

Abstract
The glutamate transporter gene, EAAT2/GLT-1, is induced by epidermal growth factor (EGF) and downregulated by tumor necrosis factor α (TNFα). While TNFα is generally recognized as a positive regulator of NF-κB-dependent gene expression, its ability to control transcriptional repression is not well characterized. Additionally, the regulation of NF-κB by EGF is poorly understood. Herein, we demonstrate that both TNFα-mediated repression and EGF-mediated activation of EAAT2 expression require NF-κB. We show that EGF activates NF-κB independently of signaling to IκB. Furthermore, TNFα can abrogate IKKβ- and p65-mediated activation of EAAT2. Our results suggest that NF-κB can intrinsically activate EAAT2 and that TNFα mediates repression through a distinct pathway also requiring NF-κB. Consistently, we find that N-myc is recruited to the EAAT2 promoter with TNFα and that N-myc-binding sites are required for TNFα-mediated repression. Moreover, N-myc overexpression inhibits both basal and p65-induced activation of EAAT2. Our data highlight the remarkable specificity of NF-κB activity to regulate gene expression in response to diverse cellular signals and have implications for glutamate homeostasis and neurodegenerative disease.
Keywords: EAAT2, EGF, N-myc, NF-κB, TNFα
Introduction
The excitatory amino-acid transporter, EAAT2/GLT-1, is responsible for the majority of clearance of the neurotransmitter glutamate from neuronal synapses in the central nervous system (CNS) (Tanaka et al, 1997). Impaired glutamate uptake by glial cells can result in cell death from excessive levels of glutamate and overstimulation of glutamate receptors (Choi, 1988). Indeed, glutamate toxicity is implicated in a wide variety of neurodegenerative disorders, including Alzheimer's disease, amyotrophic lateral sclerosis and multiple sclerosis (Doble, 1999). The importance of regulating glutamate transport is underscored by the observation that mice lacking EAAT2 develop progressive neurodegeneration and epilepsy as a result of aberrant glutamate homeostasis (Rothstein et al, 1996; Tanaka et al, 1997). Therefore, it is extremely important to understand the regulation of EAAT2 expression in glial cells. Interestingly, EAAT2 is positively regulated by epidermal growth factor (EGF) and negatively regulated by the proinflammatory cytokine, tumor necrosis factor α (TNFα) (Zelenaia et al, 2000; Schluter et al, 2002; Su et al, 2003). However, the requirements for growth factor- and cytokine-mediated regulation of EAAT2 expression have not been clearly elucidated.
TNFα activates the NF-κB family of transcription factors, which are ubiquitously expressed and are pivotal in controlling diverse cellular processes, including immune responses, cell proliferation and differentiation (Israel, 2000; Baldwin, 2001; Silverman and Maniatis, 2001; Ghosh and Karin, 2002; Li and Verma, 2002). Increasingly, it has become evident that NF-κB also plays important roles in the CNS (O'Neill and Kaltschmidt, 1997). In most cells, the predominant induced NF-κB complex is a p65/RelA and p50 heterodimer, which is generally a positive regulator of gene-specific transcription. Activation of NF-κB typically involves regulating the stability of the inhibitory protein, IκB. Signal-induced phosphorylation, ubiquitination and degradation of IκB induce NF-κB nuclear accumulation and subsequent target-specific DNA binding. Phosphorylation of IκB is carried out by the IκB kinase (IKK) complex, which is comprised of IKKα, β and γ subunits (Israel, 2000; Ghosh and Karin, 2002; Yamamoto and Gaynor, 2004). Notably, IKKβ is the primary kinase responsible for cytokine-induced IκB phosphorylation and NF-κB activation (Ghosh and Karin, 2002).
The regulation of NF-κB activity by growth factors such as EGF is less well understood. For example, EGF has been reported to stabilize IκB and block NF-κB activation (Mehta and Besner, 2003; Banan et al, 2004a, 2004b). However, in carcinoma cells that overexpress EGF receptor family members, EGF has been shown to induce IκBα degradation and NF-κB DNA binding (Sun and Carpenter, 1998; Biswas et al, 2000). Additionally, NF-κB-inducing kinase (NIK) has been reported to be complexed with the EGF receptor, which potentiates EGF activation of NF-κB (Chen et al, 2003). Consistent with EGF-controlled activation of NF-κB, Anest et al (2004) have recently reported positive regulation of the immediate-early gene c-fos by NF-κB through a mechanism involving constitutive association of p65 with the promoter. Clearly, further studies are necessary to understand the regulation of EGF-responsive genes by NF-κB.
The opposing regulation of EAAT2 expression by cytokines and growth factors provides a unique opportunity to study how different physiological or pathological signals elicit distinct transcriptional responses from a single promoter. In this work, we establish that both positive and negative regulation of EAAT2 gene expression are controlled by NF-κB. We show that EGF induces NF-κB recruitment to the EAAT2 promoter in a manner that does not involve IκB degradation or enhanced p65/RelA nuclear accumulation. Furthermore, our data reveal that NF-κB is an intrinsic positive regulator of EAAT2 gene expression and that TNFα-mediated repression involves another transcription factor, N-myc. These experiments have important implications for understanding differential gene expression as well as the altered glutamate homeostasis associated with different CNS disorders.
Results
EGF and TNFα differentially regulate EAAT2 expression
Previous reports have demonstrated that diverse signals can positively or negatively regulate the expression of EAAT2 in astrocytes and we sought to determine whether this dual regulation occurred in human H4 astroglioma cells. Using quantitative real-time RT–PCR analysis in wild-type H4 cells, we observed induction of EAAT2 mRNA in response to treatment with EGF (Figure 1A). In contrast, in response to TNFα treatment and consistent with previous reports (Su et al, 2003), EAAT2 mRNA expression was decreased.
Figure 1.
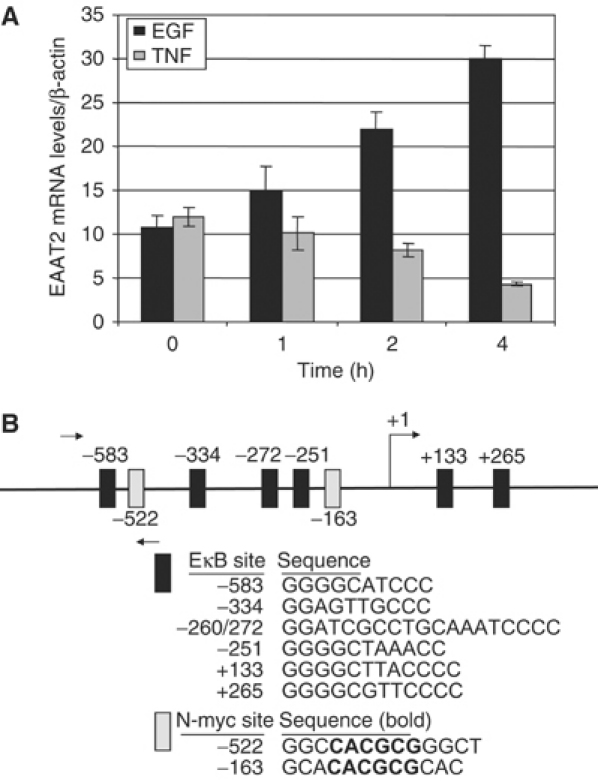
EAAT2 expression is differentially regulated by TNFα and EGF. (A) Quantitative real-time RT–PCR analysis of EAAT2 mRNA levels in H4 glioma cells treated with EGF or TNFα. PCR reactions were performed in triplicate and EAAT2 mRNA levels were normalized to β-actin as an endogenous control. (B) NF-κB and N-myc consensus sites in the EAAT2 promoter. The relative positions and sequences of putative NF-κB and N-myc DNA-binding sites are indicated. Arrows indicate the position of primers used for ChIPs (Figures 3 and 7).
