

Abstract
Cyclin E, an activator of phospho-CDK2 (pCDK2), is important for cell cycle progression in metazoans and is frequently overexpressed in cancer cells. It is essential for entry to the cell cycle from G0 quiescent phase, for the assembly of prereplication complexes and for endoreduplication in megakaryotes and giant trophoblast cells. We report the crystal structure of pCDK2 in complex with a truncated cyclin E1 (residues 81–363) at 2.25 Å resolution. The N-terminal cyclin box fold of cyclin E1 is similar to that of cyclin A and promotes identical changes in pCDK2 that lead to kinase activation. The C-terminal cyclin box fold shows significant differences from cyclin A. It makes additional interactions with pCDK2, especially in the region of the activation segment, and contributes to CDK2-independent binding sites of cyclin E. Kinetic analysis with model peptide substrates show a 1.6-fold increase in kcat for pCDK2/cyclin E1 (81–363) over kcat of pCDK2/cyclin E (full length) and pCDK2/cyclin A. The structural and kinetic results indicate no inherent substrate discrimination between pCDK2/cyclin E and pCDK2/cyclin A with model substrates.
Keywords: cyclin E, cyclin-dependent kinases, protein kinase regulation, oncogenes
Introduction
Cyclins play a key role in the orderly progression of the cell division cycle through their timed expression and their ability to bind, activate and enhance substrate affinity of their associated cyclin-dependent protein kinases (CDKs). Human cyclin E1 was first identified as a G1 cyclin on the basis of its ability to complement mutations in the CLN genes in budding yeast (Koff et al, 1991; Lew et al, 1991). A second isoform, cyclin E2, was later reported that is 61% identical to cyclin E1 (Lauper et al, 1998; Zariwala et al, 1998). Both E-type cyclins are activators of cyclin-dependent kinase 2 (CDK2) (reviewed in Moroy and Geisen, 2004). In mammalian cells, the mitogen-stimulated activities of CDK4 or CDK6 in association with a D-type cyclin promote transcription of cyclin E during G1 phase and activation of CDK2/cyclin E kinase activity through the redistribution of cell cycle inhibitors (Sherr and Roberts, 1999). Levels of cyclin E decline during S phase in response to auto- and other phosphorylation events that target cyclin E for ubiquitination by the Skp1/cullin/Cdc4 F-box protein (SCFCdc4) complex and subsequent degradation by the proteasome (Clurman et al, 1996; Won and Reed, 1996; Strohmaier et al, 2001). As cyclin E levels decline, those of cyclin A increase and it is thought that it is the activity of the CDK2/cyclin A complex that drives cells through S phase. As cells enter mitosis, cyclin A associates with and activates CDK1 but shortly after cyclin A is targeted for degradation by the anaphase-promoting complex ubiquitination. Cyclin B in complex with CDK1 drives cells through M phase. Cyclin E binds preferentially to CDK2, and CDK1/cyclin E complexes are not normally found in cells (Koff et al, 1992; Arooz et al, 2000). High concentrations of cyclin A, but not high concentrations of cyclin E, are able to trigger entry into mitosis (Strausfeld et al, 1996). The origins of the different specificities of cyclin A and cyclin E for the different CDKs are not understood.
The most important role of cyclin E is to promote the assembly of the prereplication complex, through the loading of Cdc6 to the origins of replication and recruitment of the Mcm2–7 proteins, during the transition from quiescent G0 to G1 phases of the cell cycle and subsequently to trigger replication origin firing in order for cells to enter S phase (Su and O'Farrell, 1998; Cook et al, 2002; Coverley et al, 2002). CDK2/cyclin E substrates also include proteins involved in the regulation of transcription (such as pRb and pRb-related proteins), pre-mRNA splicing, histone biosynthesis, gene expression control, centrosome duplication and cell cycle progression (for example, p27Kip1 and CDC25) (Moroy and Geisen, 2004). Although some substrates appear to be specific for CDK2/cyclin E in vivo, almost all can be phosphorylated by CDK2/cyclin A in vitro. This has raised the question of why cells need both cyclin E and cyclin A. In a dramatic development in 2003, it was shown from knockout mouse embryo fibroblast cells, in which both cyclin E1 and cyclin E2 were ablated (Geng et al, 2003; Parisi et al, 2003), that cyclin E is dispensable for cell cycling but is essential for cells to re-enter the cell cycle from the quiescent G0 state and for endoreduplication, highlighting the important role of cyclin E in the assembly of the prereplication complex.
Recent knockout and other experiments have also shown that CDK2 is dispensable for cell cycle progression and cell division (Ortega et al, 2003; Tetsu and McCormick, 2003). The cyclin E and CDK2 ablation results demonstrate the robustness of the cell cycle but raise questions of the essential function of cyclin E since the major partner of cyclin E in mouse cells is CDK2. In the CDK2−/− cell extracts, immunoprecipitation with cyclin A also brought down kinase activity (presumably CDK1) but immunoprecipitation with cyclin E showed no kinase activity. The CDK2−/− mice are viable but cyclin E1−/−, E2−/− mice die in utero. This suggests that cyclin E may have a role that is independent of kinase activity, such as a scaffold or assembly protein. Evidence for a CDK2-independent role has come from recent work (Matsumoto and Maller, 2004) in which a 20-amino-acid sequence in cyclin E was identified as a centrosome localisation signal that is essential for centrosomal targeting and promotion of DNA synthesis in a CDK2-independent manner.
Cyclin E deregulation is directly implicated in cancer (Bortner and Rosenberg, 1997). Unusually high and persistent levels of cyclin E have been observed in human tumour cells, especially in the most aggressive cancers (Porter et al, 1997; Donnellan and Chetty, 1999; Malumbres et al, 2003). Deregulation in some naturally occurring cancers has been associated with mutations in the hCDC4 gene (Strohmaier et al, 2001; Spruck et al, 2002; Rajagopalan et al, 2004) leading to the lack of the ability of cells to degrade cyclin E shortly after S-phase entry. The consequences of some of these mutations can be understood from the structure of the Skp1/Cdc4 complex with the cyclin E pThr380 phospho-peptide (Orlicky et al, 2003). Deregulation of cyclin E accelerates the entry of cells into S phase but causes inefficient progression through S phase. The untimely presence of cyclin E has been shown to interfere with the replication complex assembly as cells exit mitosis (Ekholm-Reed et al, 2004). Forced overexpression of cyclin E (but not A) results in chromosome instability (Spruck et al, 1999). Remarkably, cyclin E-deficient cells are resistant to oncogenic transformation by Ras (Geng et al, 2003). There is evidence that cyclin E might have a CDK2-independent role in transformation of normal cells. Geisen and Moroy (2002) have shown that some cyclin E1 mutants, which were defective in their ability to form an active kinase complex, were able to malignantly transform rat embryonic fibroblasts in cooperation with Ha-Ras.
In addition to association with cyclin A or E, activation of CDK2 requires post-translational phosphorylation on residue Thr160 (numbering in the human enzyme) by the activating CDK kinase complex CDK7/cyclin H/Mat1. Structural studies on CDK2 in association with cyclin A have provided detailed structural explanations for the activation of CDK2 by cyclin A and by phosphorylation and the mode of recognition of substrates (De Bondt et al, 1993; Jeffrey et al, 1995; Russo et al, 1996b; Brown et al, 1999a, 1999b). In order to understand the structural basis by which cyclin E activates CDK2 and by which it might promote a CDK2-independent role, we have determined the structure of cyclin E1 in complex with pCDK2.
Results
Structure of pCDK2/cyclin E1
Full-length cyclin E1 was expressed in insect cells and complexed with pCDK2, but we were unable to obtain crystals. Noting the results of Porter et al (2001), who had observed low-molecular-weight forms of cyclin E in breast cancer cells and showed that cyclin E is selectively cleaved by elastase, we constructed a truncated version of cyclin E1 (residues 81–363). Cyclin E1 (residues 81–363) lacks two important recognition sites: the nuclear localisation sequence (Jackman et al, 2002; Moore et al, 2002) and the sites of phosphorylation that target cyclin E for ubiquitination (Welker et al, 2003) (Figure 1). The construct includes both cyclin boxes identified from sequence similarities with cyclin A (Brown et al, 1995). The pCDK2/cyclin E1 (81–363) crystal structure was solved by molecular replacement and refined at 2.25 Å resolution (Table I).
