and in cultured cells
p68 has previously been reported to interact with ERα (Endoh et al, 1999) as well as p300, CBP and RNA polymerase II (Rossow and Janknecht, 2003), suggesting that it may form part of a multiprotein complex to regulate transcription. We therefore investigated whether p68 could interact with p53. We first determined whether p68 and p53 can interact in vitro by performing GST pull-down experiments using purified GST-tagged p68 expressed in mammalian cells and in vitro-translated 35S-labelled p53. As shown in Figure 3A and B, GST-tagged p68, but not a GST vector control, interacted with in vitro-translated p53. Interestingly, p68 also interacted with ΔN-p53 (Figure 3B), which is the product of an alternative internal translation initiation site and lacks much of the amino-terminal transactivation domain of p53 (Courtois et al, 2002; Yin et al, 2002). We also tested whether p72 interacts with p53 in vitro; p72 does interact with p53 but much less efficiently than p68 (Figure 3A and B). To determine whether these proteins interact in the cell, we examined whether endogenous p53 and p68/p72 could co-immunoprecipitate from cell lysates. Nuclear extracts were prepared from U2OS cells, which harbour WT p53, and from SAOS-2 as a p53-null control. p53 was immunoprecipitated using DO-1; immunoprecipitated proteins were resolved by SDS–PAGE and Western blotted for the presence of p68/p72 in the immune complex. Reciprocal immunoprecipitation (IP)/Westerns were also carried out and appropriate irrelevant antibodies were included as additional controls. All nuclear extracts were treated with DNase/RNase prior to IP to exclude the possibility that any observed interaction between p68/p72 and p53 was merely via nucleic acid. As shown in Figure 3C and D, p68 co-immunoprecipitated with p53 from the U2OS extract, indicating that these proteins interact in the cell. In addition, the absence of any p68 in the p53 IP (Figure 3C) from the SAOS-2 extract confirmed that the DO-1 antibody was not immunoprecipitating p68 nonspecifically. As a further control, we performed IP/Westerns for p53/p68 using a different p53 antibody (CM1); this gave similar results (Figure 3E). To determine whether p72 also co-immunoprecipitates with p53, proteins in the p53 IP (Figure 3C) were Western blotted for p72 (Figure 3F). p72 and the alternative upstream translation initiation product p82 (Uhlmann-Schiffler et al, 2002) are both present in the p53 IP. The reciprocal IP/Western gave similar results (Figure 3G). However, in the light of the low p72/p53 in vitro interaction (Figure 3B), the presence of p72/p82 in the p53 IP may be, at least partially, due to the previously reported interaction of p72/82 with p68 (Ogilvie et al, 2003) rather than their specific interaction with p53.
Figure 3.

p68 and p72 interact with p53 in vitro and in vivo. (A) Expression of GST vector control and GST-tagged p68/p72 used in the GST pull-down experiments as shown by Western blotting of cell lysates with a GST-specific antibody. (B) GST ‘pull-down' of in vitro-translated (35S-labelled) p53, showing both input and p53 species interacting with GST-tagged p68/p72. (C) Co-IP of p53 and p68 from nuclear extracts. p53 in U2OS extract was immunoprecipitated with the mouse monoclonal antibody (DO-1) and p68 and p53 in the IP were detected by Western blotting with rabbit polyclonal antibodies 2907 (p68) and CM1 (p53). (D) Reciprocal co-IP of p53 and p68. In this case, p68 was immunoprecipitated using the rabbit polyclonal antibody 2907 and immunoprecipitated p68 and p53 were detected by Western blotting with monoclonal antibodies PAb204 (p68) and DO-1 (p53). (E) Co-IP of p53 and p68 using a different p53-specific immunoprecipitating antibody. p53 in U2OS extract was immunoprecipitated with polyclonal antibody (CM1) and p68 and p53 were detected by Western blotting with monoclonal antibodies PAb204 (p68) and DO-1 (p53). (F) Co-IP of p53 and p72. Proteins immunoprecipitated by the p53 antibody (DO-1) (shown in (C)) were also Western blotted for p72 using the rabbit polyclonal antibody K14. (G) Reciprocal co-IP of p53 and p72. p72 was immunoprecipitated with the K14 antibody and p72 and p53 were detected by Western blotting with K14 and DO-1. Note that since only one p72 antibody is available, the same antibody had to be used for IP and Western blotting, giving a strong crossreaction with heavy chain (H). NE: nuclear extract; molecular weight markers (in kDa) are indicated. A nuclear extract from the p53-null cell line SAOS-2 and an irrelevant mouse or rabbit IgG (as appropriate; Cont. IP) were used as controls for IP.
p68 is required for the p53 DNA damage response
To determine whether p68 was required for p53 function in a physiological context, we ‘knocked down' p68 expression by RNAi in MCF-7 cells (which express WT p53) and determined whether loss of p68 affected the p53 response to DNA damage. Using a p68-specific siRNA, we achieved an 80–90% knock-down of p68 protein expression (Figure 4A). Treatment with the DNA-damaging agent etoposide had no effect on p68 expression but, as expected, clearly induced p53 protein expression. Strikingly, however, while suppression of p68 expression had no effect on the induction of p53 protein levels, there was a significant reduction in the ability of p53 to stimulate expression of the p53 target genes p21 and mdm2 following etoposide treatment, as observed by Western blotting for the respective proteins (Figure 4A). These findings therefore indicate that p68 is required for the induction of p53 transcriptional activity in response to DNA damage. (Similar results were obtained with U2OS cells; data not shown.) In addition, the p68 RNAi knock-down specifically suppressed p68 expression, as there was no reduction in expression of the highly related RNA helicase, p72 (Lamm et al, 1996); we instead observed a minor increase in p72 expression (Figure 4A). To rule out the possibility that the effects seen by the p68 siRNA were due to the specific siRNA oligonucleotide chosen, we repeated the p68 knock-down with a second p68 siRNA directed against a different region of the p68 coding region (see Materials and methods). This gave results identical to those obtained with the first siRNA oligonucleotide (data not shown).