Given the strong role of NF-κB in TNFα-induced gene expression and the less well-established role of this transcription factor in EGF-induced gene expression, we analyzed the EAAT2 proximal promoter sequence for potential NF-κB-dependent regulatory elements. We identified six putative NF-κB consensus binding sites, including two sites (+133 and +265) in the region corresponding to the 5′ untranslated region (5′UTR) (Figure 1B). The −583 and −272 sites were previously reported and a reporter construct containing these NF-κB sites in the EAAT2 promoter was shown to remain responsive to both EGF- and TNFα-mediated regulation (Su et al, 2003). Results from our initial studies found that changes in EAAT2 mRNA levels were not due to alterations in NF-κB p65 or p50 protein levels resulting from EGF or TNFα treatments (data not shown), raising the potential that alteration of NF-κB transcriptional activity is associated with the differential control of EAAT2 gene expression by growth factors and cytokines.
NF-κB is recruited to the EAAT2 promoter in response to TNFα and EGF
To address the potential role of NF-κB in regulating EAAT2 gene expression, we asked whether TNFα and EGF induce NF-κB DNA binding to the consensus sites in the EAAT2 promoter. Electrophoretic mobility shift assays (EMSAs) were performed with nuclear extracts from H4 cells treated with TNFα or EGF. TNFα strongly induced DNA/protein complex formation at the −583 position, which consists of the consensus sequence 5′-GGGGCATCCC-3′ (Figure 2A). The binding of complex 1 was rapidly induced after 15 min of treatment and persisted for at least 4 h. Complex 2 was induced with slightly slower kinetics. We observed that EGF treatment also induced binding to this element, albeit weakly as compared to that of TNFα. EGF induction of complex 1 occurred at 1 h and complex 2 was also more weakly induced (Figure 2A). Experiments were also performed with a probe in which the −583 consensus site was mutated in order to determine whether the induced binding was specific for NF-κB. Indeed, both TNFα- and EGF-induced NF-κB complexes did not form with the mutated probe (Figure 2B). The EGF-induced increase in NF-κB DNA binding to the −583 site was further investigated using a DNA affinity purification assay (DAPA) where nuclear proteins were allowed to bind to an immobilized, biotinylated oligo containing the −583 sequence (see Materials and methods). Bound proteins were eluted and subsequently analyzed by Western blot with a p65-specific antibody. We found that NF-κB p65 DNA binding to the −583 site of the EAAT2 promoter was strongly increased in response to TNFα and weakly increased in response to EGF (Figure 2C). Results from these experiments clearly confirmed our EMSA results and also identified the NF-κB p65 subunit as part of both TNFα- and EGF-induced DNA-binding complexes.
Figure 2.
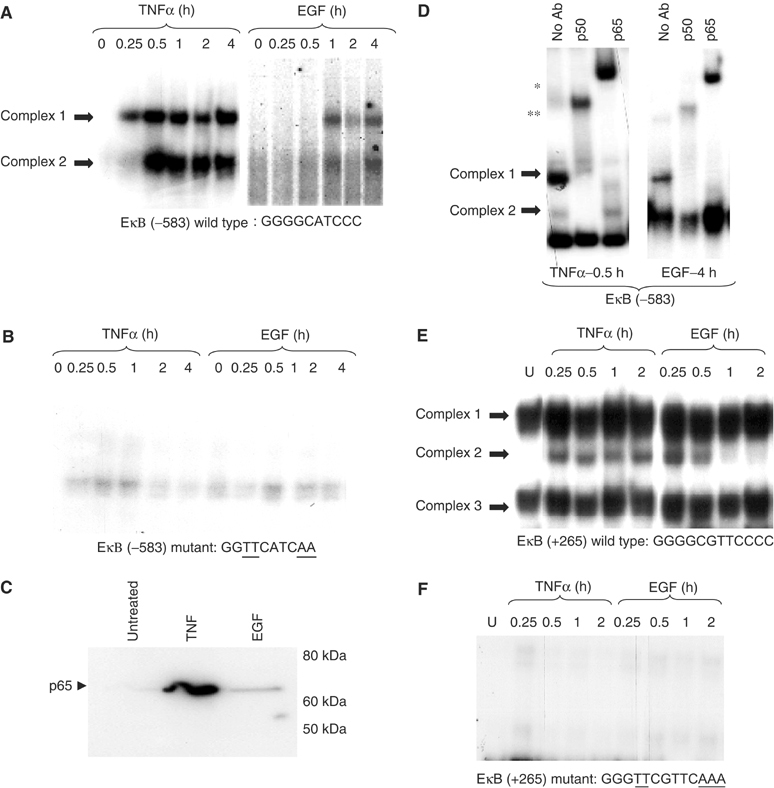
TNFα and EGF induce NF-κB DNA binding at the EAAT2 promoter. (A) EMSAs were performed with nuclear extracts from H4 cells treated with TNFα or EGF over a 4 h time course using a probe corresponding to the wild-type −583 site (EκB (−583)). A darker gel exposure is shown in the EGF panel to better visualize weak induction of DNA binding. (B) NF-κB binding using the same extracts from (A) was compared to a mutant EκB (−583) site. Mutated sequences are underlined. (C) DAPAs were performed by incubating H4 nuclear extracts with an immobilized, biotinylated EκB (−583) double-stranded oligo. Bound protein complexes were washed, eluted with 1 × sample buffer and subjected to PAGE and Western blot analysis with a p65-specific antibody. (D) EMSAs were performed using nuclear extracts from cells treated with TNFα (0.5 h) or EGF (4 h) in the presence or absence of p65 or p50 antibodies. Supershifted complexes are indicated with asterisks (*p65, **p50). (E) EMSA was performed with a probe corresponding to the +265 site in the EAAT2 5′UTR using H4 nuclear extracts treated with TNFα or EGF over a 2 h time course. (F) EMSA was performed using a mutant +265 probe. Mutated sequences are underlined.
To further characterize the proteins in the EMSA complexes, we performed supershift assays with antibodies to specific NF-κB family members. In TNFα-treated cells, complex 1 was completely supershifted with α-p65 and α-p50 antibodies and complex 2 was partially supershifted with α-p50 (Figure 2D). In EGF-treated cells, although weak supershifted bands were formed, we observed complete loss of complex 1 binding by incubating nuclear extracts with α-p65 or α-p50, indicating that these antibodies did not generate a clear supershift but nevertheless, specifically interfered with complex 1 formation. Results from these experiments demonstrated that the TNFα- and EGF-induced NF-κB complex 1 primarily consists of the p65–p50 heterodimer (Figure 2D). The TNFα-induced complex 2 contains p50 as well as the p52 subunit, whereas c-Rel and RelB are absent (see Supplementary data).
Next, we examined NF-κB binding to one of the putative consensus NF-κB sites in the region of the EAAT2 gene corresponding to the 5′UTR. Interestingly, we observed a different pattern of binding to the +265 site, which consists of the sequence 5′-GGGGCGTTCCC-3′. Results from EMSAs showed that EGF strongly induced NF-κB binding in a manner comparable to that of TNFα (Figure 2E). Additionally, EGF induced binding at 30 min that was diminished by 4 h post-treatment. In contrast, TNFα rapidly induced NF-κB binding 15 min after the addition of EGF and this binding persisted for up to 4 h, which is a pattern similar to that seen with TNFα at the −583 site (Figure 2A). We observed three specific complexes that bind to the +265 site, and mutation of this site eliminated complex formation (Figure 2F). Supershift analyses indicated that the inducible complex 2 is a heterodimer of the p65 and p50 NF-κB subunits (see Supplementary data). We are currently investigating the nature of the additional bands that form at the +265 site. Additionally, we observed binding to other NF-κB sites in the EAAT2 promoter, including −334 as well the three sites at −272, −251 and +133 (see Supplementary data).