Figure 1.
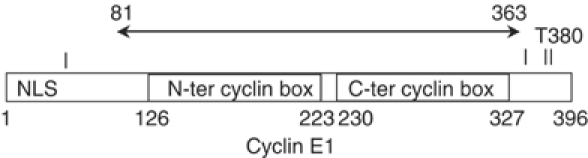
Schematic diagram of cyclin E1 showing the relative positions of the nuclear localisation sequence (NLS) residues 12RSRKRK (Moore et al, 2002), the major site of phosphorylation (Thr380) and other phosphorylation sites (Thr62, Ser372 and Ser384 shown as bars) that are required for targeting cyclin E for ubiquitination and turnover (Welker et al, 2003), the construct used for the crystals of pCDK2/cyclin E1 (residues 81–363) and the positions of the N- and C-terminal cyclin box folds.
Table 1.
Summary of pCDK2/cyclin E1 data collection and refinement statistics
| Data collection | ||
| X-ray source | ESRF station BM14, λ=0.9537 Å | ESRF station 14.4, λ=1.009 Å |
| Space group and unit cell | P41212, a=b=100.3 Å, c=150.5 Å | P41212, a=b=99.6 Å, c=150.0 Å |
| Resolution range (last shell) (Å) | 38.5–3.30 (3.48–3.30) | 83.1–2.25 (2.37–2.25) |
| Rmerge | 0.108 (0.348) | 0.079 (0.417) |
| Number of observations | 44 340 (6425) | 143 647 (21 059) |
| Number of unique observations | 11 651 (1680) | 35 727 (5118) |
| Mean I/σ(I) | 11.3 (3.3) | 13.6 (2.6) |
| Completeness (%) | 97.1 (98.2) | 98.9 (98.5) |
| Multiplicity | 3.8 (3.8) | 4.0 (4.1) |
| Wilson B-factor (Å2) | 65.2 | 45.2 |
| Refinement | ||
| Protein atoms | 4617 | |
| Water molecules | 592 | |
| R (Rfree) | 0.184 (0.246) | |
| R.m.s.d. bond lengths (Å) | 0.018 | |
| R.m.s.d. bond angles (deg) | 1.69 |
Cyclin E1 (81–363) (hereafter referred to as cyclin E1) associates tightly with pCDK2. The complex shows pCDK2 structure in its active conformation, characterised by the movement of the C-(PSTAIRE) helix and the correct location of the activation segment. The overall structures of the active pCDK2/cyclin E1 and active pCDK2/cyclin A are similar, despite sequence differences between cyclin E1 and cyclin A, but there are also significant differences (Figure 2). The conformation and sequence of the C-terminal cyclin box of cyclin E1 differs significantly from that of cyclin A and makes additional interactions with pCDK2 that are not made by cyclin A, thus strengthening the pCDK2/cyclin E1 interface.
Figure 2.
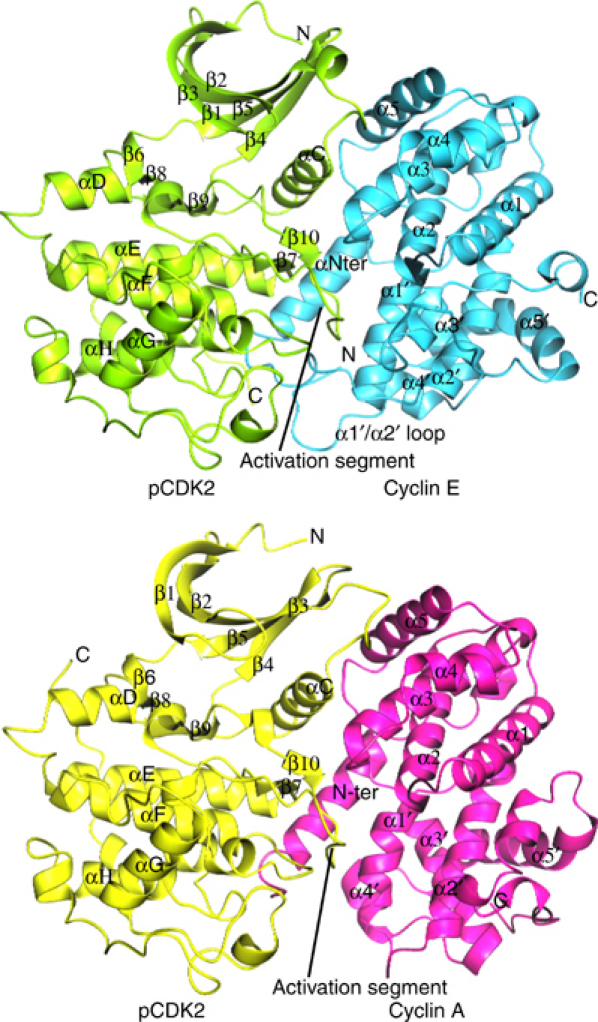
pCDK2/cyclin E1 and pCDK2/cyclin A complexes. pCDK2/cyclin E1 is shown in the top panel with pCDK2 in green and cyclin E1 in cyan. The activation segment of pCDK2 and the α1′/α2′ loop of cyclin E1 are labelled. pCDK2/cyclin A is shown in the lower panel with pCDK2 in yellow and cyclin A in magenta. Secondary structural elements are labelled. It is apparent that cyclin E1 makes more contacts to pCDK2 that involve the α1′/α2′ loop than does cyclin A.
Structure of pCDK2 in the pCDK2/cyclin E1 complex. The typical kinase fold of pCDK2 comprises an N-terminal lobe (residues 1–82), which is mostly β-sheet with one helix, the C-helix, which contains the sequence PSTAIRE. The C-terminal lobe (residues 83–297) is mostly α-helix. It contains the catalytic residues responsible for promoting phosphorylation and the activation segment (residues 145 (DFG)–172 (APE)) that includes the phospho-threonine residue pT160. The ATP binding site is located between the two lobes and ATP recognition involves residues from both lobes. The peptide substrate binding site is associated with the C-terminal lobe and the correct orientation of the activation segment plays the key role for selective recognition of the sequence motif SPxK (Brown et al, 1999a). In the crystal structure of pCDK2/cyclin E1, the PSTAIRE (C-helix) is in its rotated inner conformation that favours a hydrogen bond between Glu51 from the PSTAIRE helix and Lys33 and hence allows Lys33 to adopt a conformation that is ready to stabilise the correct position of the triphosphate moiety of ATP for catalysis. Despite no nucleotide being bound in the pCDK2/cyclin E1 crystals, all the residues that are involved in the recognition of ATP are in similar positions as those observed in the productive complex of pCDK2/cyclin A/AMPPNP/peptide substrate. The only exception is the glycine-rich loop (CDK2 residues Glu8–Tyr19), which is displaced away from the ATP binding site by ∼1–2 Å, presumably because of the lack of bound AMPPNP in the crystal. The r.m.s.d. in Cα positions between pCDK2 in the pCDK2/cyclin E1 and pCDK2/cyclin A complexes is 0.9 Å for residues 1–288 (excluding residues 38–40).
Surprisingly, the pCDK2 C-terminal residues 289–296 are in very different positions in the pCDK2/cyclin E1 and pCDK2/cyclin A complexes (Figure 2). In the pCDK2/cyclin A complex, the C-terminal residues wrap around the outside of the kinase and come close to the αD helix where Leu296 stacks against Phe90. In the pCDK2/cyclin E1 complex, the direction of the polypeptide chain from residue 289 onwards is almost 180° from that in the pCDK2/cyclin A complex and residues 289–297 make no contact with pCDK2 molecule. Residue Leu296 reaches across to a symmetry-related molecule and docks into the hydrophobic RxL binding pocket on cyclin E1. This change in the CDK2 C-terminal residues does not affect the important catalytic residues. It appears that these eight CDK2 C-terminal residues are easily distorted in response to lattice contacts.