Figure 4.
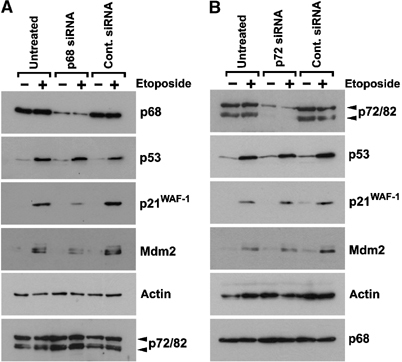
RNAi depletion of p68, but not p72, inhibits expression of p53 target genes in response to DNA damage. Western blots showing expression of p68, p53, p21 and Mdm2 in MCF-7 cells, which had been transfected with (A) p68-specific or (B) p72-specific siRNA oligonucleotides. In both cases, a control siRNA was used and untransfected (UN) cells served as an additional control. In each case, the effect of treatment with the DNA-damaging agent etoposide (100 μM for 4 h) was examined. Equal amounts of protein (as determined by Bradford reagent (Sigma)) were loaded and detection of actin in the lysates was used as a loading control. Moreover, Western blots showing the levels of p68 and p72/82 in the reciprocal ‘knock-downs' confirm the specificity of the siRNAs. (The antibodies used for Western blotting are described in Materials and methods; 2907, K14 and DO-1 were used to detect p68, p72 and p53, respectively.)
p72 has been reported to also act as a coactivator of ERα (Watanabe et al, 2001) and to interact with p68 in the cell (Ogilvie et al, 2003). We therefore examined whether a knock-down of p72 similarly affected the p53 DNA damage response. Using a p72-specific siRNA, we achieved efficient suppression of expression of both p72 and the alternative upstream translation initiation product p82, which appears to have similar functions to p72 (Figure 4B) (Uhlmann-Schiffler et al, 2002). Strikingly, however, the p72 RNAi knock-down appeared to have no effect on the ability of p53 to induce p21 and Mdm2 expression upon etoposide treatment (Figure 4B), suggesting that the effect on the p53 DNA damage response is specific to p68. This finding is consistent with the results obtained from the cotransfection experiments, which showed that p72 is not such a potent coactivator of p53 transcription activity (see above and Figure 1B). As expected, the p72 knock-down had no significant effect on p68 expression (Figure 4B).
In order to confirm that the lack of induction of p21 and Mdm2 expression was occurring at the mRNA level and to determine whether induction of other cellular p53 target genes in response to etoposide was similarly affected, the p68 RNAi experiment was repeated, RNA was extracted from cells and the levels of mRNA for a range of p53 target genes were determined by quantitative RT–PCR. These included p21 (cell cycle arrest) and mdm2 as well as the apoptosis-promoting genes Fas/APO1 and PIG3. GAPDH was used as a control and in each case the values obtained were normalised against β-actin to avoid discrepancies from differences in overall RNA levels between samples. As shown in Figure 5A and B, suppression of endogenous p68 expression resulted in a lack of induction of p21 and mdm2 mRNA in response to etoposide, consistent with a defect in the ability of p53 to induce transcription of the respective genes. Similar defects were observed in the induction of Fas/APO1 and PIG3 (Figure 5C and D), while there were no significant effects on the levels of GAPDH mRNA either as a result of etoposide treatment or knock-down of p68 (Figure 5E). These findings suggest that depletion of p68 results in a defect in the ability of p53 to induce expression of both cell cycle arrest and apoptosis-promoting genes in response to DNA damage, while it has no significant effect on genes that are not affected by p53, such as GAPDH.
Figure 5.
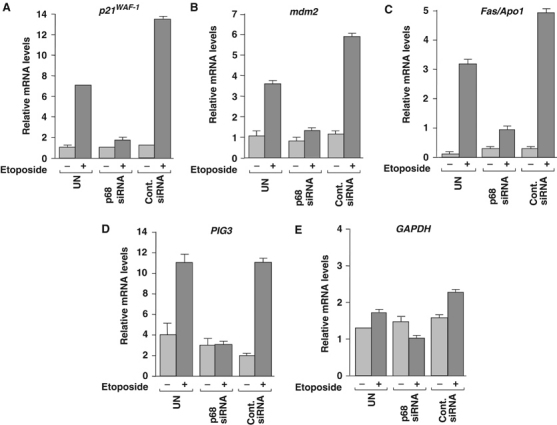
RNAi depletion of p68 inhibits expression of cell cycle arrest and apoptosis-promoting p53 target genes. Quantitative RT–PCR of mRNA extracted from MCF-7 cells, which had been transfected with p68-specific siRNA. In both cases, a control siRNA was used and untransfected (UN) cells served as an additional control. Etoposide treatment was performed as in Figure 4. RT–PCR reactions to measure (A) p21, (B) mdm2, (C) Fas/APO1, (D) PIG3 and (E) GAPDH mRNA levels, in each case relative to β-actin, are shown. The average values from three independent RT–PCR reactions are shown.