To examine the kinetics of in vivo recruitment of NF-κB proteins to the EAAT2 promoter in untreated and EGF- or TNFα-treated H4 cells, we performed chromatin immunoprecipitation (ChIP) assays using a p65-specific antibody (Figure 3A and B). The kinetics of detectable p65 recruitment to the EAAT2 promoter is greater with TNFα than EGF treatment over the course of 4 h. Moreover, the overall levels of p65 recruitment are approximately three times greater with TNFα compared with that of EGF. Although ChIP analysis does not itself measure affinity or the extent of promoter occupancy, the kinetics of p65 promoter recruitment by this approach is consistent with results from the EMSAs and real-time PCR analysis of EAAT2 expression. Overall, these data indicate that TNFα and EGF induce NF-κB recruitment to the EAAT2 promoter in vivo and that the subunit composition of NF-κB complexes is similar for both inducers.
Figure 3.
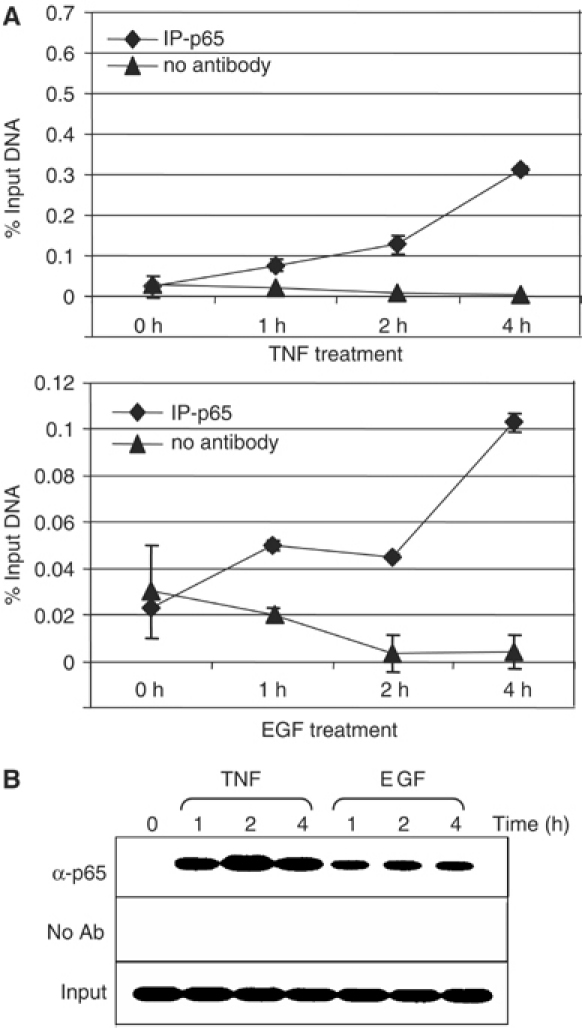
NF-κB p65 is present at the EAAT2 promoter in vivo in response to EGF and TNF. (A) H4 cells were treated with TNFα (20 ng/ml) or EGF (50 ng/ml) for the indicated time points. ChIP assays were performed using a p65-specific antibody. IgG precipitations are shown as a negative control. Prior to immunoprecipitation, a portion of samples was reserved for input controls. Immunoprecipitated chromatin was prepared and subjected to quantitative real-time PCR analysis using primers that amplify the −583 region (see Figure 1B and Materials and methods). Reactions were performed in triplicate and values are represented as a percentage of input DNA. (B) DNA from real-time PCR analysis was visualized by agarose gel electrophoresis with EtBr.
Positive and negative regulation of EAAT2 gene expression requires NF-κB
Thus far, our data demonstrate in vitro and in vivo that EGF and TNFα induce the formation of NF-κB DNA-binding complexes that are correlated with either transcriptional activation or repression. To determine whether regulation of EAAT2 expression required NF-κB, we tested the activity of an EAAT2 promoter-driven luciferase reporter and compared this to EAAT2 reporters that had mutations in NF-κB consensus sites (Figure 4A). Mutation of the −583 site significantly impaired the constitutive activation of EAAT2, indicating that NF-κB plays a role in controlling uninduced levels of gene expression. Indeed, the activity of a triple mutant reporter (−583, −272 and −251 sites) was completely abolished. Interestingly, TNFα-mediated repression of reporter activity was only partially abrogated with the −583 mutant reporter, whereas EGF-mediated activation was completely lost. The fact that TNFα-mediated repression is not completely abolished with the −583 mutant likely reflects the contribution of additional NF-κB sites that were not mutated. When we examined the activity of an EAAT2 reporter in which the NF-κB sites in the 5′UTR (+133/+265) were mutated, we again observed lower basal activity. However, in this context, we observed complete loss of TNFα-mediated repression, whereas EGF was still able to activate (Figure 4A). These results suggest that distinct NF-κB sites may contribute preferentially to activation or repression of EAAT2. Next, we monitored EAAT2 mRNA levels in H4 astroglial cells expressing either vector or IκBα super-repressor (SR), a nondegradable, dominant-negative inhibitor of all NF-κB complexes. Stable expression of IκBα-SR resulted in loss of NF-κB DNA-binding activity as assayed by EMSAs (data not shown). H4 cells stably expressing IκBα-SR were no longer sensitive to TNF-mediated inhibition of EAAT2 expression (Figure 4B) as compared with cells expressing vector alone. Additionally, induction of EAAT2 gene expression by EGF was also abrogated in H4 IκBα-SR cells. These results definitively show that NF-κB activity is required for both TNFα-mediated inhibition and EGF-mediated induction of EAAT2 expression in glial cells.
Figure 4.
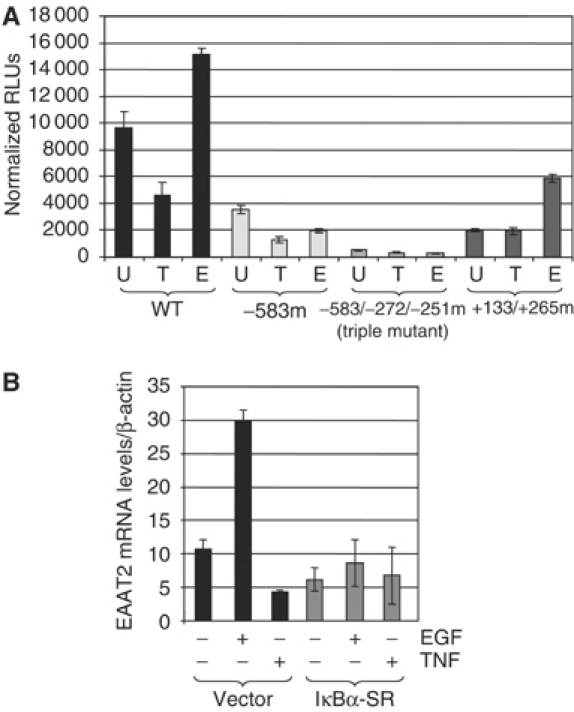
NF-κB is required for regulation of EAAT2 expression. (A) Luciferase reporter assays were performed in H4 cells with wild-type and mutant EAAT2 promoter constructs that had mutations in three NF-κB consensus sites (see Materials and methods). Treatments were performed with 20 ng/ml TNFα or 50 ng/ml EGF. Transfections were performed in triplicate. Normalized RLUs (relative luciferase units) represent luciferase activity normalized to a Renilla reporter. (B) Wild-type and IκBα-SR-expressing H4 cells were treated with TNFα or EGF for the indicated time points. Total RNA was isolated, reverse-transcribed with MMLV-RT and quantitative real-time PCR analysis of EAAT2 cDNA was subsequently performed. EAAT2 mRNA levels were normalized to β-actin mRNA as an internal control.