Cyclin E1 structure. Cyclin E1, like cyclin A, is organised around two five-helical domains with additional helices at the N- and C-termini (Figure 3). The first domain, the N-terminal cyclin box fold, comprises the N-terminal α-helix followed by five helices with identical topology and structure to the N-terminal cyclin box fold of cyclin A (Figure 3). Helix α3 forms an organising hydrophobic core of the motif and its central portion is surrounded by the other four helices. After structural alignment of the N-terminal cyclin box folds, the sequence identity between cyclin E1 (residues 126–225: α1–α5) and cyclin A (residues 207–305) is 42% and the r.m.s.d. in Cα atoms is 0.8 Å for 100 atoms. In particular, the conserved small amino acids Ala154 and Ala184 (corresponding to Ala235 and Ala264 in cyclin A) allow close packing of the α2 and α3 helices.
Figure 3.
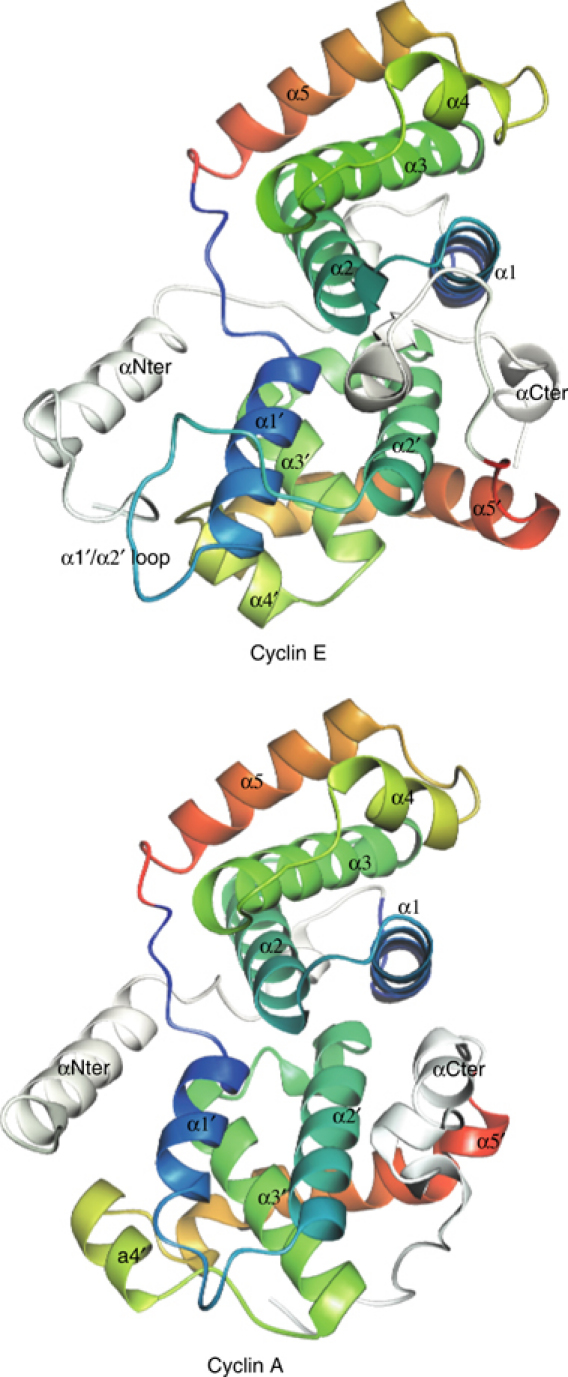
Structures of cyclins E1 and A. The diagrams are colour coded following the spectrum (blue to red) from α1 to α5 of the N-terminal cyclin box fold and then from α1′ to α5′ of the C-terminal cyclin box fold. The N- and C-terminal helices are shown in white. The N-terminal cyclin box folds of cyclin E1 and cyclin A are very similar but there are significant differences in the C-terminal cyclin box folds. For further details, see text.
The C-terminal cyclin box of cyclin E1 superficially resembles that of cyclin A in the arrangement of the five α-helices, but there are significant differences in the length of the α1′ helix, which is longer in cyclin E1, in the extended loop in cyclin E1 between α1′ and α2′, in the relative disposition of the α3′ and α4′ helices, in the direction and kinking of the α5′ helix and at the C-terminal region, where in cyclin E1 there is a short region of 310 helix that interacts with the C-terminal cyclin box followed by the C-terminal α-helix that runs antiparallel to the C-terminal helix of cyclin A (Figure 3). The loop α1′/α2′ (residues 244–254) has the highest temperature factors of the cyclin E1 molecule, but the loop is well located in the electron density. The sequence identity between cyclin E1 (residues 226–330 (α1′–α5′)) and cyclin A (residues 306–401) is 13% and the r.m.s.d. in Cα atoms is 1.4 Å for 72 atoms. The two alanine residues (Ala333 and Ala363) that facilitate the close packing of the α2′ and α3′ helices in cyclin A are not conserved in cyclin E1. The corresponding residues are Ile263 and Phe292 and these make an intimate hydrophobic packing following the relative displacement of α2′ and α3′ in cyclin E1. The two cyclin boxes of cyclin E1 show only a limited similarity (r.m.s.d. between residues 126–222 and 226–327 is 2.2 Å for 40 atoms). In contrast, in cyclin A, the two cyclin boxes (residues 207–302 and 310–401) superimpose with an r.m.s.d. of 1.7 Å for 70 residues.
Despite the differences in sequence, the internal packing of the two cyclins is generally similar with many complementary changes. Cyclin E1 Arg130 (arginine of the conserved MRAIL motif in α1) makes a buried ion pair with Asp159 (α2), similar to that formed by the equivalent residues in cyclin A. The mutation Arg130 to Ala in cyclin E1 results in a cyclin that is unable to bind CDK2 (Clurman et al, 1996; Geisen and Moroy, 2002). The structure suggests that disruption of this ion pair and an uncompensated buried charged group would lead to some protein unfolding. There is an additional buried ion pair in cyclin E1 between Arg114 (end of N-terminal helix) and Asp156 (α2), which is not made in cyclin A because of sequence changes (R114 is equivalent to Pro195 in cyclin A).
Structure of the pCDK2/cyclin E1 interface
The pCDK2/cyclin E1 interface is the key to the activating properties of the cyclin. The interface is similar to that of pCDK2/cyclin A, but there are additional interactions. The molecular surface area buried is 3252 Å2 for the pCDK2/cyclin E1 complex and 2839 Å2 for the pCDK2/cyclin A complex, an increase of 14% for the pCDK2/cyclin E1 complex. The interacting residues for the cyclins and for pCDK2 are summarised in Figure 4. As in cyclin A, cyclin E1 binds pCDK2 predominantly through the C-(PSTAIRE) helix and activation segment. In cyclin E1, the principal contact residues include those from the N-terminal helix, helices α3, α4, α5 and α1′, the loop between α5 and α1′, the loop between α1′ and α2′, and residues from the C-terminal region (Figure 4A). The additional contacts made by the α1′/α2′ loop and the C-terminal region contribute to increased buried molecular surface area in pCDK2/cyclin E1 compared to pCDK2/cyclin A.
Figure 4a.
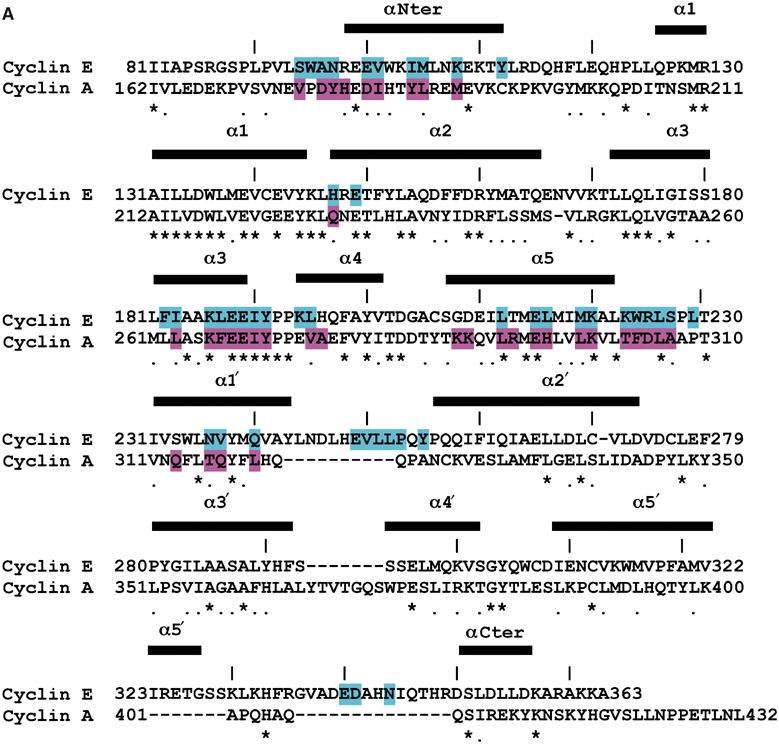
Sequence alignments showing residues in contact at the pCDK2/cyclin interfaces. (A) Cyclin E1 and cyclin A aligned on the basis of structure for the construct used for cyclin E1 expression (residues 81–363). The secondary structure elements for cyclin E1 are indicated. Residues coloured in cyan from cyclin E1 are in contact with pCDK2 in the pCDK2/cyclin E1 complex and residues coloured in magenta from cyclin A are in contact with pCDK2 in the pCDK2/cyclin A complex. Contact is defined as any atoms <3.5 Å in separation from cyclin and pCDK2. Every 10th residue is marked with a line for cyclin E1.