RNAi depletion of p68 has no significant effect on induction of NF-κB transcriptional activity
p68 has also been shown to coactivate ERα (Endoh et al, 1999; Watanabe et al, 2001) and to interact with CBP/p300 and RNA polymerase II (Rossow and Janknecht, 2003). Therefore, it was important to determine whether p68 was required for the induction of transcriptional activity of other transcription factors, such as NF-κB. For these experiments, we used a HeLa cell line (HeLa57A), which has an integrated copy of the NF-κB-inducible reporter 3Enhancer-κB-conA-Luc and of the control reporter pRC/RSV-β-galactosidase (Rodriguez et al, 1999). We knocked down p68 expression in these cells by RNAi and examined whether inhibition of p68 expression affected the induction of NF-κB in response to treatment with TNFα, as measured by luciferase activity. Cells transfected with a control siRNA and untreated cells served as controls. We confirmed that p68 was efficiently knocked down and that TNFα treatment had no effect on p68 levels (Supplementary data 2). Interestingly, cells in which p68 expression had been knocked down by RNAi showed no defect in the induction of NF-κB activity in response to TNFα (Figure 6A). In fact, we consistently observed a minor increase in the induction of luciferase activity in these cells compared with controls (Figure 6A). As expected, there was no significant difference in the relative activity of the control pRC/RSV-β-galactosidase reporter in the p68 knock-down and control cells in the presence or absence of TNFα treatment (Figure 6B). These findings thus indicate that p68 is not a general transcriptional coactivator but, instead, acts as a specific coactivator for certain inducible transcription factors, which include ERα (Endoh et al, 1999; Watanabe et al, 2001) and, as shown in the present study, p53.
Figure 6.
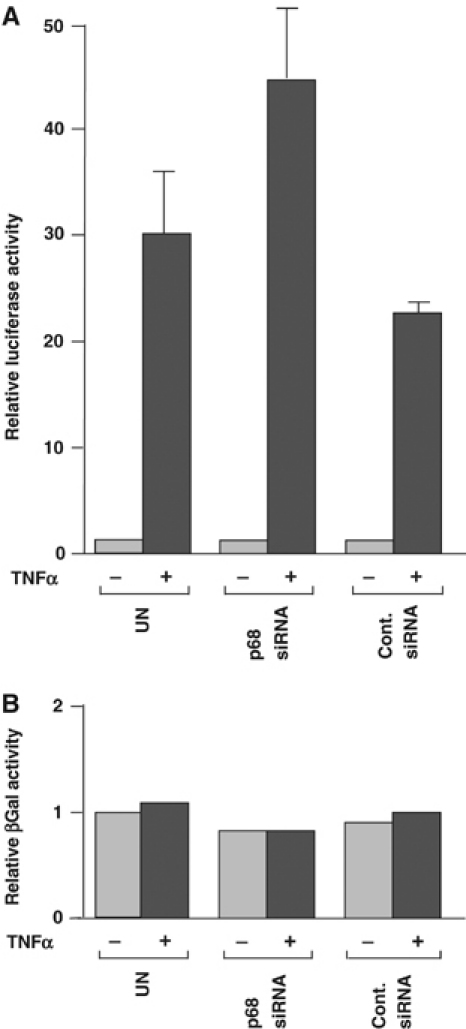
RNAi depletion of p68 does not affect NF-κB activation by TNFα. (A) Induction of NF-κB activity by TNFα in cells transfected with p68 or control siRNA, compared with untransfected cells, as determined by measuring relative luciferase activity. The luciferase activity of untransfected cells, which had not been treated with TNFα, was taken as 1. (B) β-Galactosidase activity in cells. The levels shown represent values relative to that of untransfected and untreated cells, which were taken as 1. Graphs represent the average values from two independent experiments, and reactions were, in each case, performed in duplicate.
p68 is recruited to the p21 promoter
In order to investigate the mechanism by which p68 stimulates p53 transcriptional activity of p53 target genes, we examined whether p68 is recruited to the p21 promoter and whether, as for p53, the recruitment is affected by DNA damage. For this, we performed ChIP–PCR of MCF-7 cells using p68- and p53-specific antibodies, and p21 promoter-specific primers. GAPDH promoter-specific primers were used to confirm specificity of recruitment of p68/p53 to the p21 promoter. Cells were treated with etoposide to determine the effect of DNA damage on recruitment of p68 and p53 to the p21 promoter, with untreated cells serving as a control. As expected, p53 was specifically recruited to the p21 promoter and the amount recruited increased concomitantly with the induction of p53 expression in response to etoposide (Figure 7A) (Szak et al, 2001; Espinosa et al, 2003). Strikingly, p68 is similarly recruited to the p21 promoter and the amount increases in response to etoposide treatment (Figure 7A). Neither p53 nor p68 was found to be recruited to the control GAPDH promoter (Figure 7A), indicating that these proteins are specifically recruited to the p21 promoter and that, like p53, p68 is specifically recruited to the p21 promoter in a DNA damage-dependent manner. To determine whether the recruitment of p68 to the p21 promoter was dependent on p53, we performed similar ChIP experiments with the osteosarcoma cell lines U2OS (WT p53) and SAOS-2 (p53-null). As shown in Figure 7b, p68 and p53 are recruited to the p21 promoter in U2OS cells but not in SAOS-2 cells, showing that the recruitment of p68 to the promoter is indeed p53-dependent. As for MCF-7 cells (Figure 7A), neither p53 nor p68 was recruited to the GAPDH promoter in U2OS cells (data not shown). Our findings thus establish that p68 is recruited to the p21 promoter through interaction with p53 and are consistent with p68 promoting transcriptional initiation of p53 target genes in response to DNA damage.
Figure 7.