TNFα and EGF use different signaling pathways to regulate NF-κB
Given the clear quantitative and kinetic differences in the ability of EGF and TNFα to elicit the formation of NF-κB DNA-binding complexes at the EAAT2 promoter, we set out to address whether these signals utilized the same pathways to regulate NF-κB activity. We asked whether both EGF and TNFα employed the classical mechanism of signal-induced IκBα phosphorylation and degradation to trigger NF-κB nuclear localization. In H4 cells treated with TNF, the typical kinetics of IκBα phosphorylation and degradation at 15–30 min were observed, which is consistent with induction of DNA binding at that time point (Figure 5A). However, treatment with EGF altered neither the phosphorylation nor the stability of IκBα. Similar results were also observed with IκBβ and IκBɛ (Figure 5B). In order to confirm activation of the EGF signaling pathway, we assessed whether downstream targets of that pathway were activated with treatment. Indeed, we observe induction of MEK1/2 phosphorylation after EGF treatment (Figure 5C). To determine whether EGF-induced stabilization of IκB affected NF-κB levels in the nucleus, we performed Western blot analysis of nuclear extracts from H4 cells. Results from these experiments showed that TNFα treatment results in greatly increased nuclear p65 levels, whereas EGF did not significantly alter the pre-existing, constitutive levels of nuclear p65 (Figure 5D). Together, these data suggest that TNFα acts through the classical IκB degradation pathway to induce NF-κB DNA binding and transcriptional repression, whereas EGF regulates the activity of constitutively nuclear NF-κB complexes independently of IκB to regulate positively EAAT2 expression.
Figure 5.
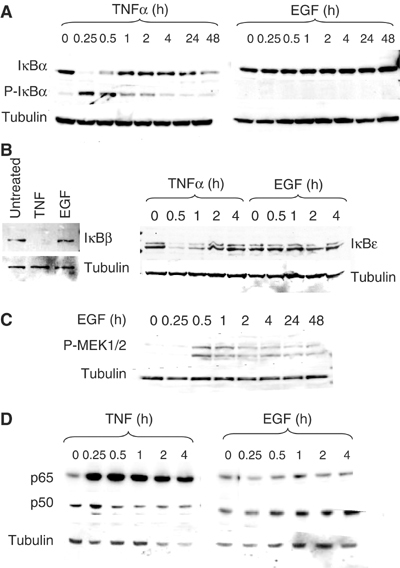
EGF and TNFα signal to NF-κB via different signaling pathways. Western blot analysis was performed with protein extracts from H4 cells that were treated with either TNF or EGF for the indicated time points. Cytoplasmic fractions were used unless otherwise indicated. Blots were reprobed with β-tubulin to demonstrate equal loading of protein. (A) Total and phosphorylated IκBα proteins. (B) IκBβ and IκBɛ proteins. (C) Phosphorylated MEK1/2 in whole cell lysates. (D) p65 protein levels in nuclear extracts from cells treated with TNF or EGF.
TNFα-mediated repression overcomes intrinsic activation of EAAT2 by NF-κB
We next asked whether the activating or repressive signal would be dominant with cotreatment of H4 cells with EGF and TNFα. Results from transient transfection assays with the wild-type EAAT2-luciferase reporter show that the repressive signal by TNFα prevents EGF-mediated activation of EAAT2. We sought to determine whether a signal that elicits NF-κB activation alone would activate or repress EAAT2 gene expression. Thus, we tested whether overexpression of p65 or IKKβ, which is essential for TNFα-mediated activation of NF-κB, would positively or negatively regulate EAAT2. Our results demonstrated that p65 and IKKβ expression dramatically increased wild-type EAAT2 reporter activation (Figure 6A), suggesting that NF-κB has an inherent ability to activate EAAT2. Interestingly, TNFα treatment resulted in repression of both p65- and IKKβ-mediated EAAT2 activation. Mutation of the −583, −272 and −251 NF-κB sites resulted in a significant decrease in basal EAAT2 expression and loss of the ability of IKKβ to induce gene expression (Figure 6B). Additionally, we find that TNFα activates a reporter containing NF-κB consensus sites from the MHC class I promoter, demonstrating that the repressive effects of TNFα are promoter specific (Figure 6C). Based on these data, we suggest that TNFα functions at two distinct levels to regulate NF-κB activity and repress EAAT2 gene expression: (i) inducing NF-κB DNA binding through IκB signaling and (ii) regulating different signaling pathways and/or transcriptional regulators that ultimately convert NF-κB to a transcriptional repressor (see Figure 9).
Figure 6.
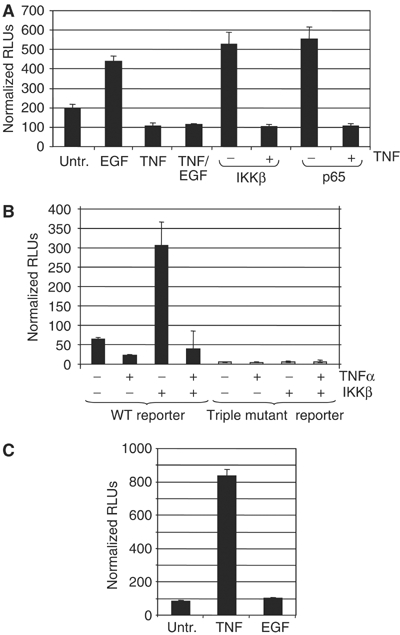
TNFα-mediated repression overcomes activation of EAAT2 by NF-κB. (A) The effect of TNFα on EGF-, IKKβ- or p65-mediated activation of the wild-type EAAT2 reporter was tested. Normalized RLUs represent luciferase units normalized to Renilla control reporter activity. (B) The ability of IKKβ to regulate the triple mutant EAAT2 reporter was tested and compared to that of wild type. (C) TNFα-mediated repression is specific to the EAAT2 promoter. The activity of a 3x-κB-luciferase promoter containing consensus NF-κB sites from the MHC class I promoter was tested in the presence of TNFα or EGF.
Figure 9.
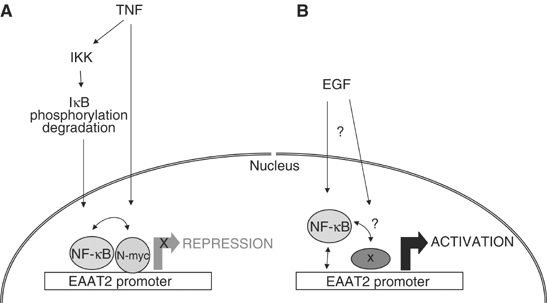
A model for the differential regulation of EAAT2 expression by NF-κB. (A) TNFα signals to IKK to trigger IκB phosphorylation and degradation, thereby eliciting NF-κB nuclear translocation, DNA binding and transcriptional repression of EAAT2. We propose that TNFα functions to induce NF-κB DNA binding as well as regulate different signaling pathways and/or other regulators, such as N-myc, to convert NF-κB to a transcriptional repressor. (B) EGF signaling regulates the activity of pre-existing NF-κB nuclear complexes to induce transcriptional activation. EGF may also regulate the activity or recruitment of other regulators at the promoter that cooperate with NF-κB to regulate positively EAAT2 transcription.