Interactions with the PSTAIRE helix. The pCDK2 C-helix (PSTAIRE) packs on one side almost parallel to the α5 cyclin E1 helix and is nearly perpendicular to the C-terminal end of the cyclin E1 α3 helix, as observed in pCDK2/cyclin A (Figure 5A). There are extensive hydrophobic interactions especially with pCDK2 Ile49 and Ile52. The interface is reinforced by hydrogen bonds that are similar to those in the pCDK2/cyclin A complex, despite some sequence changes (Figure 5A). Slightly different interactions are made by pCDK2 Lys56. In pCDK2/cyclin A, the side chain of Lys56 contacts Asp305, but in cyclin E1, the corresponding residue is Arg225, and in the pCDK2/cyclin E1 complex, both basic groups (pCDK2 Lys56 and cyclin E1 Arg225) are exposed and turned away from one another (Figure 5A).
Figure 5.
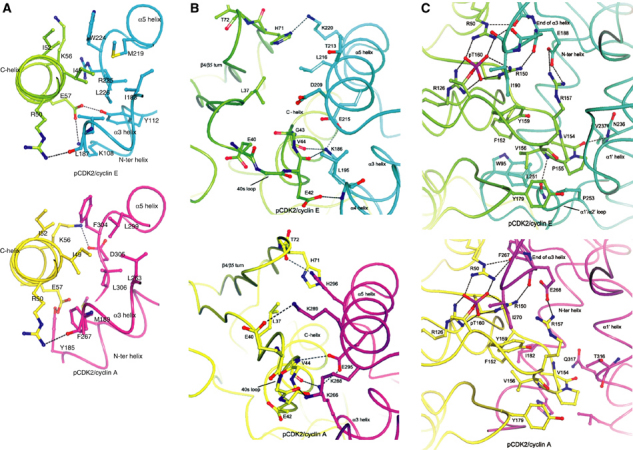
Details of the interactions between pCDK2/cyclin E1 and pCDK2/cyclin A. pCDK2/cyclin E1 is shown in the top panel and pCDK2/cyclin A in the lower panel. The colour scheme is the same as in Figure 1. (A) Interactions with the C-(PSTAIRE) helix. The hydrophobic face at the end of the cyclin E1 or cyclin A α5 helix interacts with hydrophobic residues Ile49 and Ile52 on pCDK2. The interface is further stabilised with hydrogen bonds from pCDK2 Arg50 to main-chain carbonyl of cyclin E1 Leu187, an interaction that is equivalent to that in pCDK2/cyclin A. With cyclin E1, there are also hydrogen bonds from pCDK2 Glu57 to cyclin E1 Tyr112 and Lys108 that are not made in pCDK2/cyclin A, while in pCDK2/cyclin A, there is a hydrogen bond from pCDK2 Lys56 to cyclin A Asp305, a contact that is not made in pCDK2/cyclin E because of sequence changes. For further details, see text. (B) Contacts at the 40s loop preceding the C-helix and the 70s loop at the β4/β5 turn. In the cyclin E1 complex, pCDK2 Glu42 hydrogen bonds to the main-chain nitrogen of cyclin E1 Leu195 at the start of α4, while in pCDK2/cyclin A, this residue makes no contacts because of a difference in conformation of the 40s loop. The pCDK2 main-chain carbonyls of Glu42 and Val44 contact cyclin E1 Lys186. Lys186 is localised by an ion pair with Glu215. At the pCDK2/cyclin A interface, Val44 main-chain carbonyl oxygen contacts Lys288, which in turn forms an ion pair with Glu295. In the pCDK2/cyclin A complex, pCDK2 Glu40 contacts cyclin A Lys289 and pCDK2 Thr41 main-chain carbonyl receives a hydrogen bond from cyclin A Lys289. These contacts are not possible in cyclin E1 because of sequence changes. At the β4/β5 turn (the 70s loop) in the pCDK2/cyclin A complex, there are contacts from pCDK2 His71 side chain and Thr72 main-chain oxygen to cyclin A His296 from the α5 helix. Because of sequence changes, the 70s loop adopts a different conformation and His71 interacts with Lys220. (C) Contacts at the pCDK2 activation segment and the α1′/α2′ loop of cyclin E1. The contacts to the phospho-Thr160 are described in the text. The α1′/α2′ loop makes additional interactions. The main chain oxygen of pCDK2 Val 154 hydrogen bonds to cyclin E1 Val237 (α1′ helix) main-chain nitrogen. Leu251 main-chain oxygen hydrogen bonds to the main-chain nitrogen of pCDK2 Val156 and Leu251 side chain makes van der Waals interactions with the pCDK2 residues Phe152 and Val156. These hydrophobic interactions are supported by cyclin E1 Trp95 from the region before the N-terminal helix, a region with a different conformation in cyclin E1 from that in cyclin A. For further details, see text.
Interactions with the pCDK2 40s and 70s loop. In the loop (residues 33–41) immediately preceding the PSTAIRE helix (the 40s loop), the contacts in the pCDK2/cyclin E1 complex are different from those of the pCDK2/cyclin A complex as a result of sequence changes (Figure 5B). The 40s loop shows conformational variability in a number of CDK2/cyclin A ligand complexes. Cyclin A residues Lys288 and Lys289 are changed to Gly208 and Asp209 in cyclin E1 and this results in a change of pCDK2 loop conformation and hydrogen bonding partners at the interface in order to avoid a close contact between cyclin E Asp209 and pCDK2 Glu40. Details are described in Figure 5B. There is also a conformational shift in the pCDK2 loop region 71–74 (β4/β5 loop; the 70s loop) compared with pCDK2/cyclin A. The change of His296 in cyclin A to Leu216 in cyclin E1 causes a shift of the 70s loop to avoid contact of the leucine with the pCDK2 His71 and Thr72. In the shifted position of the 40s loop, Leu216 in cyclin E1 can contact Leu37 of pCDK2 (Figure 5B). Thus, despite sequence differences between cyclin A and cyclin E1 at the interface region with pCDK2, the sequence changes can be accommodated by alternative conformations of the pCDK2 40s and 70s loop regions without affecting the pCDK2 catalytic residues. The cyclin remains rigid, assisted by the cyclin E1 internal ion pair between Lys186 (α3) and Glu215 (α5) (Figure 5B) that mimics the ion pair of Lys266 to Glu295 in cyclin A, a hall mark of cyclin folds (Jeffrey et al, 1995).