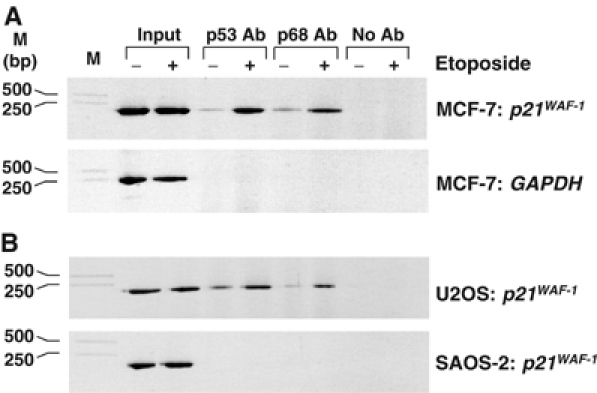
p68 is recruited to the p21 promoter, in a p53-dependent manner, in response to DNA damage. ChIP assays showing recruitment of p53 and p68 to the p21 promoter. (A) Enhancement of p53 and p68 recruitment to the p21 promoter in response to etoposide treatment. PCR reactions were performed using p21-specific and control (GAPDH-specific) primers to confirm specificity. (B) p68 to the p21 promoter in U2OS (p53 WT) but not in SAOS-2 (p53-null) cells. For all ChIP assays, samples of the input DNA, prior to IP with p53- or p68-specific antibodies (Ab), were used in the PCR reactions to confirm that equal amounts of DNA were present in the untreated and etoposide-treated samples. A control IP reaction with no antibody was also included. The cell line and the promoter-specific primers used for each assay are indicated as are DNA molecular weight markers.
In order to examine whether the efficiency of interaction between p68 and p53 is affected by DNA damage, we performed further p68/p53 co-immunoprecipitation (co-IP) experiments in U2OS cells that had been treated by etoposide, with untreated cells as control. As shown in Figure 8A, we observed a small increase in the amount p53 co-immunoprecipitating with p68 as a result of DNA damage, although this could be explained by the fact that the levels of p53 in the cell are much higher in etoposide-treated cells. IP of p53 from etoposide-treated cells did not appear to co-precipitate increased levels of p68 (Figure 8B). These findings indicate that there is no significant increase in the interaction between p68 and p53 as a result of etoposide treatment, suggesting that the observed increase of p68 recruitment to the p21 promoter in response to DNA damage is due to higher levels and/or recruitment of p53.
Figure 8.
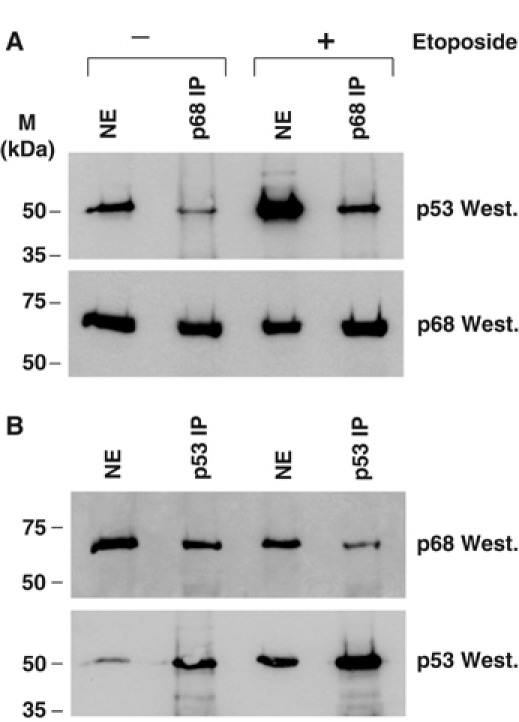
DNA damage does not have a significant effect on the interaction between p68 and p53. Western blots showing co-IP of p68 and p53 from nuclear extracts of U2OS cells, which had been treated with 100 μM etoposide, with untreated cells as controls. (A) p68 IP. (B) p53 IP.
RNAi depletion of p68 causes a reduction in p53-dependent apoptosis
To determine whether the observed effect of p68 RNAi depletion on expression of p53 target genes resulted in a biological effect, we examined whether a p68 knock-down affects p53-dependent apoptosis in SAOS-tetWTp53 cells, which stably express tetracycline-inducible WT p53 and undergo apoptosis in response to doxycycline. We used these cells to avoid possible complications arising from stresses inducing p53-independent apoptosis. p68 was knocked down by RNAi and cells were treated with doxycycline to induce p53 expression. We confirmed that p68 was knocked down efficiently and that, as expected, doxycycline treatment induced p53 expression (Supplementary data 3). For measurement of apoptosis, cells were harvested, stained for active caspases and detected by FACS as described in Materials and methods. Representative FACS profiles are shown in Figure 9A. Since cells transfected with either the control or the p68 siRNA had a somewhat higher background of apoptosis in the absence of doxycycline treatment when compared with untransfected cells (presumably due to the transfection), we determined the % increase in apoptosis resulting from doxycycline treatment and thus p53 induction. As shown in Figure 9B, p68 depletion resulted in a significant decrease in apoptosis resulting from p53 induction, when compared with either cells treated with the control siRNA or the untransfected control (approximately 29–19%), suggesting that p68 plays an important role in p53-dependent apoptosis.
Figure 9.
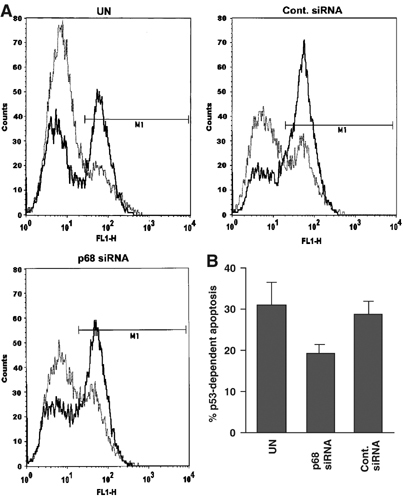
RNAi depletion of p68 causes a reduction in p53-dependent apoptosis. (A) Representative FACS profiles of SAOS-tetWTp53 transfected with a p68 siRNA or a control siRNA, with untransfected (UN) cells as a control. Cells were treated with 1 μM doxycycline for 36 h to induce p53 expression and thus apoptosis, with untreated cells acting as a control in each case. Apoptotic cells were stained as in Materials and methods and analysed by FACS. Graphs show number of cells (counts: y-axis) plotted against the fluorescence shift (FL1-H: x-axis). In each case, untreated cells are shown as a faint dotted line, while doxycycline-treated cells are shown as a bold solid line. (B) % apoptosis in doxycycline-treated cells minus % background apoptosis in untreated cells (i.e. p53-dependent apoptosis) for SAOS-tetWTp53 cells shown in (A). Graphs represent the average values from two independent experiments.
