

Abstract
Connector enhancer of KSR (CNK), an essential component of Drosophila receptor tyrosine kinase/mitogen-activated protein kinase pathways, regulates oppositely RAF function. This bimodal property depends on the N-terminal region of CNK, which integrates RAS activity to stimulate RAF and a bipartite element, called the RAF-inhibitory region (RIR), which binds and inhibits RAF catalytic activity. Here, we show that the repressive effect of the RIR is counteracted by the ability of Src42 to associate, in an RTK-dependent manner, with a conserved region located immediately C-terminal to the RIR. Strikingly, we found that several cnk loss-of-function alleles have mutations clustered in this area and provide evidence that these mutations impair Src42 binding. Surprisingly, the derepressing effect of Src42 does not appear to involve its catalytic function, but critically depends on the ability of its SH3 and SH2 domains to associate with CNK. Together, these findings suggest that the integration of RTK-induced RAS and Src42 signals by CNK as a two-component input is essential for RAF activation in Drosophila.
Keywords: CNK, MAPK module, RAF, RAS, SFK
Introduction
The mitogen-activated protein kinase (MAPK) pathway is a critical route used by numerous receptor tyrosine kinases (RTKs) to convey proliferation, differentiation and survival signals (for review, see Widmann et al, 1999). At its core, this pathway comprises one isoform of each of the RAF, MEK and ERK/MAPK family of kinases, respectively, which form an evolutionarily conserved signaling unit also referred to as the MAPK module.
Given its position in the module, RAF is the entry point for RTK-induced upstream events. Three RAF proteins exist in mammals, namely, A-RAF, B-RAF and Raf-1/C-RAF, and one member is present in Drosophila or Caenorhabditis elegans (for review, see Dhillon and Kolch, 2002; Chong et al, 2003). RTK-induced activation of the small GTPase RAS was recognized early on as a critical event in RAF activation. RAS triggers plasma membrane anchoring of RAF through a direct contact between GTP-loaded RAS and RAF. However, this step is insufficient to induce RAF activation, but is a prerequisite for a complex series of regulatory events. For example, Ste20-like kinases and Src family kinases (SFKs) have been shown to collaborate with RAS in RTK-induced Raf-1 activation, owing to their ability to directly phosphorylate Raf-1 serine 338 (S338) and tyrosine 341 (Y341), respectively. However, these particular events are probably specific to Raf-1 as the equivalent S338 residue in B-RAF is constitutively phosphorylated, whereas the Y341-like residue is not conserved in B-RAF or in Drosophila and C. elegans RAF. Nonetheless, it remains possible that these kinases use different means to regulate RAF members. This would be consistent with genetic findings in Drosophila, which suggest that RAF is also regulated by an RTK-induced but RAS-independent pathway linked to SFKs (Hou et al, 1995; Li et al, 2000).
In addition to kinases and phosphatases regulating RAF activity, a number of apparently nonenzymatic proteins also modulate RAF function. One of these is Connector eNhancer of KSR (CNK), an evolutionarily conserved multidomain-containing protein originally identified in a KSR-dependent genetic screen in Drosophila (Therrien et al, 1998). Genetic experiments in flies indicated that CNK activity is required downstream of RAS, but upstream of RAF, thus suggesting that CNK regulates RAF activity. In agreement with this interpretation, CNK was found to interact directly with the catalytic domain of RAF and to modulate its function (Therrien et al, 1998; Anselmo et al, 2002; Douziech et al, 2003). The role of CNK, however, is probably not restricted to the MAPK pathway. Indeed, although mammalian CNK proteins have also been found to modulate the RAS/MAPK pathway (Lanigan et al, 2003; Bumeister et al, 2004), recent studies indicated that they also control other events, including membrane/cytoskeletal rearrangement (Bumeister et al, 2004), Rho-mediated SRF transcriptional activity (Jaffe et al, 2004) and RASSF1A-induced cell death (Rabizadeh et al, 2004). Given their ability to influence distinct signaling events, it is possible that CNK proteins act as signal integrators to orchestrate crosstalks between pathways.