A role for N-myc in TNFα-mediated repression of EAAT2
To address the mechanism by which TNFα induces repression, we investigated whether other regulatory factors were involved. The EAAT2 promoter contains consensus binding sites for several transcription factors, including N-myc (see Figure 1B), a basic helix–loop–helix transcription factor that is necessary for neurogenesis (Knoepfler et al, 2002) and is frequently amplified in neuroblastoma (Schwab, 1993). Using ChIP assays, we found that N-myc recruitment to the EAAT2 promoter is significantly increased with TNFα treatment as compared with EGF treatment (Figure 7A and B). Furthermore, using DNA purification assays, we observed increased TNFα-induced N-myc DNA binding to a probe spanning the −534 to −505 region of the EAAT2 promoter, which contains a putative CACGCG N-myc site (Figure 7C). These data suggest that N-myc may be involved in TNFα-mediated repression of EAAT2. To test this possibility, we constructed an EAAT2-luciferase reporter that had both N-myc consensus sites mutated and tested its activity in transient transfection assays (see Materials and methods). We observed that TNFα was unable to repress the activity of the N-myc mutant reporter, while EGF remained able to activate (Figure 8A). Next, we examined whether overexpression of N-myc would affect the basal activity of the EAAT2 promoter or the ability of NF-κB to activate EAAT2. Indeed, in luciferase reporter assays, we observed that N-myc repressed both basal and p65-induced activation of the wild-type EAAT2 reporter in a dose-response manner (Figure 8B). Importantly, the loss of NF-κB activation was not due to decreased p65 or p50 protein levels by N-myc (Figure 8B). Together, these data indicate that the preferential, early recruitment of N-myc to the EAAT2 promoter with TNFα treatment contributes to the repression of intrinsic NF-κB-mediated activation.
Figure 7.
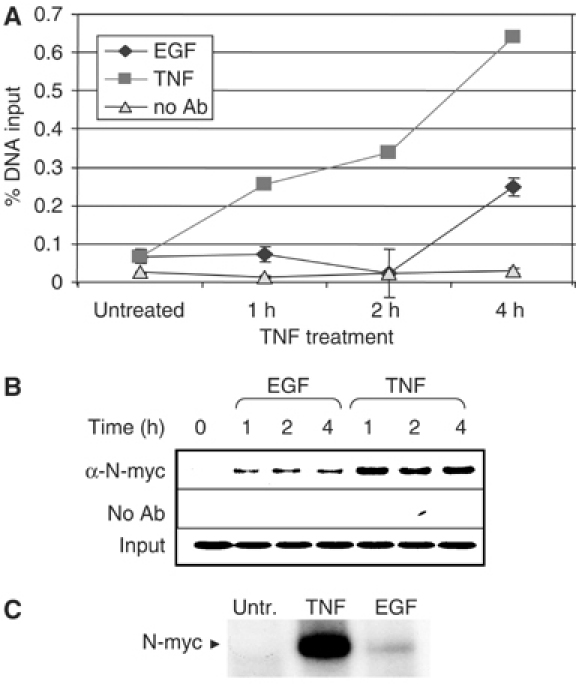
N-myc is recruited to the EAAT2 promoter in vivo. Using an N-myc antibody, ChIP assays were performed in H4 cells at various time points in the presence or absence of TNFα or EGF. Immunoprecipitated DNA was analyzed by (A) quantitative real-time PCR and (B) agarose gel electrophoresis. (C) DAPAs were performed by incubating H4 nuclear extracts with an immobilized, biotinylated double-stranded oligo that contains an N-myc consensus site (see Materials and methods). Bound protein complexes were washed, eluted with 1 × sample buffer and subjected to PAGE and Western blot analysis with an N-myc-specific antibody.
Figure 8.
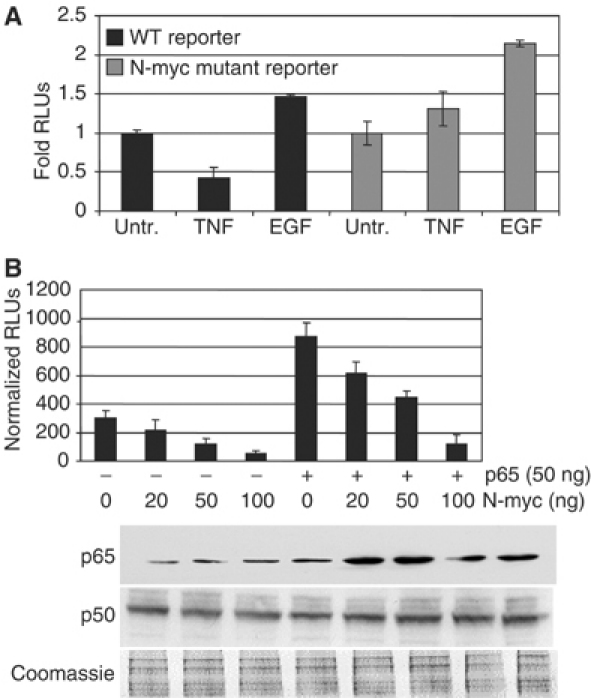
N-myc-binding sites in the EAAT2 promoter are required for N-myc- and TNFα-mediated repression. (A) Wild-type or N-myc mutant reporters were transfected into H4 cells. Untreated and TNFα- and EGF-treated cells were assayed for luciferase activity. (B) The wild-type EAAT2-luciferase reporter in the absence or presence of p65 was cotransfected H4 cells with increasing amounts of N-myc. Western blot analysis with p65- or p50-specific antibodies was performed on whole cell lysates from transfected cells.
Discussion
Here we investigate the regulation of EAAT2/GLT-1 expression by growth factors and cytokines. We show that EAAT2 gene expression is negatively regulated by TNFα and positively regulated by EGF in H4 astroglioma cells. Previous work suggesting that regulation of EAAT2 expression was mediated by NF-κB was based on inhibiting NF-κB activity using antioxidants, such as PDTC (Schreck et al, 1992; Sato et al, 1996; Zelenaia et al, 2000; Su et al, 2003). However, it has recently been demonstrated that these antioxidants have pleiotropic effects and do not function as specific NF-κB inhibitors (Hayakawa et al, 2003). Therefore, it was important to determine the precise role of NF-κB in regulating EAAT2 in response to growth factor and cytokine signals. Here, we demonstrate that NF-κB is recruited to the EAAT2 promoter in vitro and in vivo in response to both EGF and TNFα and that both of these signals induce the predominant p65–p50 heterodimer (Figures 2 and 3). Our results from experiments using IκBα-SR to prevent NF-κB activity definitively establish that NF-κB is important for basal activation of EAAT2 expression, and that both the positive regulation of EAAT2 expression by EGF and negative regulation by TNFα are dependent on NF-κB. The ability of IκBα-SR to abrogate NF-κB-dependent transcriptional responses reflects the ability of this dominant-negative inhibitor to bind and sequester all NF-κB complexes, regardless of whether they are normally regulated in an IκBα-dependent manner. Interestingly, we were unable to recapitulate fully EGF-mediated activation of EAAT2 in fibroblast cells (data not shown), suggesting that the opposing regulation of EAAT2 by TNFα and EGF may be cell type specific. Overall, our results highlight the ability of NF-κB to integrate diverse physiological or pathological signals to specify either transcriptional activation or repression.