Discussion
In this report, we demonstrate a novel function for the DEAD box RNA helicase p68, namely as a potent coactivator of the tumour suppressor p53. We show that p68 synergises with p53 to stimulate transcription from p53-responsive promoters but not from promoters that lack p53-binding sites (Figure 1) and that this synergism is dependent on transcriptionally active p53 (Figure 2A). Our studies also revealed that p68 is an important element of the p53 transcriptional response to DNA damage. This represents a novel mechanism for regulating p53 function and defines a new physiological role for p68 in a critical pathway that typically protects organisms from tumorigenesis. Notably, suppression of endogenous p68 expression has no discernible effect on the induction of endogenous p53 protein in response to DNA damage (Figure 4A), that is, its stabilisation, which occurs through post-translational events. Strikingly, however, the data show that induced p53 requires the presence of p68 in order to effectively stimulate expression of key downstream genes involved in growth arrest (p21), apoptosis (Fas and PIG3) and negative feedback (mdm2). This was observed both at the protein level for p21 and Mdm2 (Figure 4A) and at the mRNA level by quantitative RT–PCR for p21, mdm2, Fas and PIG3 (Figure 5), consistent with the defect being in the ability of p53 to stimulate transcription of these genes. These effects are clearly specific since a promoter lacking p53-binding elements is not affected by p68 (Figure 1E) and the p68 siRNA does not suppress p72 or GAPDH expression (Figures 4A and 5E). Furthermore, an RNAi knock-down of the highly related p72 RNA helicase had no significant effect on the ability of p53 to induce p21 or Mdm2 expression following etoposide treatment (Figure 4B), suggesting that the effect on p53 is specific to p68. In addition, we show that a p68 RNAi knock-down has no effect on the induction of NF-κB in response to TNFα (Figure 9), indicating that p68 is not simply a general transcriptional coactivator. Importantly, we also show that RNAi depletion of p68 results in a significant reduction in the ability of cells to undergo apoptosis in response to p53 induction (Figure 6), indicating that p68 is important for the biological consequences of p53 induction and highlighting a role for p68 in p53-dependent apoptosis.
Our findings also show that p68 interacts with p53 in vitro (Figure 3A and B) and that endogenous p68 and p53 co-immunoprecipitate from nuclear extracts, indicating that the interaction is physiological (Figure 3C–E). Previous reports have shown that p68 can also interact with components of the transcription machinery including CBP/p300 and RNA polymerase II itself (Endoh et al, 1999; Rossow and Janknecht, 2003). These findings would therefore be consistent with p68 stimulating p53 function by assisting its association with the transcription complex. Moreover, p68 has been shown to interact with and stimulate ERα transcriptional activity (Endoh et al, 1999; Watanabe et al, 2001), possibly in a p53-independent manner. There are several potential mechanisms by which p68 could coactivate gene expression through single or combinational interactions with these, and perhaps other, transcription factors. Defining this mechanism precisely will be the next challenge in understanding p68 function. In this respect, it is of interest to note that other DEAD/DEAH box RNA helicases have been shown to act as transcriptional coactivators and to be associated with transcription complexes. These include RHII/Gu, which acts as a coactivator for c-Jun (Westermarck et al, 2002), and RNA helicase A, which stimulates transcription mediated by the cAMP-responsive factor (CREB) by acting as a bridge between CBP and the RNA polymerase II holoenzyme (Nakajima et al, 1997).
In an effort to explore potential mechanisms by which p68 could stimulate p53 transcriptional activity, we examined whether p68 is recruited to the promoter of the p53 target gene p21. Our ChIP experiments (Figure 7) show that, like p53, p68 is recruited to the p21 promoter but not to the control GAPDH promoter (Figure 7A), consistent with p68 coactivating p53 transcriptional activity by promoting initiation. In this respect, it is interesting to note that p68 has been shown to be recruited to the promoter of the ERα target promoter pS2 (Metivier et al, 2003). Interestingly, this recruitment of p68 to the p21 promoter is significantly enhanced in response to DNA damage (Figure 7A) and is dependent on p53 (Figure 7B). As shown in Figure 4, while etoposide treatment, as expected, causes a marked increase in the level of p53 protein, it has no effect on the levels of p68 protein. Therefore, while the increased recruitment of p53 to the p21 promoter could be explained by the increased level of p53 protein in the cell after etoposide treatment, the increase in p68 at the promoter suggests, instead, an increased level of recruitment to the p21 promoter in response to DNA damage, perhaps through interaction with p53 since the recruitment is also dependent on p53. However, we did not observe an increase in p53/p68 co-IP due to etoposide treatment, suggesting that DNA damage does not significantly enhance p53/p68 interaction per se (Figure 8). Additionally, for both p53 and p68, there is a low level of protein present at the promoter prior to treatment with etoposide, suggesting the presence of low levels of these proteins in a preassembled complex at the promoter prior to DNA damage-induced p53 activation, and consistent with results from a previous study on p53 recruitment to the p21 promoter before and after DNA damage (Espinosa et al, 2003). However, for both p53 and p68, there is a significant increase in recruitment after etoposide treatment (Figure 7A), suggesting an enhancement in the reinitiation of transcription in response to DNA damage.