Interactions with the activation segment. The pCDK2 activation segment is the key to protein substrate recognition. In the pCDK2/cyclin E1 complex, the activation segment (pCDK2 residues 145–171) is in the active conformation as observed in the complex of pCDK2/cyclin A with peptide substrate. The cyclin in concert with phosphorylation plays a critical role in the correct alignment of this region. The phospho-Thr160, acting as an organising centre, makes contacts with three arginine residues as in pCDK2/cyclin A: Arg50 from the PSTAIRE helix, Arg126 from the catalytic loop and Arg150 from the start of the activation segment (Figure 5C). Two of these arginines, Arg50 and Arg150, hydrogen bond to main-chain carbonyl groups of cyclin E1 residues Leu187 and Glu188. This arrangement is similar to that in pCDK2/cyclin A complex. In both complexes, a nonpolar cyclin residue (Ile190 in cyclin E1 and Ile270 in cyclin A) shields the phospho-threonine (Figure 5C). There are additional contacts made by cyclin E1 to the activation segment that appear to further localise the segment (Figure 5C). The α1′/α2′ loop in the region of residues 248–255 threads under the activation segment in the region of residues 152–156 and makes a number of interactions. The most important are those from cyclin E Leu251 and are described in more detail in Figure 5C legend. Additional interactions are also made in the cyclin E1 complex after the end of the activation segment before the pCDK2 αF helix where pCDK2 Tyr179 stacks with Pro253 from the cyclin E1 α1′/α2′ loop (Figure 5C).
Regions involved in potential CDK2-independent activity
Centrosome recognition site. The 20-amino-acid sequence (residues 230–249 in the human sequence) identified as essential for binding of cyclin E to centrosomes and promoting DNA synthesis in a CDK2-independent manner (Matsumoto and Maller, 2004) is located on helix α1′ and the start of the α1′/α2′ loop (Figure 6). There are few contacts to pCDK2 in this region (Figure 4A). Some of the conserved residues whose mutation prevents the centrosome interaction (Ser233Ala, Trp234Ala and Asn237Ala) are located in the α1′ helix and are buried, suggesting that their mutation might alter conformation. Gln240 (the fourth residue of the tetra mutation that prevents interaction) orients part of the α1′/α2′ loop in the region of residues 252–254 through hydrogen bonds from the glutamine side chain to Leu252 main-chain nitrogen and Gln254 main-chain oxygen. Mutation of this residue would disrupt the loop. Residues 243–249 are exposed and form a strong potential protein recognition site. It has been suggested that the centrosome localisation sequence may be essential for transformation through promotion of S-phase entry independently of CDK2. The interacting partner on the centrosome and its downstream signalling have yet to be identified.
Figure 6.
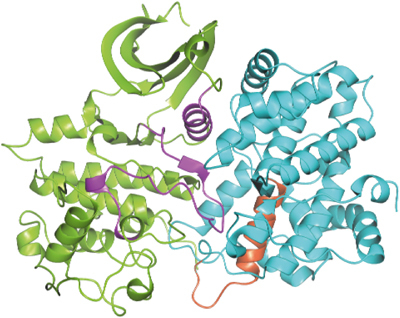
Structure of pCDK2 (in green) in complex with cyclin E1 (in cyan) with the 20-amino-acid sequence (residues 230–249) that are involved in centrosome localisation (Matsumoto and Maller, 2004) shown in red. The PSTAIRE (C-) helix and the activation segment, the two major regions of contact between pCDK2 and cyclin E1, are shown in magenta.
Potential regions involved in malignant transformation. In an effort to distinguish the roles of cyclin E1 in cells, Geisen and Moroy (2002) assayed the ability of a number of mutant cyclin E1s to activate CDK2 and to malignantly transform rat embryonic fibroblasts in cooperation with Ha-Ras. Some cyclin E1 mutants were defective in their ability to form an active kinase complex and promote G1 to S transition but were still able to malignantly transform cells. These mutants, one with a deletion of residues 129–216 and another with deletion of residues 161–220, lack almost all the N-terminal cyclin box fold up to and including the α5 helix (Figure 4A) and hence it is not surprising that they do not activate CDK2. However, their ability to transform suggests that parts of cyclin E1 of the C-terminal cyclin box fold may perform as yet unidentified functions that contribute to malignant transformations.
Binding sites on pCDK2 and cyclin E1
Kinase-associated phosphatase and Cks1 sites. pCDK2 contains a number of binding sites for other molecules such as the regulatory protein Cks1 (Bourne et al, 1996) and the kinase-associated phosphatase KAP (Song et al, 2001). Examination of these binding sites, which include the L14 loop and part of the G helix of CDK2, shows that both the Cks1 and the KAP binding sites are fully available in the pCDK2/cyclin E1 complex.
The p27Kip1 inhibitor recognition site. The CDK inhibitor p27 is an important regulator of CDK activity and in cancer cells is frequently downregulated. The complex of p27 with pCDK2/cyclin A binds to both cyclin A and pCDK2 causing a dramatic change in conformation of the kinase (Russo et al, 1996a). Binding to cyclin A is achieved through the p27 sequence motif RxLFG, which docks into the cyclin A hydrophobic pocket making contacts with the α1 helix residues Met210, Ile213, Leu214, Trp217 and Glu220 and α3 helix residues Arg250, Leu253 and Gln254. Each of these residues is conserved both in sequence and in conformation in cyclin E1, with the exception of only one residue: Arg250 in cyclin A is changed to Lys170 in cyclin E1. Thus, recognition at the RxL site is likely to be identical in cyclins A and E. Although there are a few sequence changes between cyclin A and cyclin E1 in the other residues that contact p27 as the inhibitor chain heads in a helix across the cyclin α5 helix towards pCDK2, the positions of these residues are similar and it appears that p27 could bind in an identical manner as an inhibitor to the pCDK2/cyclin E1 complex as to the pCDK2/cyclin A complex.
The VxCxE motif. In a search for regions that might confer different substrate binding properties of CDKs in complex with different cyclins, it was noted (Kelly et al, 1998) that cyclin E contains the sequence VDCLE, which is similar to the motif LxCxE that is recognised by pRb and other family member substrates and is present in the N-terminal region of cyclin D and in viral oncogenes. The authors explored the hypothesis that the VxCxE motif in cyclin E might be a putative pRb interaction motif. Mutation of residues DVDCLE to AVACLA or to DVDALE eliminated the ability of cyclin E to promote cell progression but did not interfere with the kinase activity of the pCDK2/cyclin E mutant on histone H1 (Kelly et al, 1998). The crystal structure of pCDK2/cyclin E1 shows that the sequence DVDCLE (residues 273–278) occurs at the short loop between α2′ and α3′ and is remote from the pCDK2 binding site and hence is unlikely to affect catalytic activity. Val274, Cys276 and Leu277 are buried, while Asp273, Asp275 and Glu278 are exposed and available for interaction. The mutations of the two aspartates and the glutamate to alanines would remove the negatively charged cluster at the surface of the cyclin and this could affect a putative recognition site. The crystal structure of the pRb pocket domain in complex with a nine-residue peptide from the conserved region of HPV E7 containing the LxCxE sequence showed that the motif binds in an extended conformation to a shallow groove in the B cyclin box fold of pRb (Lee et al, 1998). The alternating residues of the LxCxE motif (leucine (Leu22), cysteine (Cys24), glutamate (Glu26) and a further leucine (Leu28)) point into the B-box groove and make intermolecular interactions, while the remaining residues point towards the solvent. It is apparent that the turn conformation of the motif in cyclin E1 would not allow such interactions with pRb. We conclude that the VxCxE motif in cyclin E1 is unlikely to interact with pRb in the same way as the viral oncogene LxCxE motif.
Comparison of cyclin E1 and cyclin E2
The two isoforms of cyclin E, cyclin E1 and cyclin E2, are 61% identical over the construct seen in the crystal structure (cyclin E1 residues 88–357). The residues of cyclin E1 that contact pCDK2 (atoms <4.5 Å separation) are 78% identical and 89% similar in cyclin E2. Modelling by SwissModel (Schwede et al, 2003) predicts a structure of cyclin E2 that can bind to pCDK2 in a manner very similar to that of cyclin E1, with just slight adjustments in the region of Arg225 (cyclin E1), which is a glutamate in cyclin E2. In order to observe the effects of cyclin E knockout in mice (Geng et al, 2003), it was necessary to knockout both cyclin E1 and cyclin E2, suggesting that either isoform can carry out necessary functions of a cyclin E, a view reinforced by the crystal structure.
Why does cyclin E form complexes with CDK2 and not CDK1?