The presence of NF-κB consensus sites in the region corresponding to the 5′UTR of EAAT2 evokes the interesting possibility of alternative mechanisms regarding both positive and negative control of gene expression. Others have reported a transcriptional regulatory role for the region between the transcription and translation start sites of several genes, including HIV-1, HIV-2, adenovirus, human β-globin and aggregan (Mansour et al, 1986; Jones et al, 1988; Amrolia et al, 1995; Valhmu et al, 1998). Additionally, NF-κB binding in the 5′ intragenic regions of genes was recently reported (Martone et al, 2003). Our finding that both TNFα and EGF elicit strong NF-κB DNA binding at the +265 site suggests that it is a functional regulatory element, which may contribute to regulating gene expression in conjunction with NF-κB sites present in the promoter (Figure 2). Indeed, our results suggest that the distinct NF-κB sites in the promoter proximal and 5′UTR regions may differentially contribute to gene activation or repression (Figure 4). Although the mechanism of regulation by the NF-κB sites in the EAAT2 5′UTR has yet to be elucidated, it is possible that TNFα-mediated NF-κB DNA binding in this region may serve to hinder RNA polymerase (PolII) processivity and elongation, thereby contributing to transcriptional repression.
Interestingly, we found that TNFα and EGF utilize distinct signaling pathways to regulate NF-κB activity (Figure 5). TNFα induces the classical IκB degradation pathway to trigger NF-κB nuclear translocation and DNA binding to repress EAAT2 expression. In contrast, EGF activates EAAT2 expression independently of IκB degradation and NF-κB nuclear translocation. Therefore, our data suggest that EGF induces the recruitment of pre-existing, constitutively nuclear NF-κB complexes to the EAAT2 promoter. Generally, the complex spatial and temporal regulation of gene expression in higher eukaryotes is achieved through combinatorial regulation of transcription factors. Likewise, EGF may regulate the activity of other regulators that cooperate with NF-κB to activate expression of EAAT2 thereby allowing cells to finely control gene transcription and integrate multiple signal transduction pathways (Figure 9). The identity of EGF-induced regulators is currently under investigation.
Although the mechanisms of NF-κB activation by TNFα are well established, TNFα-mediated repression is poorly understood. Transcriptional repression by NF-κB proteins is probably best exemplified by Dorsal, a Drosophila Rel family member that can either activate or repress gene expression through the recruitment of coactivators, such as CBP, or corepressors such as Groucho (Belvin and Anderson, 1996). Examples of NF-κB-mediated repression in mammals include TNFα-mediated downregulation of collagen gene expression, which involves activation of the Jun-N-terminal kinase (JNK) and c-Jun transcription factor (Verrecchia et al, 2002). Additionally, NF-κB has been reported to repress transcription by inhibiting the transactivating function of other regulators, such as SF-1 (Hong et al, 2003). It has also been shown that other inducers, such as chemotherapeutic agents, can elicit NF-κB transcriptional repression in a manner that is dominant to TNFα-induced activation (Campbell et al, 2004). Our results demonstrating the ability of TNFα to overcome p65- and IKKβ-mediated activation of EAAT2 expression suggest the acquisition of a repressive complex through a distinct signaling pathway to block intrinsic NF-κB-mediated gene activation. Indeed, the preferential recruitment of N-myc to the EAAT2 promoter by TNFα and the ability of N-myc to repress NF-κB-mediated activation of EAAT2 suggest a mechanism by which TNFα may overcome intrinsic NF-κB-mediated activation. Notably, N-myc does not significantly affect the expression of NF-κB (Figure 8B). Therefore, in contrast to an earlier report that describes N-myc-mediated repression of MHC class I gene expression through downregulation of p50 (van ‘t Veer et al, 1993), our data suggest that N-myc blocks NF-κB transcriptional activity at the EAAT2 promoter. Preliminary immunoprecipitation experiments in transiently transfected cells overexpressing N-myc indicate that repression occurs through protein–protein interaction (data not shown). Given that N-myc recruitment is weakly induced with EGF at later time points in a manner similar to NF-κB, we cannot rule out the possibility that N-myc plays a role in activation of EAAT2. Alternatively, delayed recruitment of N-myc with EGF could be involved in turning off gene expression at later time points. Experiments addressing these possibilities are currently underway.
N-myc is a transcription factor that is critical for development as N-myc null mice are embryonic lethal (Sawai et al, 1991; Stanton et al, 1992), and conditionally deleting N-myc in neuronal progenitor cells prevents proper nervous system development (Knoepfler et al, 2002). However, the function of this regulator in glial cells is not known. Moreover, very few N-myc target genes have been identified to date. Our data suggest that N-myc may play a role in regulating EAAT2 expression and glutamate uptake (Figures 7 and 8). Interestingly, increased N-myc expression is observed in reactive astrocytes of brains of patients with Alzheimer's, Parkinson's and Huntington's disease (Ferrer and Blanco, 2000), suggesting that aberrant N-myc activity in glial cells may contribute to neurodegenerative pathologies.
Finally, the ability of NF-κB to regulate EAAT2 expression has important implications for the regulation of glutamate homeostasis in the CNS. To prevent the overstimulation of neuronal glutamate receptors that can trigger excitotoxic mechanisms and cell death, extracellular concentrations of excitatory amino acids are tightly controlled by transport systems on both neurons and glial cells (Danbolt, 2001). EAAT2 is critical for rapid clearance of synaptically released glutamate for proper neurotransmission (Rothstein et al, 1996, 2004). Accumulation of excessive glutamate levels in neuronal synapses can lead to excitotoxic neuronal death, which has been implicated in the pathogenesis of numerous neurodegenerative diseases, as well as CNS injury resulting from stroke and ischemia (Doble, 1999). Notably, these conditions have been associated with increased NF-κB activity (Mattson and Camandola, 2001), and reduced EAAT2 expression is observed after brain injury and in patients with Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis and multiple sclerosis (Trotti, 2002; Maragakis et al, 2004). Interestingly, EAAT2 may also have a role in development, as work in Drosophila demonstrates that this transporter is involved in terminal glial cell differentiation (Soustelle et al, 2002). Moreover, reduced glial cell populations are observed in mice lacking the EGF receptor (Kornblum et al, 1998), suggesting that EGF signaling is also important for glial cell differentiation. Based on our work in this study, we propose that positive regulation of EAAT2 by NF-κB in response to EGF may promote glial cell differentiation and uptake of synaptic glutamate by glial cells, whereas TNFα-mediated inhibition of EAAT2 by NF-κB may contribute to glutamate toxicity and cell death in neuroinflammation and disease.
Materials and methods
Cell culture and reagents
Human H4 astroglioma cells were obtained from ATCC (HTB-148) and cultured in DMEM (Sigma), 10% fetal bovine serum (FBS; Sigma) and 1 × pen-strep (Invitrogen). H4 cultures were switched to 0.5% serum for 14–24 h before treating with TNFα or EGF. TNFα (Promega) was used at a concentration of 20 ng/ml. EGF (Upstate) was used at 50 ng/ml. All oligos for EMSA, DAPA and PCR analyses were obtained from Integrated DNA Technologies. H4 cells stably expressing IκBα-SR were generated as described previously for MC615 cells (Sitcheran et al, 2003).
Quantitative real-time RT–PCR analysis
Total RNA was prepared from wild-type H4 astroglial cells or H4 cells expressing vector or IκBα-SR according to the manufacturer's protocol for RNeasy (Qiagen). cDNA was transcribed with random primers and MMLV reverse transcriptase. Quantitative real-time PCR analysis of EAAT2 cDNA was performed using Taqman® Assays-on-Demand™ for EAAT2 (Applied Biosystems assay # Hs00188189_m1) in an ABI Prism Sequence Detection System 7000.