While our findings indicate a significant and previously unknown function for p68, they also raise a number of questions and implications concerning p68 function. Firstly, it seems probable that p68 is a pleiotropic protein with a number of biological roles in different cellular pathways and with different biochemical activities. Our data from the p53/p68 cotransfection/luciferase assays (Figure 2B) and those from with respect to ERα (Endoh et al, 1999) suggest that ATPase/helicase activity can be uncoupled from the role of p68 in transcriptional transactivation. However, it is also possible that recruitment of p68 to transcriptional complexes localises other functions of p68, such as its ability to unwind RNA structures, to sites of transcriptional activity. This is important since p68 has been shown to be involved in pre-mRNA splicing (Liu, 2002) and mRNA stability (Bond et al, 2001). Thus, p68 may play a role in coupling the processes of transcription and pre-mRNA processing and RNA helicase activity may be required for the post-transcriptional functions of p68. Such effects may not be detected in reporter gene assays, which utilise a heterologous unspliced mRNA encoding the luciferase protein.
Another prediction from our results is that, if p68 is required for p53 transactivation function, loss of p68 coactivator function may be one of the many mechanisms that could attenuate p53 action during tumorigenesis. We previously reported that p68 is overexpressed in colon carcinomas in a heavily post-translationally modified form (mainly ubiquitylated protein) (Causevic et al, 2001). Interestingly, in that study, we observed a gradual disappearance of the ‘normal' p68 species during tumour progression and suggested that the loss of the normal p68, rather than the appearance of the modified forms, may be important in tumour development (Causevic et al, 2001). That report did not investigate whether this might influence p68-dependent effects on transcription, but it certainly raises the possibility that alteration of p68 function in this manner could be important during cancer development. Our finding that p68 appears to be required for p53 to function as a transcriptional activator in response to DNA damage thus suggests a potential role for p68 as a tumour cosuppressor.
Materials and methods
Cell lines
The cells lines used included the p53-null SAOS-2 (osteosarcoma) and H1299 (lung carcinoma) as well as U2OS (osteosarcoma) and MCF-7 (breast adenocarcinoma), both of which harbour WT p53. NF-κB induction experiments were performed with a HeLa cell line (HeLa57A) expressing the NF-κB-inducible reporter 3Enhancer-κB-conA-Luc and the control reporter pRC/RSV-β-galactosidase (Rodriguez et al, 1999), which was a gift from Ron Hay. Apoptosis assays were performed using SAOS-tetWTp53, an SAOS-2 cell line stably expressing a tetracycline-inducible WT p53, which was a gift from Carol Midgley.
Plasmids
Plasmids expressing WT or an ATPase/RNA helicase-inactive mutant (generated by mutating the DEAD motif to NEAD) p68 and WT or a transcriptionally inactive (L22Q/W23S) mutant of p53 under the control of the CMV promoter were obtained by cloning the respective cDNAs in the vector pcDNA3 (Invitrogen). Myc-tagged versions of p68 and p72 were in pcDNA3 and pSG5 (Stratagene), respectively. The plasmid encoding the L22Q/W23S p53 mutant was a gift from Ted Hupp.
Luciferase reporter plasmids under the control of p53-responsive promoters included pPG13LUC (the polyomavirus early promoter and 13 copies of a synthetic consensus p53-binding site; (el-Deiry et al, 1993), the p21 promoter (el-Deiry et al, 1993), the Bax promoter (Miyashita and Reed, 1995) and pRasH-Adluc, containing the intron 1 sequence of the human c-Ha-Ras gene upstream of the minimum adenovirus major late promoter sequence of pAdluc. pAdluc itself acted as a non-p53-responsive control (Deguin-Chambon et al, 2000).
Antibodies
The following antibodies were used: p68—PAb204 (Ford et al, 1988) and 2907 (a rabbit polyclonal antibody raised against the C-terminal 15 amino acids of p68); p72—K14 (a rabbit polyclonal antibody raised against amino acids 597–609 of p72); p53—CM1 (a rabbit polyclonal antibody raised against recombinant—p53), DO1 (Santa Cruz) and PAb421 (Harlow et al, 1981); p21—C-19 (Santa Cruz); Mdm2—SMP14 and D12 (Santa Cruz); actin—A2066 (Sigma); GST—27-4577-01 (Amersham). Appropriate anti-mouse, anti-rabbit and anti-goat secondary antibodies were obtained from DAKO.
Luciferase/β-galactosidase assays
H1299 cells were transfected by the calcium phosphate method described by Webster and Perkins (1999) with plasmids encoding p68, p53 and appropriate promoters/luciferase reporters as indicated in the figure legends, and luciferase activity was measured 24 h later using the E1501 kit from Promega. In each case, results are from three independent experiments, except for the p68/p53 titres, where duplicate transfections were carried out. Relative luciferase activity was obtained by comparing with transfected promoter/reporter construct alone, which was set to 1 in each experiment. β-Galactosidase assays were performed as described previously (Midgley et al, 2000). For the NF-κB induction experiment, HeLa57A cells were treated with 10 nM TNFα (Sigma) for 6 h and relative β-galactosidase activity was obtained by comparing with that of untreated cells, which was set to 1.
GST pull-downs
GST-tagged p68/p72 and GST vector control were expressed in 293 cells as described previously (Ogilvie et al, 2003) and purified on glutathione beads using standard conditions. p53 was translated in vitro using the TNT kit from Promega. The GST pull-down of in vitro-translated (35S-labelled) p53 was carried out as described by Hsieh et al (1999).