Intriguingly, although CNK activity is vital for RAS/MAPK signaling in Drosophila, we recently found that it has opposite effects on RAF function (Douziech et al, 2003). A structure/function analysis revealed, on the one hand, that two domains (SAM and CRIC) located in the N-terminal region of CNK are integrating RAS signals enabling RAF to phosphorylate MEK. However, the ability of CNK to associate with the RAF catalytic domain was mapped to a short bipartite element, named the RAF-inhibitory region (RIR), that strongly antagonized MEK phosphorylation by RAF. Surprisingly, the RIR exerted its effect even in the presence of RAS signals, hence resulting in lower RAS-induced MAPK signaling output.
Here, we show that the inhibitory effect of the RIR is relieved by an RTK-induced SFK signal. Specifically, we have identified a region located immediately C-terminal to the RIR including tyrosine 1163 (Y1163) that is essential for CNK's positive function in vivo and for Sevenless (SEV) RTK-dependent MAPK activation. We found that upon SEV expression, one of the two SFKs found in Drosophila, Src42 (Takahashi et al, 1996), associates and mediates, through the Y1163 region of CNK, RTK positive effects on the MAPK module. Remarkably, cnk loss-of-function mutations affecting the Y1163 region are fully compensated by inactivation of the RIR, thereby arguing that the Y1163 region is integrating the RTK-induced Src42 signal to counteract the RIR inhibitory function. Unexpectedly, genetic and molecular evidence revealed that it is not Src42 catalytic function per se, but rather its binding capacity that is the key event in this process. Taken together, these results provide compelling evidence that CNK regulates RAF function by integrating both RAS and Src42 signals elicited by an RTK.
Results and discussion
Endogenous CNK is tyrosine phosphorylated upon RTK activation
Coexpression of a CNK construct and activated Sevenless (SEVS11) in S2 cells was previously shown to induce tyrosine phosphorylation of CNK (Therrien et al, 1998). To assess the specificity of this event, we verified whether endogenous CNK also becomes tyrosine phosphorylated upon SEVS11 expression. As shown in Figure 1A (left panel), SEVS11 expression led to sustained tyrosine phosphorylation of endogenous CNK, which paralleled MAPK kinase activation. Similar results were obtained by stimulating the Drosophila Egf or insulin RTKs (Figure 1A, center and right panels). Together, these results indicated that CNK is a bona fide and common RTK-dependent tyrosine phosphorylation target.
Figure 1.
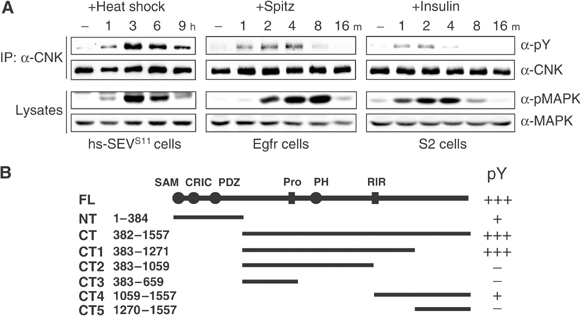
Activation of RTKs induces tyrosine phosphorylation of endogenous CNK. (A) Stable S2 cells expressing heat-inducible SEVS11 (left panel), WT Egfr (middle panel) or plain S2 cells (right panel) were stimulated by heat shock, Spitz or human insulin (10 μg/ml), respectively, and harvested at the indicated time (h (hours) or m (minutes)). Total and tyrosine-phosphorylated CNK levels were assessed by immunoblots following immunoprecipitation (IP) of endogenous CNK. Total and activated MAPK (pMAPK) levels were determined from cell lysates. Results presented here and thereafter are representative of at least three similar experiments. (B) Schematic (top line) of FL-CNK (1557 amino acid) and its conserved domains/elements (Douziech et al, 2003). Representations and amino-acid position of various Flag-tagged CNK variants are shown below, and arbitrary + or − signs to the right indicate their relative SEVS11-dependent tyrosine phosphorylation levels.
CNK tyrosine phosphorylation and activity depend on its Y1163 region
To map the region(s) associated with tyrosine phosphorylation, we transfected S2 cells with SEVS11 and various Flag-tagged CNK deletion constructs (Figure 1B). A C-terminal (CT) CNK variant was almost as efficiently phosphorylated as full-length (FL) CNK, whereas an N-terminal (NT) CNK protein was three to five times less efficiently phosphorylated (Supplementary Figure S1). Because of its apparently greater phosphorylation stoichiometry, we decided to narrow down the region(s) of CT-CNK associated with tyrosine phosphorylation. Five additional CT-CNK truncations (CT1–CT5; Figure 1B) were coexpressed with SEVS11 and their respective tyrosine phosphorylation level was determined. These experiments revealed that sequences between amino-acid positions 1059 and 1271 are the most critical for CNK tyrosine phosphorylation (Figure 1B and Supplementary Figure S1).