We examined putative structures of CDK1/cyclin A and CDK1/cyclin E1, in which CDK1 was modelled by SwissModel on pCDK2. CDK1 is 65% identical in sequence to CDK2 (Figure 4B). Of the 41 residues that are in contact with cyclin A and CDK2, 29 (70%) are conserved in CDK1 and a further five are similar. Analysis of the seven residues that are different indicates that these differences are unlikely to disrupt the putative CDK1/cyclin A interface. Of the 38 residues in CDK2 that are in contact with cyclin E1, 29 are identical (76%) in CDK1 and a further three are similar. Analysis of the putative CDK1/cyclin E1 interface in the regions of the six differences reveals that changes Val154Ile and Val156Ile could cause some crowding with cyclin E. The change of Tyr179, from the loop that connects the activation segment to αF in CDK2, to an arginine in CDK1 causes a bad contact. The model shows that the arginine would clash with cyclin E1 Pro253 (Figure 5C). Since Pro253 is in the α1′/α2′ loop of cyclin E1, the region that is specifically different between cyclin A and cyclin E1, this clash appears significant. However, it is possible to relieve the bad contact by adjustment of the CDK1 arginine side chain so that it may contact Glu340 from the cyclin E1 C-terminal region. Hence, although the structure indicates that cyclin E1 may bind less well to CDK1 compared with CDK2, which is consistent with the early evidence (Koff et al, 1992), the differences at the interface seem relatively minor compared with the overall strength of the CDK2/cyclin interaction. The structural studies indicate a higher affinity of cyclin E1 for CDK2 than cyclin A and a possibly weaker interaction of cyclin E1 with CDK1. These differences might explain the partners observed in cells and cell extracts where CDKs are in considerable excess of cyclins and almost all of cyclin E is observed in complex with CDK2 (Arooz et al, 2000). A more definite explanation must await a structure of CDK1.
Figure 4b.
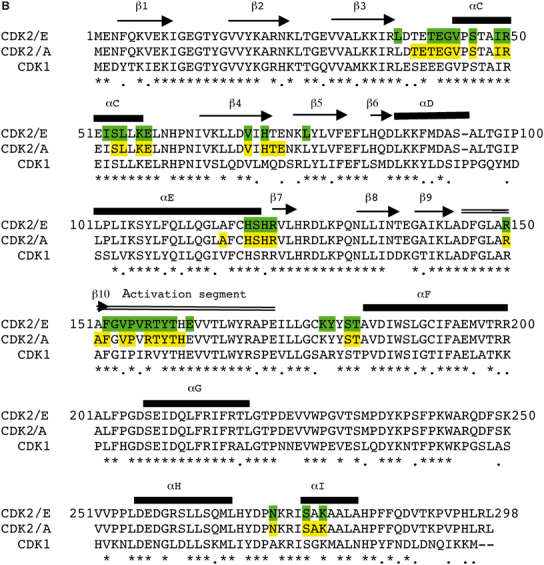
(B) Sequences of CDK2 showing secondary structural elements and CDK1. Residues coloured in green are in contact with cyclin E1 in the pCDK2/cyclin E1 complex and residues coloured in yellow are in contact with cyclin A in the pCDK2/cyclin A complex.
Cyclin E1 fully activates CDK2
In order to compare the activities of pCDK2/cyclin E1 (full length) and pCDK2/cyclin A with model substrates and the effect of the truncation on cyclin E1, we measured the kinetic properties with the model peptide substrate sequence HHASPRK (Brown et al, 1999a). The results (Table II) show that pCDK2/cyclin E1 (full length) is an effective enzyme for phosphorylation of this peptide substrate and has similar kinetic parameters as pCDK2/cyclin A. The complex of pCDK2/cyclin E1 (81–363) showed a higher kcat, a higher Km and a slightly higher specificity constant kcat/Km than the pCDK2/cyclin E1 (full length) complex, indicating that the truncated cyclin E1 is a slightly more effective CDK2 activator than the full-length cyclin E1.
Table 2.
Kinetic parameters for pCDK2/cyclin A and pCDK2/cyclin E1
| Enzyme | Substrate | kcat (s−1) | Km (μM) | kcat/Km (s−1 μM−1) |
|---|---|---|---|---|
| pCDK2/cyclin A | HHASPRK | 34.2±2.5 | 793±89 | 0.046 |
| pCDK2/cyclin E1 (full length) | HHASPRK | 30.5±5.6 | 766±102 | 0.039 |
| pCDK2/cyclin E1 (81–363) | HHASPRK | 53.2±4.5 | 1005±127 | 0.053 |
| pCDK2/cyclin A | CDC6-modified peptidea | 18.9±2.4 | 74±24 | 0.25 |
| pCDK2/cyclin E1 (81–363) |
CDC6-modified peptidea |
22.6±1.7 |
181±31 |
0.125 |
| Sequence: HHASPRKQGKKENGPPHSHTLKGRRLVFDN (for further details, see text). | ||||
We also compared the activities with a modified peptide substrate derived from the natural substrate CDC6 residues 71–100. In this modified peptide (sequence HHASPRKQGKKENGPPHSHTLKGRRLVFDN), the phosphorylation site of CDC6 (PPCSPPK) is replaced by the sequence of HHASPRK, as in the heptapeptide substrate above, and the remainder of the peptide corresponds to the sequence from CDC6 including the RxL motif. The RxL motif is a remote site substrate recognition motif contained in many important CDK2 substrates. In pCDK2/cyclin A, the RxL motif docks into a nonpolar site on the cyclin with affinities of the order of micromolar (Lowe et al, 2002). The kinetic results with pCDK2/cyclin A on the modified CDC6 peptide showed a 10-fold reduction in Km and a five-fold increase in kcat/Km compared with the simple heptapeptide substrate. With pCDK2/cyclin E1 (81–363), there was a six-fold reduction in Km and a two-fold increase in kcat/Km (Table II). The results demonstrate the ability of the remote site recognition motif to improve the catalytic efficiency of both pCDK2/cyclin A and pCDK2/cyclin E1. The approximate two-fold reduction in kcat with both enzyme complexes probably results from the higher apparent affinity of the substrate leading to a reduction in the rate of product release, which is the rate-limiting step in protein kinase reactions. The kinetic results demonstrate that pCDK2/cyclin E1 (81–363) is as effective a catalyst as pCDK2/cyclin A with model peptide substrates and that there is no substrate discrimination between the two enzymes.
Discussion
The structural studies have defined the conformation of a truncated version of cyclin E1 (residues 81–363) bound to pCDK2. This shortened version of cyclin E1 is an effective activator of pCDK2 producing a slightly (1.6-fold) higher kcat than the full-length cyclin E1 and higher activity than pCDK2/cyclin A. In cells, this shortened version of cyclin E1 would be oncogenic because it lacks the sites for phosphorylation that target cyclin E for destruction. Cyclin E1 activates pCDK2 through promoting changes on association that are similar to those previously observed in the pCDK2/cyclin A complex. Our kinetic results with model peptide substrates show that there is no inherent discrimination in substrate recognition between pCDK2/cyclin E1 and pCDK2/cyclin A. In cells and with intact substrates, some discrimination does exist but this might reflect the different timing of expression and destruction of different cyclins and their subcellular localisation.
Major differences between cyclin E1 and cyclin A occur in the C-terminal cyclin box fold, especially in the more extended loop between the α1′ and α2′ helices and the fold of the C-terminal region. These differences result in further contacts to pCDK2 that are not observed in pCDK2/cyclin A. They strengthen the contacts at the start of the activation segment between the DFG motif and pThr160 and towards the end of the activation segment just prior to the APE motif. It is significant that regions in the C-terminal cyclin box fold are those that are involved in CDK2-independent interactions with centrosomes (Matsumoto and Maller, 2004) and in transforming properties (Geisen and Moroy, 2002).
It has been suggested (Tetsu and McCormick, 2003) that since CDK2 is dispensable for cell proliferation, selective CDK2 inhibition may not be promising for cancer therapy. However, in cases where tumorigenesis is driven through genomic instability engineered by untimely expression of cyclin E, such a strategy may be effective since CDK2 is the preferred partner of cyclin E in cells. If on the other hand, it is cyclin E in a CDK2-independent role that gives rise to oncogenic properties, then alternative strategies are necessary such as for example targeting the protein/protein interactions involved in the α1′/α2′ loop. The structure of cyclin E1 bound to CDK2 does not distinguish these roles but it does allow the differences between cyclin E1 and cyclin A to be defined and it offers definitive information for further work.