Nuclear extract preparation and co-immunoprecipitation
Nuclear extracts were prepared from U2OS and SAOS-2 cells as described by Dignam et al (1983) (except that the NaCl concentration was reduced to 330 mM, then diluted to 150 mM NaCl and treated with RNase/DNase). The extract was precleared with protein Sepharose G beads and IP was carried out in IP buffer (20 mM Hepes (pH 7.9), 150 mM NaCl, 0.5 mM DTT, 20% (v/v) glycerol, 10 mM NaF and protease inhibitor cocktail (Roche)) with antibody/protein G Sepharose beads for 1 h at 4°C. After washing in IP buffer plus 0.8% Igepal (Sigma), immunoprecipitated proteins were Western blotted.
Western blotting
Cell lysates were prepared in 50 mM Tris pH 8, 150 mM NaCl, 0.1% SDS, 1% Igepal (Sigma) and protease inhibitor cocktail (Roche). Proteins were separated by SDS–PAGE and Western blotted using standard conditions and appropriate primary and secondary antibodies. Immunoreactive proteins were detected using the ECL method (Amersham).
RNAi depletion of p68 and p72
Expression of endogenous p68/p72 was suppressed using specific siRNA duplexes (see Supplementary data 4 for sequence) and Scramble I duplex oligonucleotide as control from Dharmacon Research Inc. The depletion was carried out in MCF-7, U2OS and HeLa47A cells using two sequential transfections as recommended (http://www.mpibpc.gwdg.de/abteilungen/100/105/sirna.html) over 5 days. Cells were treated with 100 μM etoposide (MCF-7, U2OS) for 4 h, 10 nM TNFα (HeLa57A) for 6 h or 1 μM doxycycline SAOS-tet WT p53 for 36 h and, in each case, untreated cells served as additional controls. Cells were lysed in SDS/Igepal buffer (see above) for Western blotting, or Promega Passive Lysis Buffer for luciferase assays.
Quantitative RT–PCR
Total RNA was extracted from MCF-7 cells using the RNeasy kit (Qiagen), treated with RQ1 DNase (Promega) and reverse transcribed using RNase-free Superscript reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Quantitative (TaqMan) PCR was performed using the ABI-Prism 7700 sequence detection system and probes were labelled with a 5′ reporter 6-carboxyfluorescein (FAM) and 3′ quencher 6-carboxy-tetramethyl-rhodamine (TAMRA). The p21, mdm2, Fas/APO1 and PIG3 primer and probe sequences and PCR cycles used are given in Supplementary data 4. GAPDH and β-actin primers/probes (PE Applied Biosystems) were used as controls and all measurements were normalised against β-actin.
Chromatin immunoprecipitation–PCR
Cells (2.5 × 106) were seeded in 10 cm plates and the following day were treated with 100 μM etoposide for 2 h; untreated cells served as controls. Cells were washed twice in PBS; chromatin was crosslinked with 1.5% formaldehyde for 5 min at 37°C and cells were washed again twice in PBS. Cells were collected in 1 ml of 100 mM Tris–HCl (pH 9.4) and 10 mM DTT and the ChIP procedure was performed as described previously (Metivier et al, 2003) using the monoclonal antibodies PAb421 (p53) and PAb204 (p68) and the washing conditions described for PAb204 (Metivier et al, 2003). A control IP with no antibody was included. The p21 and GAPDH promoter primers and PCR cycles used are given in Supplementary data 4. In each case, a sample of the input DNA from untreated and etoposide-treated cells was taken prior to IP and included in the PCR reactions to compare the relative amounts of the relevant DNA in the different cell lysates. PCR products were separated on 10% polyacrylamide (Laemmli) gels without SDS.
Apoptosis assays
These were performed using the carboxyfluorescein FLICA apoptosis detection kit (Immunochemistry Technologies) according to the manufacturer's instructions. This kit employs a fluorescein-labelled caspase inhibitor, which binds to active caspases, thus labelling apoptotic cells. These are then detected by FACS.
Supplementary Material
Supplementary data 1
Supplementary data 2
Supplementary data 3
Supplementary data 4
Acknowledgments
We thank Ken Fernandes for help with the RT–PCR, Ron Hay for providing the HeLa57A cells and David Meek for helpful discussions. This work was supported by Tenovus Scotland, The Royal Society, the Biotechnology and Biological Sciences Research Council (PhD studentship to BJW) and the Association for International Cancer Research (PhD studentship to A-MFJ).