Electrophoretic mobility shift assays
EMSAs were performed as previously described (Mayo et al, 2001). Briefly, 4–5 μg of nuclear extracts was prepared following cell stimulation and incubated with a radiolabeled DNA probe containing an NF-κB consensus site from the EAAT2 promoter (described below). For supershifts, 1 μl of anti-p65 antibody (Rockland) or 2 μl of anti-p50 antibody (Santa Cruz, SC-7178) was added and the binding reaction was allowed to proceed for an additional 15 min. Protein–DNA complexes were resolved on a nondenaturing polyacrylamide gel and visualized by autoradiography. The following oligo pairs were annealed in STE buffer (10 mM Tris pH 8, 50 mM NaCl, 1 mM EDTA) and klenow-labeled with [α-32P]CTP: EκB (−583) wild type, 5′-GGCGGAGACGGAGGGGCATCCCGGGTCTCCGC GCGGTC-3′ and 5′-GACCGCGCGGAGACCCGGGATGCC-3′; EκB (−583) mutant, 5′-GGCGGAGACGGAGGTTCATCAAAGGGTCTCCG CGCGGTC-3′ and 5′-GACCGCGCGGAGACCCTTTGATGA-3′; EκB (+265) wild type, 5′-GCTGAAGAGGAGGGGGCGTTCCCAGACCATGC ATC-3′ and 5′-GATGCATGGTCTGGGAACGCCCCC; EκB (+265) mutant, 5′-GCTGAAGAGGAGGGTTCGTTCAAAGACCATGC ATC-3′ and 5′-GATGCATGGTCTTTGAACGAACCC-3′. NF-κB consensus sequences are underlined and mutated sites are indicated in bold.
DNA affinity purification assays
DAPA experiments were performed using the μMACS Streptavidin Kit (Miltenyi Biotec) according to the manufacturer's specifications. Briefly, 1 μg of 5′-biotinylated, duplexed oligo corresponding to the EκB (−583) wild-type site (described above for EMSA) or the N-myc −522 site (GTGACCTGGGGCCACGCGGGCTTCAGGGGA) was coupled to streptavidin microbeads for 30 min at 4°C. N-myc consensus E-box site is indicated in bold. A 100 ng portion of nuclear extracts was added and allowed to bind for an additional 30 min. DNA–protein complexes were immobilized magnetically and washed four times with 20 mM HEPES–KOH pH 7.9, 100 mM KCl and 2.5 mM MgCl2. Specifically bound proteins were eluted with 20 mM HEPES–KOH pH 7.9 and 1 M NaCl. Eluates were run on denaturing polyacrylamide gels for subsequent Western blot analysis.
Chromatin immunoprecipitation assays
ChIP analysis was performed as previously described (ref) with some modifications. Briefly, protein–DNA complexes were crosslinked with formaldehyde (10 min at room temperature) and cells were harvested. Cells were either snap-frozen in dry ice/EtOH and stored at −80°C or immediately used. DNA was sonicated to lengths 500–1000 bp and a p65 antibody (Rockland) was used for immunoprecipitation overnight at 4°C. Quantitative real-time PCR was performed and values were expressed as a percent of total input DNA for each time point. Primers to amplify the EAAT2 −583 promoter region were designed using PrimerExpress (ABI): 5′-ATGTCAGCTCTCGACGAAAATAGA-3′ (forward) and 5′-GGAGGGATTGCAAGGTTTAGC-3′ (reverse). The same primer set was used for ChIP experiments with N-myc antibody (Santa Cruz, SC-791).
Transfection and reporter assays
H4 cells were cultured in F12-DMEM containing 10% charcoal-stripped FBS, penicillin and streptomycin. A total of 1 × 104 cells per well were seeded in 24-well plates and were transfected with 500 ng total DNA using FuGene (Roche) or DuoFect (Novagen) according to the manufacturer's protocol. In all, 100 ng of EAAT2-luciferase reporter and 50 ng of pRL-SV40 (Renilla reporter control) DNA were used for transfection. Media containing 0.5% FBS and EGF (50 ng/ml) or TNFα (20 ng/ml) were added to the wells for next 48–72 h. Cells were harvested after 24–48 h of treatment. Luciferase assays were performed using the Dual Luciferase Assay System (Promega). Activity was measured using Lmax luminometer (Molecular Devices). All transfections were performed in triplicate.
Mutagenesis
The wild-type EAAT2 promoter from −954 to +45 has been previously described (Su et al, 2003) and was used for further studies and mutation analysis. To create mutation at the −583 NF-κB-binding sequence, the sequence 5′-GGGGCATCCCGG-3′ was changed to 5′-CATCTGGATTCA-3′ by PCR-based mutagenesis. To create a triple mutant, the −583 mutant construct was used as a template and the prospective NF-κB-binding sites at −272 and −251 were mutated using an oligo having mutation at both the points. The sequence at −272 site 5′-GGGATCGCCTGCAAATCCCC-3′ was changed to 5′-GATACTACTAGATAGTACTA-3′ and the sequence at −245 site 5′-GGGGCTAAACC-3′ was changed to 5′-TGGTCTAAACC-3′. To generate the mutations in the N-myc consensus sites, we used the Transformer site-directed mutagenesis kit (Clontech) according to the manufacturer's protocol using the following oligos: −522 site (5′-GTGACCTGGGGCCCCGGGGGCTTCAGGGG-3 ′) and −163 site (5′-CACACACCCGCACCCGGGCACGCACGTC-3 ′). Mutated residues are indicated in bold. In both cases, the wild-type CACGCG site was changed to CCCGGG, creating a SmaI site, which was used to analyze mutant clones. The following oligo was used to mutate the NheI site in the vector to select for mutagenic events: 5′-GCTCTTACGCGTGGCAGCCCGGGCTCGAGATC -3′). All reporters were sequenced to confirm mutations.
Western blot analysis
A 10–20 μg portion of protein from whole cell, nuclear or cytoplasmic lysates was run on precast NuPage gels, blotted onto nitrocellulose and blocked in TBST+5% nonfat dry milk and 1% BSA. The following antibodies were used: p65 (Rockland), p50 (Santa Cruz, SC-7178), IκBα (SC-1643), IκBβ (SC-8014), IκBγ (SC-8330), β-tubulin (SC-9104), phospho-IκBα (Cell Signaling Technology #9246L) and phospho-MEK1/2 (Cell Signaling #9120).
Supplementary Material
Supplemental Figure 1
Acknowledgments
We thank Stephanie Brennan for technical assistance and members of the Baldwin lab for helpful discussions. This work was supported by the Samuel Waxman Cancer Research Foundation (PBF and ASB), the Chernow Endowment (PBF) and NIH grants NS31492 (PBF), AI35908 (ASB) and CA73756 (ASB). RS was supported in part by the Cancer Research Institute. PBF is the Michael and Stella Chernow Urological Cancer Research Scientist and PBF and ASB are SWCRF Investigators.