The 1059–1271 area contains three tyrosine residues. Interestingly, these residues are located within an ∼90-amino-acid stretch that is highly conserved between Drosophila melanogaster and Anopheles gambiae CNK (Figure 2A). The conserved stretch has two other striking features: (1) the first half corresponds to the so-called RIR that associates with the kinase domain of RAF and inhibits its catalytic function (Douziech et al, 2003); (2) two previously characterized alleles of cnk (E-1222 and E-1756) have amino-acid changes within the second half of conservation (Therrien et al, 1998; Figure 2). As these are loss-of-function alleles (Therrien et al, 1998, and data not shown), it suggested that the second half of homology plays a positive role in signaling. During the course of this work, we characterized other cnk alleles to identify additional functionally relevant areas. Remarkably, of the 11 alleles that were sequenced, three (one stop codon and two amino-acid changes) were specifically found in this area (Figure 2A and B).
Figure 2.
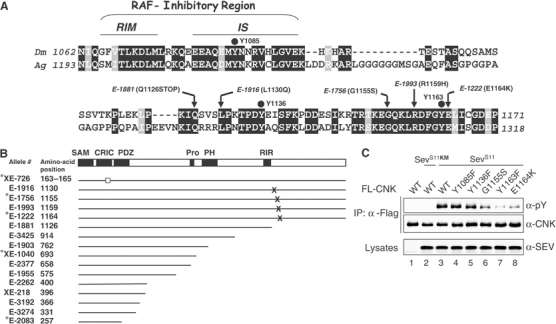
The Y1163 region is essential for CNK function and tyrosine phosphorylation. (A) Amino-acid comparison of the 1062–1171 region of D. melanogaster (Dm) CNK to an equivalent region (positions 1193–1318) in A. gambiae (Ag) CNK. Identical and conserved residues are in black and gray boxes, respectively. The RIR is a negatively acting bipartite region made of a RAF-interacting motif (RIM) and an adjacent inhibitory sequence (IS; Douziech et al, 2003). Black dots highlight the three tyrosine residues of the area. The position and amino-acid change found in five cnk alleles are also indicated. (B) Schematic representation of CNK is shown as a reference to position cnk mutations characterized in this study, or previously (Therrien et al, 1998; denoted by a star). Allele numbers and affected amino-acid positions are shown to the left. The hollow square indicates a three-amino-acid in-frame deletion. X denotes missense mutations, whereas a truncated line indicates premature ORF termination caused by either a frame-shift mutation (XE-1040 and XE-218) or by a nonsense mutation (remaining alleles). (C) S2 cells were transfected with the indicated FL-CNK variants (0.5–0.7 μg), kinase-inactivated SEVS11KM (0.5 μg) or SEVS11 (0.5 μg). The CNK variants were expressed for a total of 36 h under the control of the constitutive cnk promoter. WT denotes wild type.
To examine the relevance of each tyrosine within the 1059–1271 area, we independently changed them to a phenylalanine residue and tested their respective impact on SEVS11-induced CNK tyrosine phosphorylation. We also tested CNK variants that carried the mutation found in cnkE-1756 (G1155S) and cnkE-1222 (E1164K). Mutagenesis of the first two tyrosines (Y1085 and Y1136) did not affect phosphorylation (Figure 2C, lanes 4 and 5). However, mutation of Y1163 severely decreased it (Figure 2C, lane 7), thus identifying Y1163 as a critical residue in this event. Interestingly, the G1155S and E1164K mutations also impaired, albeit to a lesser degree, CNK phosphorylation, thus suggesting that not only the Y1163 residue, but also the second half of homology is involved in this event.