Materials and methods
Human phospho-Thr160-CDK2 (pCDK2) and human cyclin A2 (residues 174–432) (referred to as cyclin A) were prepared as previously described (Brown et al, 1999a).
Expression and crystallisation
Recombinant baculoviruses encoding full-length human cyclin E1 (a gift from Dr David Morgan, University of California San Francisco) were used to infect Sf9 cells. The cells were harvested postinfection, lysed in a homogeniser and the lysate clarified by centrifugation. GST-pCDK2, expressed in Escherichia coli cells, was immobilised on a glutathione–Sepharose column. The supernatant from the lysed insect cells containing the expressed full-length cyclin E1 was passed down the column. The GST-pCDK2/cyclin E1 (full length) complex was eluted with 20 mM glutathione in HBS buffer (0.2 M NaCl, 20 mM HEPES (pH 7.0), 0.01% MTG) and cleaved with GST-3C protease at 4°C overnight. The cleaved protein complex, pCDK2/cyclin E1 (full length), was gel filtered on a preparative Superdex SD75 column to yield the purified complex. Full-length cyclin E1 was subjected to limited digestion with porcine pancreatic elastase (Sigma). The major product was identified by N- and C-terminal amino-acid sequencing as residues 81–363. DNA fragments encoding cyclin E1 (residues 81–363) were produced by PCR using the following primers: 5′TCTGGATCCATTATTGCACCATCCAGAGGCTCC C and 5′CTCGAATTCTATTAGGCTTTCTTTGCTCGGGC. The amplified cDNA was digested with BamHI and EcoRI and cloned into the pGEX-6P1 plasmid.
Cyclin E1 (residues 81–363) was expressed in E. coli B834(DE3) pLys S cells. Overnight cultures were diluted 1:100 and grown at 37°C until OD600 reached 0.8–0.9. The temperature was lowered to 20°C and cells were grown for a further 1 h. Expression was induced with 0.1 mM IPTG and cells were further incubated for 20 h. Cells were pelleted, resuspended in HBS buffer (10 mM HEPES (pH 7.5), 0.15 M NaCl, 0.01% MTG) containing the nonionic detergent (octylphenoxy)polyethoxyethanol (IGEPAL) 0.1% and sonicated on ice. After removal of cell debris by centrifugation, the GST-cyclin E1 was bound to glutathione–Sepharose 4B column equilibrated with HBS buffer. Purified pCDK2 was passed down the glutathione–Sepharose column prebound with GST-cyclin E1. After elution with 20 mM glutathione, pCDK2/GST-cyclin E1 was digested with GST-3C protease overnight. The pCDK2/cyclin E1 complex was separated from monomeric cyclin E1 on a Superdex 75 column. Fractions containing the CDK2/cyclin E1 complex were loaded onto a glutathione column to remove GST-3C protease and GST. The flow through was reconcentrated using a vivaspin concentrator (Vivascience). The yield of the complex was 3 mg from 4 l of cyclin E1 cell culture.
Crystallisation conditions were screened with the Tecan crystallisation robot using available commercial screens and improved with manual screening. Optimal conditions were found with pCDK2/cyclin E1 (6–8 mg/ml), 1 mM AMPPNP, 10–15% PEG3350 and 0.2 M tri-sodium citrate (pH 7.5–8.25). Subsequently, it was found that no AMPPNP was bound in the crystals and crystals could be obtained equally well without AMPPNP.
X-ray crystallography
Initial X-ray crystallographic data were collected at station BM14 at ESRF, Grenoble to 3.3 Å resolution. A further data set to 2.15 Å resolution was collected at station ID 14.4. The crystals are tetragonal space group P41212 with one pCDK2/cyclin E1 molecule per asymmetric unit. Data were processed with MOSFLM (Leslie, 1999) and programs from the CCP4 suite (CCP4, 1994). The structure was solved by molecular replacement with the program MOLREP (CCP4, 1994) using the structure of active pCDK2 and the N-terminal cyclin box of cyclin A from the coordinates of the pCDK2/cyclin A complex (Protein Data Bank (PDB) ID 1JST) as the search object. Initial refinement was carried out at 3.3 Å resolution using the program BUSTER (Blanc et al, 2004), which allowed the modelling of the missing C-terminal cyclin box. The structure was further refined with REFMAC at 2.15 Å resolution (Murshudov et al, 1997) to yield a final structure with R=0.181 and Rfree=0.246 (Table I). The structure contains the complete pCDK2 molecule and residues 88–357 of cyclin E1. Coordinates have been deposited in the PDB code 1W98.
Kinetics
Peptide substrates were chemically synthesised by GB Bloomberg (Bristol University). The phosphorylation reactions were measured using the spectrophotometric assay (Cook et al, 1982; Adams et al, 1995) in which ADP production is coupled to NADH oxidation by pyruvate kinase (PK) and lactate dehydrogenase (LDH) and assumes a molar extinction coefficient for NADH of 6220 M−1 cm−1 at 340 nm. The assay mixture (volume 0.4 ml; temperature 30°C) contained 30 U/ml LDH, 12 U/ml PK, 1 mM phosphoenolpyruvate, 0.25 mM NADH, 0.5 mg/ml bovine serum albumin, 50 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM KCl, 10 mM MgCl2 and 2 mM DTT. The concentration of the peptide substrate, sequence HHASPRK (Brown et al, 1999a), was varied from 0.07 to 0.7 mM. After incubation for 1–5 min, the reaction was initiated by the simultaneous addition of 0.86 mM ATP and either pCDK2/cyclin A (1.4 μg/ml) or pCDK2/cyclin E1 (full length) (2 μg/ml) or pCDK2/cyclin E1 (residues 81–363) (1.9 μg/ml). Aliquots (0.1 ml) were withdrawn at 3, 6 and 9 min and transferred into 0.1 ml sodium dodecyl sulphate (0.2%). Control reactions in the absence of peptide substrate were performed to monitor any ATPase activity. Reaction rates were found to be linear with enzyme concentration. Measurements were also made with a longer peptide based on a modified CDC6 peptide (sequence HHASPRKQGKKENGPPHSHTLKGRRLVFDN). Reaction volumes were 0.6 ml, peptide concentrations varied from 0.02 to 0.4 mM and reactions were stopped at 3.5 and 12 min. The kinetic data were analysed by the nonlinear regression program GraFit (Leatherburrow, 1992).
Acknowledgments
We thank Tim Hunt for stimulating discussions. We also thank Pietro Roversi for help with BUSTER and John Sinclair for help with the early stages of this work. We acknowledge the assistance of the ESRF beam line scientists at BM14 and ID14.4 and the support from the MRC, the Wellcome Trust and European Community's Human Potential Programme under contract HPRN-CT-2002-00252 (CAMKIN). We declare that we have no financial interests related to this work.