References
- Amrolia PJ, Cunningham JM, Ney P, Nienhuis AW, Jane SM (1995) Identification of two novel regulatory elements within the 5′-untranslated region of the human A gamma-globin gene. J Biol Chem 270: 12892–12898 [DOI] [PubMed] [Google Scholar]
- Anest V, Cogswell PC, Baldwin AS (2004) IKKalpha and p65/RelA contribute to optimal EGF-induced c-fos gene expression independent of Ikappa Balpha degradation. J Biol Chem 279: 31183–31189 [DOI] [PubMed] [Google Scholar]
- Baldwin AS (2001) Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest 107: 241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Farhadi A, Fields JZ, Shaikh M, Keshavarzian A (2004a) PKC-beta1 isoform activation is required for EGF-induced NF-kappaB inactivation and IkappaBalpha stabilization and protection of F-actin assembly and barrier function in enterocyte monolayers. Am J Physiol Cell Physiol 286: C723–C738 [DOI] [PubMed] [Google Scholar]
- Banan A, Zhang LJ, Shaikh M, Fields JZ, Farhadi A, Keshavarzian A (2004b) Inhibition of oxidant-induced nuclear factor-kappaB activation and inhibitory-kappaBalpha degradation and instability of F-actin cytoskeletal dynamics and barrier function by epidermal growth factor: key role of phospholipase-gamma isoform. J Pharmacol Exp Ther 309: 356–368 [DOI] [PubMed] [Google Scholar]
- Belvin MP, Anderson KV (1996) A conserved signaling pathway: the Drosophila toll-dorsal pathway. Annu Rev Cell Dev Biol 12: 393–416 [DOI] [PubMed] [Google Scholar]
- Biswas DK, Cruz AP, Gansberger E, Pardee AB (2000) Epidermal growth factor-induced nuclear factor kappa B activation: a major pathway of cell-cycle progression in estrogen-receptor negative breast cancer cells. Proc Natl Acad Sci USA 97: 8542–8547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND (2004) Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell 13: 853–865 [DOI] [PubMed] [Google Scholar]
- Chen D, Xu LG, Chen L, Li L, Zhai Z, Shu HB (2003) NIK is a component of the EGF/heregulin receptor signaling complexes. Oncogene 22: 4348–4355 [DOI] [PubMed] [Google Scholar]
- Choi DW (1988) Glutamate neurotoxicity and diseases of the nervous system. Neuron 1: 623–634 [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1–105 [DOI] [PubMed] [Google Scholar]
- Doble A (1999) The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther 81: 163–221 [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R (2000) N-myc and c-myc expression in Alzheimer disease, Huntington disease and Parkinson disease. Brain Res Mol Brain Res 77: 270–276 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109 (Suppl): S81–S96 [DOI] [PubMed] [Google Scholar]
- Hayakawa M, Miyashita H, Sakamoto I, Kitagawa M, Tanaka H, Yasuda H, Karin M, Kikugawa K (2003) Evidence that reactive oxygen species do not mediate NF-kappaB activation. EMBO J 22: 3356–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CY, Park JH, Seo KH, Kim JM, Im SY, Lee JW, Choi HS, Lee K (2003) Expression of MIS in the testis is downregulated by tumor necrosis factor alpha through the negative regulation of SF-1 transactivation by NF-kappa B. Mol Cell Biol 23: 6000–6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel A (2000) The IKK complex: an integrator of all signals that activate NF-kappaB? Trends Cell Biol 10: 129–133 [DOI] [PubMed] [Google Scholar]
- Jones KA, Luciw PA, Duchange N (1988) Structural arrangements of transcription control domains within the 5′-untranslated leader regions of the HIV-1 and HIV-2 promoters. Genes Dev 2: 1101–1114 [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN (2002) N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev 16: 2699–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblum HI, Hussain R, Wiesen J, Miettinen P, Zurcher SD, Chow K, Derynck R, Werb Z (1998) Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J Neurosci Res 53: 697–717 [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM (2002) NF-kappaB regulation in the immune system. Nat Rev Immunol 2: 725–734 [DOI] [PubMed] [Google Scholar]
- Mansour SL, Grodzicker T, Tjian R (1986) Downstream sequences affect transcription initiation from the adenovirus major late promoter. Mol Cell Biol 6: 2684–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragakis NJ, Dykes-Hoberg M, Rothstein JD (2004) Altered expression of the glutamate transporter EAAT2b in neurological disease. Ann Neurol 55: 469–477 [DOI] [PubMed] [Google Scholar]
- Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M (2003) Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci USA 100: 12247–12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Camandola S (2001) NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest 107: 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Norris JL, Baldwin AS (2001) Ras regulation of NF-kappa B and apoptosis. Methods Enzymol 333: 73–87 [DOI] [PubMed] [Google Scholar]
- Mehta VB, Besner GE (2003) Inhibition of NF-kappa B activation and its target genes by heparin-binding epidermal growth factor-like growth factor. J Immunol 171: 6014–6022 [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C (1997) NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci 20: 252–258 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Hoberg MD, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su Z-z, Gupta P, Fisher PB (2004) B Lactam antibiotics increase neurotransmitter glutamate transporter expression and function by gene activation. Nature 433: 73–77 [DOI] [PubMed] [Google Scholar]
- Sato M, Miyazaki T, Nagaya T, Murata Y, Ida N, Maeda K, Seo H (1996) Antioxidants inhibit tumor necrosis factor-alpha mediated stimulation of interleukin-8, monocyte chemoattractant protein-1, and collagenase expression in cultured human synovial cells. J Rheumatol 23: 432–438 [PubMed] [Google Scholar]
- Sawai S, Shimono A, Hanaoka K, Kondoh H (1991) Embryonic lethality resulting from disruption of both N-myc alleles in mouse zygotes. New Biol 3: 861–869 [PubMed] [Google Scholar]
- Schluter K, Figiel M, Rozyczka J, Engele J (2002) CNS region-specific regulation of glial glutamate transporter expression. Eur J Neurosci 16: 836–842 [DOI] [PubMed] [Google Scholar]
- Schreck R, Grassmann R, Fleckenstein B, Baeuerle PA (1992) Antioxidants selectively suppress activation of NF-kappa B by human T-cell leukemia virus type I Tax protein. J Virol 66: 6288–6293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M (1993) Amplification of N-myc as a prognostic marker for patients with neuroblastoma. Semin Cancer Biol 4: 13–18 [PubMed] [Google Scholar]
- Silverman N, Maniatis T (2001) NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev 15: 2321–2342 [DOI] [PubMed] [Google Scholar]
- Sitcheran R, Cogswell PC, Baldwin AS Jr (2003) NF-kappaB mediates inhibition of mesenchymal cell differentiation through a posttranscriptional gene silencing mechanism. Genes Dev 17: 2368–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soustelle L, Besson MT, Rival T, Birman S (2002) Terminal glial differentiation involves regulated expression of the excitatory amino acid transporters in the Drosophila embryonic CNS. Dev Biol 248: 294–306 [DOI] [PubMed] [Google Scholar]
- Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF (1992) Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev 6: 2235–2247 [DOI] [PubMed] [Google Scholar]
- Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB (2003) Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc Natl Acad Sci USA 100: 1955–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Carpenter G (1998) Epidermal growth factor activation of NF-kappaB is mediated through IkappaBalpha degradation and intracellular free calcium. Oncogene 16: 2095–2102 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699–1702 [DOI] [PubMed] [Google Scholar]
- Trotti D (2002) A role for glutamate transporters in neurodegenerative diseases. Adv Exp Med Biol 513: 225–248 [DOI] [PubMed] [Google Scholar]
- Valhmu WB, Palmer GD, Dobson J, Fischer SG, Ratcliffe A (1998) Regulatory activities of the 5′- and 3′-untranslated regions and promoter of the human aggrecan gene. J Biol Chem 273: 6196–6202 [DOI] [PubMed] [Google Scholar]
- van ‘t Veer LJ, Beijersbergen RL, Bernards R (1993) N-myc suppresses major histocompatibility complex class I gene expression through down-regulation of the p50 subunit of NF-kappa B. EMBO J 12: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrecchia F, Wagner EF, Mauviel A (2002) Distinct involvement of the Jun-N-terminal kinase and NF-kappaB pathways in the repression of the human COL1A2 gene by TNF-alpha. EMBO Rep 3: 1069–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Gaynor RB (2004) IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci 29: 72–79 [DOI] [PubMed] [Google Scholar]
- Zelenaia O, Schlag BD, Gochenauer GE, Ganel R, Song W, Beesley JS, Grinspan JB, Rothstein JD, Robinson MB (2000) Epidermal growth factor receptor agonists increase expression of glutamate transporter GLT-1 in astrocytes through pathways dependent on phosphatidylinositol 3-kinase and transcription factor NF-kappaB. Mol Pharmacol 57: 667–678 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1