The fact that cnkE-1756 and cnkE-1222 are loss-of-function alleles and that their associated amino-acid change correlates with reduced CNK phosphorylation (Figure 2C) suggested that this or a related event is required for CNK function. We investigated this possibility by first verifying whether mutagenesis of the Y1163 residue also affected CNK activity. Overexpression of FL-CNK during Drosophila eye development was previously shown to weakly antagonize RAS signaling, apparently owing to its ability to inhibit RAF catalytic function (Douziech et al, 2003). We reasoned that if the Y1163 residue is normally integrating an RTK-dependent signal, then its mutagenesis should increase CNK dominant-negative effect. To verify this, we overexpressed FL-CNKY1163F during eye development. In contrast to wild-type (WT) FL-CNK (Figure 3A and C), overexpression of one copy of the FL-CNKY1163F transgene potently antagonized normal eye development (Figure 3B and D), therefore arguing that the Y1163 residue is important for CNK activity.
Figure 3.
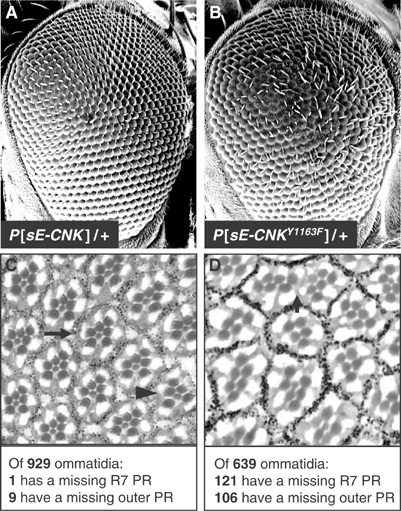
Forced expression of cnkY1163F interferes with Drosophila eye development. (A, B) Scanning electron micrographs of adult eyes. Anterior is to the right. (C, D) Apical eye sections at the plane of photoreceptor cells for the following genotypes: (C) P[sE-CNK]/+; (D) P[sE-CNKY1163F]/+. An example of a normal ommatidium is indicated by the arrow in (C): it comprises six large rhabdomeres (one for each outer photoreceptor (PR) cell) surrounding a smaller rhabdomere associated with the R7 PR cell. Dark staining granules around each ommatidium are produced by pigment cells. The arrowhead in (C) highlights one ommatidium missing an outer PR cell. Expression of CNKY1163 also affects pigment cells (arrow in (D)). Number of ommatidia analyzed and missing PR cells are indicated at the bottom.
To show that the Y1163 residue is genuinely required for CNK function, distinct cnk genomic rescue constructs were generated and tested in transgenic flies for their ability to rescue the recessive lethality associated with cnk loss-of-function. While the WT CNK construct fully restored viability to a lethal cnk allelic combination, the CNKY1163F construct was devoid of activity (Table I, and data not shown), thus confirming the functional importance of that residue. The inhibitory activity of CNK has been shown to be mediated by the RIR (Douziech et al, 2003). Because the Y1163F mutation elevated CNK's dominant-negative effect (Figure 3), we suspected that in that context, the RIR has an increased inhibitory potential. If that was the case, then mutagenesis of the RIR should counteract the effect of the Y1163F mutation. To test this, we inactivated the RIR in the Y1163F context by introducing a three-amino-acid change in the inhibitory sequence (IS) of the RIR (RIRmut; Douziech et al, 2003). Remarkably, the CNKRIRmut-Y1163F double-mutant construct rescued the recessive lethality associated with cnk loss-of-function alleles to the same extent as CNKWT (Table I, and data not shown) thereby demonstrating that disruption of the RIR can reverse the cnkY1163F mutation. Together, these findings revealed that the two conserved regions within the 1059–1271 area act oppositely and that the Y1163 region is integrating an RTK-produced signal that apparently releases the RIR inhibitory effect.
Table 1.