References
- Balint EE, Vousden KH (2001) Activation and activities of the p53 tumour suppressor protein. Br J Cancer 85: 1813–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond AT, Mangus DA, He F, Jacobson A (2001) Absence of Dbp2p alters both nonsense-mediated mRNA decay and rRNA processing. Mol Cell Biol 21: 7366–7379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causevic M, Hislop RG, Kernohan NM, Carey FA, Kay RA, Steele RJ, Fuller-Pace FV (2001) Overexpression and poly-ubiquitylation of the DEAD-box RNA helicase p68 in colorectal tumours. Oncogene 20: 7734–7743 [DOI] [PubMed] [Google Scholar]
- Courtois S, Verhaegh G, North S, Luciani MG, Lassus P, Hibner U, Oren M, Hainaut P (2002) DeltaN-p53, a natural isoform of p53 lacking the first transactivation domain, counteracts growth suppression by wild-type p53. Oncogene 21: 6722–6728 [DOI] [PubMed] [Google Scholar]
- Deguin-Chambon V, Vacher M, Jullien M, May E, Bourdon JC (2000) Direct transactivation of c-Ha-Ras gene by p53: evidence for its involvement in p53 transactivation activity and p53-mediated apoptosis. Oncogene 19: 5831–5841 [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res 11: 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825 [DOI] [PubMed] [Google Scholar]
- Endoh H, Maruyama K, Masuhiro Y, Kobayashi Y, Goto M, Tai H, Yanagisawa J, Metzger D, Hashimoto S, Kato S (1999) Purification and identification of p68 RNA helicase acting as a transcriptional coactivator specific for the activation function 1 of human estrogen receptor alpha. Mol Cell Biol 19: 5363–5372 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Espinosa JM, Verdun RE, Emerson BM (2003) p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell 12: 1015–1027 [DOI] [PubMed] [Google Scholar]
- Ford MJ, Anton IA, Lane DP (1988) Nuclear protein with sequence homology to translation initiation factor eIF-4A. Nature 332: 736–738 [DOI] [PubMed] [Google Scholar]
- Guil S, Gattoni R, Carrascal M, Abian J, Stevenin J, Bach-Elias M (2003) Roles of hnRNP A1, SR proteins, and p68 helicase in c-H-ras alternative splicing regulation. Mol Cell Biol 23: 2927–2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Crawford LV, Pim DC, Williamson NM (1981) Monoclonal antibodies specific for simian virus 40 tumor antigens. J Virol 39: 861–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirling H, Scheffner M, Restle T, Stahl H (1989) RNA helicase activity associated with the human p68 protein. Nature 339: 562–564 [DOI] [PubMed] [Google Scholar]
- Hsieh JK, Chan FS, O'Connor DJ, Mittnacht S, Zhong S, Lu X (1999) RB regulates the stability and the apoptotic function of p53 via MDM2. Mol Cell 3: 181–193 [DOI] [PubMed] [Google Scholar]
- Iggo RD, Jamieson DJ, MacNeill SA, Southgate J, McPheat J, Lane DP (1991) p68 RNA helicase: identification of a nucleolar form and cloning of related genes containing a conserved intron in yeasts. Mol Cell Biol 11: 1326–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iggo RD, Lane DP (1989) Nuclear protein p68 is an RNA-dependent ATPase. EMBO J 8: 1827–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iost I, Dreyfus M (1994) mRNAs can be stabilized by DEAD-box proteins. Nature 372: 193–196 [DOI] [PubMed] [Google Scholar]
- Lamm GM, Nicol SM, Fuller Pace FV, Lamond AI (1996) p72: a human nuclear DEAD box protein highly related to p68. Nucleic Acids Res 24: 3739–3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZR (2002) p68 RNA helicase is an essential human splicing factor that acts at the U1 snRNA-5′ splice site duplex. Mol Cell Biol 22: 5443–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F (2003) Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 115: 751–763 [DOI] [PubMed] [Google Scholar]
- Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT, Lane DP (2000) An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene 19: 2312–2323 [DOI] [PubMed] [Google Scholar]
- Miyashita T, Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299 [DOI] [PubMed] [Google Scholar]
- Nakajima T, Uchida C, Anderson SF, Lee CG, Hurwitz J, Parvin JD, Montminy M (1997) RNA helicase A mediates association of CBP with RNA polymerase II. Cell 90: 1107–1112 [DOI] [PubMed] [Google Scholar]
- Ogilvie VC, Wilson BJ, Nicol SM, Morrice NA, Saunders LR, Barber GN, Fuller-Pace FV (2003) The highly related DEAD box RNA helicases p68 and p72 exist as heterodimers in cells. Nucleic Acids Res 31: 1470–1480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Thompson J, Hay RT, Dargemont C (1999) Nuclear retention of IkappaBalpha protects it from signal-induced degradation and inhibits nuclear factor kappaB transcriptional activation. J Biol Chem 274: 9108–9115 [DOI] [PubMed] [Google Scholar]
- Rossow KL, Janknecht R (2003) Synergism between p68 RNA helicase and the transcriptional coactivators CBP and p300. Oncogene 22: 151–156 [DOI] [PubMed] [Google Scholar]
- Stevenson RJ, Hamilton SJ, MacCallum DE, Hall PA, Fuller Pace FV (1998) Expression of the ‘dead box' RNA helicase p68 is developmentally and growth regulated and correlates with organ differentiation/maturation in the fetus. J Pathol 184: 351–359 [DOI] [PubMed] [Google Scholar]
- Szak ST, Mays D, Pietenpol JA (2001) Kinetics of p53 binding to promoter sites in vivo. Mol Cell Biol 21: 3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner NK, Linder P (2001) DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell 8: 251–262 [DOI] [PubMed] [Google Scholar]
- Uhlmann-Schiffler H, Rossler OG, Stahl H (2002) The mRNA of DEAD box protein p72 is alternatively translated into an 82-kDa RNA helicase. J Biol Chem 277: 1066–1075 [DOI] [PubMed] [Google Scholar]
- Venot C, Maratrat M, Sierra V, Conseiller E, Debussche L (1999) Definition of a p53 transactivation function-deficient mutant and characterization of two independent p53 transactivation subdomains. Oncogene 18: 2405–2410 [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Watanabe M, Yanagisawa J, Kitagawa H, Takeyama K, Ogawa S, Arao Y, Suzawa M, Kobayashi Y, Yano T, Yoshikawa H, Masuhiro Y, Kato S (2001) A subfamily of RNA-binding DEAD-box proteins acts as an estrogen receptor alpha coactivator through the N-terminal activation domain (AF-1) with an RNA coactivator, SRA. EMBO J 20: 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Webster GA, Perkins ND (1999) Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol 19: 3485–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J, Weiss C, Saffrich R, Kast J, Musti AM, Wessely M, Ansorge W, Seraphin B, Wilm M, Valdez BC, Bohmann D (2002) The DEXD/H-box RNA helicase RHII/Gu is a co-factor for c-Jun-activated transcription. EMBO J 21: 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Stephen CW, Luciani MG, Fahraeus R (2002) p53 stability and activity is regulated by Mdm2-mediated induction of alternative p53 translation products. Nat Cell Biol 4: 462–467 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data 1
Supplementary data 2
Supplementary data 3
Supplementary data 4