References
- Adams JA, McGlone ML, Gibson R, Taylor SS (1995) Phosphorylation modulates catalytic function and regulation in the cAMP-dependent protein kinase. Biochemistry 34: 2447–2454 [DOI] [PubMed] [Google Scholar]
- Arooz T, Yam CH, Siu WY, Lau A, Li KK, Poon RY (2000) On the concentrations of cyclins and cyclin-dependent kinases in extracts of cultured human cells. Biochemistry 39: 9494–9501 [DOI] [PubMed] [Google Scholar]
- Blanc E, Roversi P, Vonrhein C, Flensberg C, Lea SM, Bricogne G (2004) Refinement of severely incomplete macromolecular structures with BUSTER_TNT. Acta Crystallogr D 60: 2210–2221 [DOI] [PubMed] [Google Scholar]
- Bortner DM, Rosenberg MP (1997) Induction of mammary gland hyperplasia and carcinomas in transgenic mice expressing human cyclin E. Mol Cell Biol 17: 453–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Watson MH, Hickey MJ, Holmes W, Rocque W, Reed SI, Tainer JA (1996) Crystal structure and mutational analysis of the human CDK2 kinase complex with cell cycle-regulatory protein CksHs1. Cell 84: 863–874 [DOI] [PubMed] [Google Scholar]
- Brown NR, Noble ME, Endicott JA, Garman EF, Wakatsuki S, Mitchell E, Rasmussen B, Hunt T, Johnson LN (1995) The crystal structure of cyclin A. Structure 3: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Brown NR, Noble ME, Endicott JA, Johnson LN (1999a) The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nat Cell Biol 1: 438–443 [DOI] [PubMed] [Google Scholar]
- Brown NR, Noble ME, Lawrie AM, Morris MC, Tunnah P, Divita G, Johnson LN, Endicott JA (1999b) Effects of phosphorylation of threonine 160 on cyclin-dependent kinase 2 structure and activity. J Biol Chem 274: 8746–8756 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 (Collaborative Computational Project Number 4) suite: programmes for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM (1996) Turnover of cyclin E by the ubiquitin–proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev 10: 1979–1990 [DOI] [PubMed] [Google Scholar]
- Cook FN, Neville ME, Vrana KE, Hartl FT, Roskowski R (1982) Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 21: 5794–5799 [DOI] [PubMed] [Google Scholar]
- Cook JG, Park CH, Burke TW, Leone G, DeGregori J, Engel A, Nevins JR (2002) Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. Proc Natl Acad Sci USA 99: 1347–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coverley D, Laman H, Laskey RA (2002) Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat Cell Biol 4: 523–528 [DOI] [PubMed] [Google Scholar]
- De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH (1993) Crystal structure of cyclin-dependent kinase 2. Nature 363: 595–602 [DOI] [PubMed] [Google Scholar]
- Donnellan R, Chetty R (1999) Cyclin E in human cancers. FASEB J 13: 773–780 [DOI] [PubMed] [Google Scholar]
- Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI (2004) Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol 165: 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisen C, Moroy T (2002) The oncogenic activity of cyclin E is not confined to Cdk2 activation alone but relies on several other, distinct functions of the protein. J Biol Chem 277: 39909–39918 [DOI] [PubMed] [Google Scholar]
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P (2003) Cyclin E ablation in the mouse. Cell 114: 431–443 [DOI] [PubMed] [Google Scholar]
- Jackman M, Kubota Y, den Elzen N, Hagting A, Pines J (2002) Cyclin A and cyclin E CDK2 complexes shuttle between the nucleus and the cytoplasm. Mol Cell Biol 13: 1030–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP (1995) Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature 376: 313–320 [DOI] [PubMed] [Google Scholar]
- Kelly BL, Wolfe KG, Roberts JM (1998) Identification of a substrate-targeting domain in cyclin E necessary for phosphorylation of the retinoblastoma protein. Proc Natl Acad Sci USA 95: 2535–2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koff A, Cross F, Fisher A, Schumacher J, Leguellec K, Philippe M, Roberts JM (1991) Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell 66: 1217–1228 [DOI] [PubMed] [Google Scholar]
- Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM (1992) Formation and activation of a cyclin E–cdk2 complex during the G1 phase of the human cell cycle. Science 257: 1689–1694 [DOI] [PubMed] [Google Scholar]
- Lauper N, Beck AR, Cariou S, Richman L, Hofmann K, Reith W, Slingerland JM, Amati B (1998) Cyclin E2: a novel CDK2 partner in the late G1 and S phases of the mammalian cell cycle. Oncogene 17: 2637–2643 [DOI] [PubMed] [Google Scholar]
- Leatherburrow RJ (1992) GraFit Version 3.0. Erithakus Software, Staines
- Lee JO, Russo AA, Pavletich NP (1998) Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 391: 859–865 [DOI] [PubMed] [Google Scholar]
- Leslie AGW (1999) Integration of macromolecular diffraction data. Acta Crystallogr D 10: 1696–1702 [DOI] [PubMed] [Google Scholar]
- Lew DJ, Dulic V, Reed SI (1991) Isolation of three novel human cyclins by rescue of G1 cyclin (Cln) function in yeast. Cell 66: 1197–1206 [DOI] [PubMed] [Google Scholar]
- Lowe ED, Tews I, Cheng KY, Brown NR, Gul S, Noble ME, Gamblin SJ, Johnson LN (2002) Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry 41: 15625–15634 [DOI] [PubMed] [Google Scholar]
- Malumbres M, Hunt SL, Sotillo R, Martin J, Odajima J, Martin A, Dubus P, Ortega S, Barbacid M (2003) Driving the cell cycle to cancer. Adv Exp Med Biol 532: 1–11 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Maller JL (2004) A centrosomal localization signal in cyclin E required for CDK2-independent S phase entry. Science 306: 885–888 [DOI] [PubMed] [Google Scholar]
- Moore JD, Kornbluth S, Hunt T (2002) Identification of the nuclear localisation signal in Xenopus cyclin E and analysis of its role in replication and mitosis. Mol Biol Cell 13: 4388–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroy T, Geisen C (2004) Cyclin E. Int J Biochem Cell Biol 36: 1424–1439 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagen AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F (2003) Structural basis for phosphodependent substrate selection and orientation by the SCF Cdc4 ubiquitin ligase. Cell 112: 243–256 [DOI] [PubMed] [Google Scholar]
- Ortega S, Prieto I, Odajima J, Martin A, Dubus P, Sotillo R, Barbero JL, Malumbres M, Barbacid M (2003) Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35: 25–31 [DOI] [PubMed] [Google Scholar]
- Parisi T, Beck AR, Rougier N, McNeil T, Lucian L, Werb Z, Amati B (2003) Cyclins E1 and E2 are required for endoreplication in placental trophoblast giant cells. EMBO J 22: 4794–4803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DC, Zhang N, Danes C, McGahren MJ, Harwell RM, Faruki S, Keyomarsi K (2001) Tumor-specific proteolytic processing of cyclin E generates hyperactive lower-molecular-weight forms. Mol Cell Biol 21: 6254–6269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, Roberts JM (1997) Expression of cell-cycle regulators p27Kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med 3: 222–225 [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C (2004) Inactivation of hCDC4 can cause chromosomal instability. Nature 428: 77–81 [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP (1996a) Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A–Cdk2 complex. Nature 382: 325–331 [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Pavletich NP (1996b) Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat Struct Biol 3: 696–700 [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31: 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512 [DOI] [PubMed] [Google Scholar]
- Song H, Hanlon N, Brown NR, Noble ME, Johnson LN, Barford D (2001) Phosphoprotein–protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2. Mol Cell 7: 615–626 [DOI] [PubMed] [Google Scholar]
- Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI (2002) hCDC4 gene mutations in endometrial cancer. Cancer Res 62: 4535–4539 [PubMed] [Google Scholar]
- Spruck CH, Won KA, Reed SI (1999) Deregulated cyclin E induces chromosome instability. Nature 401: 297–300 [DOI] [PubMed] [Google Scholar]
- Strausfeld UP, Howell M, Descombes P, Chevalier S, Rempel RE, Adamczewski J, Maller JL, Hunt T, Blow JJ (1996) Both cyclin A and cyclin E have S-phase promoting (SPF) activity in Xenopus egg extracts. J Cell Sci 109 (Part 6): 1555–1563 [DOI] [PubMed] [Google Scholar]
- Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI (2001) Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature 413: 316–322 [DOI] [PubMed] [Google Scholar]
- Su TT, O'Farrell PH (1998) Chromosome association of minichromosome maintenance proteins in Drosophila endoreplication cycles. J Cell Biol 140: 451–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, McCormick F (2003) Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3: 233–245 [DOI] [PubMed] [Google Scholar]
- Welker M, Singer J, Loeb KR, Grim J, Bloecher A, Gurien-West M, Clurman BE, Roberts JM (2003) Multisite phosphoryation by CDK2 and GSK3 controls cyclin E degradation. Mol Cell 12: 381–392 [DOI] [PubMed] [Google Scholar]
- Won KA, Reed SI (1996) Activation of cyclin E/CDK2 is coupled to site specific autophosphorylation and ubiquitin dependent degradation of cyclin E. EMBO J 15: 4182–4193 [PMC free article] [PubMed] [Google Scholar]
- Zariwala M, Liu J, Xiong Y (1998) Cyclin E2, a novel human G1 cyclin and activating partner of CDK2 and CDK3, is induced by viral oncoproteins. Oncogene 17: 2787–2798 [DOI] [PubMed] [Google Scholar]