Genomic rescue experiments revealed the RIR/Y1163 region interplay within CNK
| Genotype | Percent of cnkE-1088/cnkl(2)k16314 adult fliesa | No. of flies scoredb |
|---|---|---|
| cnkE-1088/cnkl(2)k16314 | 0 | 315 |
| cnkE-1088/cnkl(2)k16314; pcnk-cnkWT | 19.6 | 825 |
| cnkE-1088/cnkl(2)k16314; pcnk-cnkY1163F | 0 | 578 |
| cnkE-1088/cnkl(2)k16314; pcnk-cnkRIRmut | 21.2 | 663 |
|
cnkE-1088/cnkl(2)k16314; pcnk-cnkRIRmut-Y1163F |
19.5 |
619 |
| The fully penetrant larval lethality associated with cnkE-1088/cnkl(2)k16314 trans-heterozygous flies is completely rescued by introduction of one copy of a WT cnk genomic rescue construct (pcnk-cnkWT). | ||
| aFor a full rescue, the expected ratio of viable cnkE-1088/cnkl(2)k16314 flies over total progeny is 20%. | ||
| bThe number of flies scored represents a compilation of two independent transgenic lines. | ||
Src42 associates with CNK and mediates CNK tyrosine phosphorylation in an RTK-dependent manner
To characterize the functional connection between the RIR and the Y1163 region, we sought to identify the tyrosine kinase mediating CNK phosphorylation. As we failed to detect direct in vitro phosphorylation of CNK by SEVS11 (data not shown), we examined whether nonreceptor tyrosine kinases of the Src family could be implicated.
Drosophila has two SFKs, Src42 and Src64 (Simon et al, 1985; Takahashi et al, 1996). To verify their involvement, we separately eliminated their contribution by RNAi. In addition, we tested the effect of removing endogenous RAS as well as Tec29, a nonreceptor tyrosine kinase of the Tec family (TFKs; Smith et al, 2001). Interestingly, depletion of endogenous Src42 reduced SEVS11-induced CNK phosphorylation, whereas none of the other targets had an effect (Figure 4A). To investigate whether the inability of RAS depletion to affect CNK phosphorylation could be due to residual but sufficient RAS activity, we depleted concomitantly endogenous RAS and its exchange factor SOS to further lower the RAS-dependent signaling branch regulating MAPK. As shown in Supplementary Figure S2A, simultaneous depletion of both proteins almost eliminated SEV-induced MAPK activation, but did not decrease CNK tyrosine phosphorylation. Moreover, to determine whether Src64 or Tec29 could contribute, yet to a lower extent than Src42, to the total phosphorylation level of CNK, we verified whether removing concomitantly the two SFKs and Tec29 had a greater impact than Src42 depletion alone. As shown in Supplementary Figure S2B, no further decrease was observed. Therefore, these data indicated that Src42 acts as an intermediate kinase linking activated SEV to CNK tyrosine phosphorylation and suggested that this event is RAS-independent. Consistent with this, coexpression of activated Src42 (Src42Y511F) and the FL-CNK variants recapitulated the Y1163-dependent phosphorylation of CNK (Supplementary Figure S3). However, these results taken together are not sufficient to conclude that Src42 is specifically phosphorylating the Y1163 residue of CNK. Among various scenarios, it is possible that this residue is first phosphorylated by another kinase and then serves as a docking site for Src42 SH2 domain, thereby allowing Src42 to phosphorylate (an)other tyrosine residue(s) on CNK. Incidentally, this model turned out to be the most likely (see below).
Figure 4.
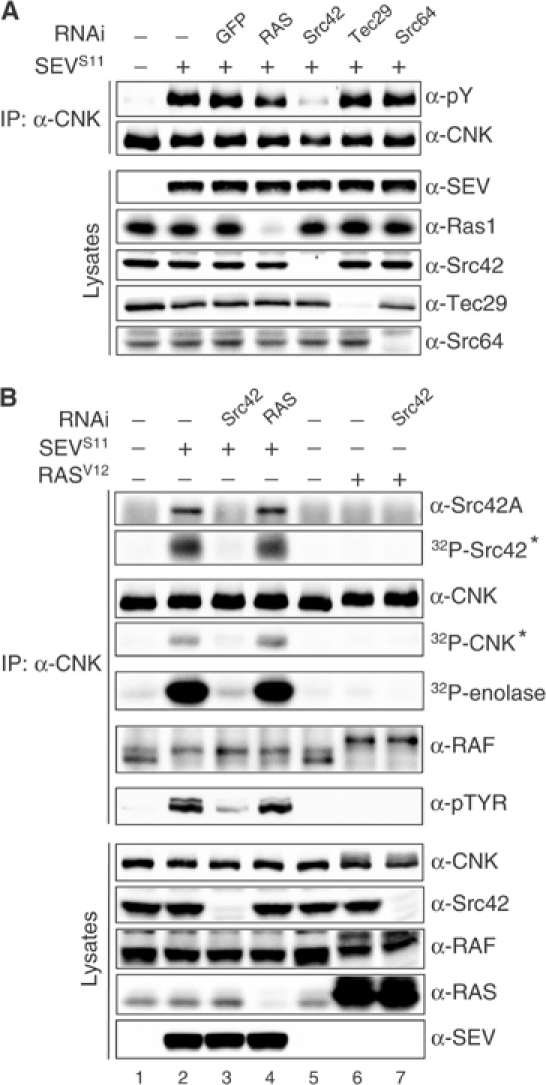
Src42 associates and mediates CNK tyrosine phosphorylation in an RTK-dependent manner. (A) SEVS11 stable cells were incubated ±the indicated double-stranded RNAs (dsRNAs) (15 μg/ml) for 4 days prior to inducing SEVS11 expression. (B) SEVS11 or RASV12 stable cells were cultured as in (A) and treated as indicated. Endogenous CNK was then immunoprecipitated (IP) using α-CNK and subjected to an in vitro kinase assay. Samples were resolved by SDS–PAGE and analyzed by autoradiography and immunoblotting. A star indicates the putative identity of the radioactive band. Src42 RNAi reversed the effects of SEVS11 expression (lane 3), thus demonstrating the involvement of Src42 in mediating the effects of SEV on CNK.
To determine whether Src42 associates with CNK, we immunoprecipitated endogenous CNK from lysates of untreated or heat-treated hs-SEVS11 cells and probed the immunoprecipitates for the presence of Src42. Indeed, Src42 was found to interact with CNK, but only upon SEV expression (Figure 4B, top panel, compare lanes 1 and 2). Immunoprecipitation of endogenous Src42 also co-precipitated CNK upon SEV expression (data not shown). Consistent with the fact that RAS is apparently not involved in SEV-induced tyrosine phosphorylation of CNK, elimination of endogenous RAS did not affect the Src42/CNK association (lane 4). Furthermore, expression of activated RAS (RASV12) did not stimulate CNK tyrosine phosphorylation nor did it induce its association with Src42 (compare lanes 5 and 6). Together, these findings demonstrated that CNK and Src42 form a complex in vivo and that their association is SEV-dependent, but RAS-independent. As expected, no SEV-induced association with CNK has been detected with either endogenous Src64 or Tec29 (data not shown).
The presence of endogenous Src42 in the CNK immunoprecipitates prompted us to ask if it was catalytically active, and if so, whether it could promote CNK phosphorylation de novo. We thus prepared anti-CNK immunoprecipitates as above and incubated them in a kinase assay cocktail containing [γ-32P]ATP and the Src substrate enolase. As shown in Figure 4B, strong phosphorylation of enolase was detected (fifth panel from the top) as well as in a band migrating at the position of Src42 (second panel from the top), which most likely corresponds to autophosphorylated Src42. Interestingly, another major radioactive band was observed at the position of CNK (fourth panel from the top) that we believe is CNK, although it is possible that this signal represents an irrelevant protein comigrating with CNK. Taken together, these results provide compelling evidence that CNK-associated Src42 is catalytically active. Furthermore, although we cannot rule out the involvement of intermediate kinase(s), these data suggest that Src42 phosphorylates CNK.
Src42 regulates positively MAPK activation through the Y1163 region of CNK
We next wanted to investigate the relationship linking Src42 and CNK and its relevance with respect to RTK-dependent MAPK activation. Using RNAi, we first depleted endogenous Src42 levels from SEVS11 cells and tested the ability of SEVS11 to activate endogenous MAPK. Similar to decreasing endogenous SOS, RAS or CNK by RNAi (Figure 5A, lanes 4, 6 and 7), depletion of Src42 impaired MAPK activation (lane 5), thus showing that Src42 plays a positive role in RTK-mediated MAPK activation. In contrast, removal of endogenous Src64 (or Tec29) had no effect (Supplementary Figure S2B, and data not shown), which suggests that the two Drosophila SFKs perform distinct roles downstream of the RTKs or respond differently depending on the RTK. Consistent with these possibilities, depletion of endogenous Src42 also affected EGFR- and InsR-dependent MAPK activation, whereas elimination of endogenous Src64 (or Tec29) had no effect (data not shown). Finally, removal of endogenous Src42 had no effect on RASV12-induced MAPK activation (Figure 5B, lane 5), thus implying that Src42 is acting upstream and/or in parallel to RAS.
Figure 5.
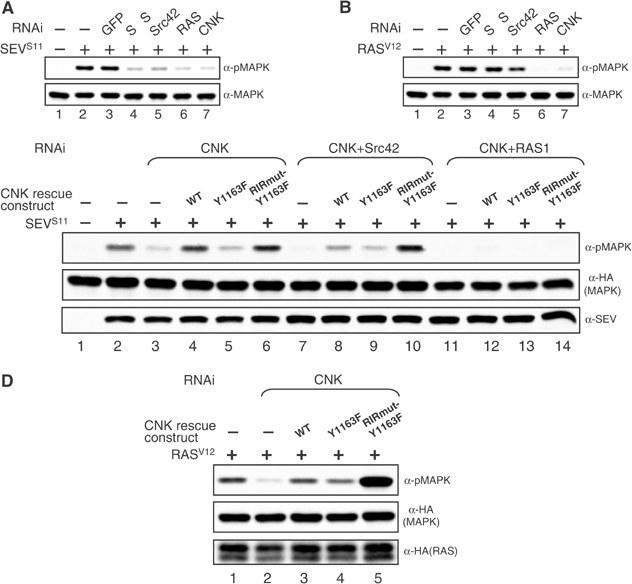
Src42 is a positive component of the MAPK pathway that acts through the Y1163 region of CNK. (A, B) SEVS11 or RASV12 stable cells were incubated ±the indicated dsRNAs (15 μg/ml) for 4 days prior to inducing SEVS11 or RASV12 expression. (C, D) Rescue of MAPK activation caused by depletion of endogenous CNK in S2 cells. For these experiments, cells were preplated ±RNAi (10 μg/ml) and then transfected 24 h later with the indicated constructs. (C) HA-tagged MAPK (0.15 μg), SEVS11 (0.1 μg) and the CNK rescue constructs (0.04 μg). SEV expression was induced 48 h post-transfection. (D) HA-MAPK (0.15 μg), HA-RASV12 (0.1 μg) and the CNK rescue constructs (0.01 μg). Transgene expression was induced 48 h post-transfection. CNK rescue constructs produced identical protein levels (not shown).
To investigate whether the Y1163 region of CNK is relevant for Src42 positive effect, we used an RNAi-based rescue assay developed previously to identify functionally relevant regions of CNK (Douziech et al, 2003). In brief, a dsRNA targeting the 3′UTR of CNK was used to deplete endogenous CNK, which was then complemented by transfecting a CNK rescue construct that uses heterologous 3′UTR sequences. In contrast to a WT construct, a CNKY1163F construct barely restored SEV-dependent MAPK activation in cells depleted of endogenous CNK (Figure 5C, lanes 2–5), thus demonstrating that the Y1163 residue is integrating an RTK signal required for full activation of the MAPK module. Given the inhibitory role of the RIR in the MAPK pathway (Douziech et al, 2003) and that its inactivation in vivo is reversing cnk loss-of-function caused by the Y1163F mutation (Table I), we verified whether inactivating the RIR in the Y1163F context would reinstate CNK activity. As shown in Figure 5C (lane 6), the CNKRIRmut-Y1163F construct was as active as CNKWT, thus providing biochemical evidence that the Y1163 region is integrating a signal that relieves the inhibitory effect of the RIR.
We then reasoned that if the RTK-induced signal integrated by the Y1163 region is mediated by Src42, then removing endogenous Src42 and CNK concomitantly should abolish CNKWT rescuing activity to the same extent as CNKY1163F, but should not affect the rescuing potential of CNKRIRmut-Y1163F as this construct should bypass the need in Src42 activity. Interestingly, we found that CNKWT was not totally devoid of activity, but could somewhat rescue Src42 depletion (Figure 5C, compare lanes 7 and 8) probably because endogenous RAS activated by SEVS11 plays a dominant role in RAF activation and as CNKWT still integrates RAS activity through its N-terminal domains, this leads to detectable phospho-MAPK. Nonetheless, the CNKY1163F construct was as active as CNKWT in this context (lane 9), which strongly suggested that Src42 effect is indeed mediated by the Y1163 region. Furthermore and as predicted, the CNKRIRmut-Y1163F construct was not affected by Src42 depletion (lane 10), thus demonstrating its ability to circumvent the need for Src42. Finally, in support of the view that RAS plays a major role in these events, abrogation of MAPK activation by depleting both endogenous CNK and RAS could not be rescued by any of the CNK constructs (Figure 5C, lanes 11–14).
Taken together, the results presented above suggest that the Y1163 region is integrating an Src42-dependent but RAS-independent signal. If this view is correct, then RASV12-induced MAPK activation should not depend on the Y1163 residue. Moreover, as the RIR is inhibitory in a RASV12-only context (Douziech et al, 2003), its inactivation should promote a stronger MAPK activation, as it would imitate the reception of an Src42 signal. This is indeed what we observed. As shown in Figure 5D, CNKY1163F was as active as CNKWT in a RASV12 context (compare lanes 2–4), whereas CNKRIRmut-Y1163F elevated RASV12-induced MAPK activation to a much greater extent (lane 5).
CNK-integrated Src42 effect on the MAPK pathway primarily depends on its binding function
Intriguingly, two previous studies suggested that Src42 negatively regulates the RAS/MAPK pathway (Therrien et al, 1998; Lu and Li, 1999). Because we found no such role in S2 cells, it is possible that these observations were cell specific and/or RTK specific. A more trivial explanation would be that the Src42 alleles used in those genetic studies were not loss-of-function, but instead had gain-of-function activity with respect to the RAS/MAPK pathway. To explore the second possibility, we compared some of the Src42 alleles used to conclude a negative role for Src42, namely Src42S-382 and Src42S-527 (Therrien et al, 1998), as well as a bona fide Src42 loss-of-function (Src42l(2)k10108), for their ability to modify dominantly a RASV12 rough eye phenotype. Surprisingly, in contrast to Src42S-382, which strongly enhanced the RASV12 rough eye phenotype (Figure 6, compare A and B), Src42l(2)k10108 did not enhance the phenotype (Figure 6, compare A and C). Src42S-527 also behaved like Src42S-382 (data not shown). Therefore, these findings suggest that the alleles used to conclude a negative function for Src42 are not loss-of-function, but act as activated alleles with respect to the RAS/MAPK pathway. In support of this claim, we found that the Src42S-382 and Src42S-527 alleles efficiently rescued the lethality associated with two independent hypomorphic alleles of raf, whereas the Src42l(2)k10108 allele could not (Supplementary Table S1). These results not only provide additional evidence that Src42S-382 and Src42S-527 act as gain-of-functions with respect to the RAS/MAPK pathway, but also suggest that the role of Src42 in this pathway is not restricted to the eye.
Figure 6.
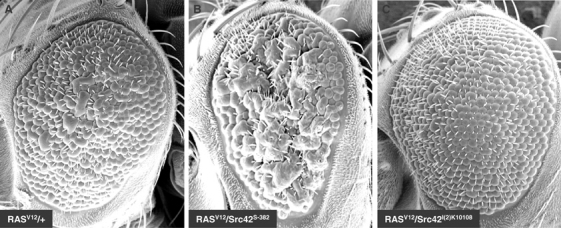
Src42S-382 acts as a gain-of-function allele in the presence of RASV12. Scanning electron micrographs of adult eyes of the following genotypes: (A) CyO, P[sev-RASV12]/+; (B) CyO, P[sev-RASV12]/Src42S-382; (C) CyO, P[sev-RASV12]/Src42l(2)k10108. Anterior is to the left.
To determine the molecular basis responsible for the apparent gain-of-function effect of Src42S-382 and Src42S-527, we isolated their genomic DNA and sequenced the Src42 exons. Src42S-382 has a point mutation that changes tryptophan 241 to an arginine (W241R). Strikingly, the equivalent residue (W260) in mammalian Src and Hck, which resides at the junction between the SH2 domain-kinase domain (SH2-KD) linker and the kinase domain (Figure 7A), has been predicted from crystallographic and functional studies to be structurally important for maintaining SFKs in an autoinhibited conformation (for review, see Hubbard, 1999), thus supporting the possibility that the W241R mutation is a gain-of-function. Unexpectedly, however, the Src42S-527 allele has a single point mutation that changes aspartate 370 (D370) to a valine residue. As D370 corresponds to the critical ‘catalytic base' found in kinase subdomain VI, this mutation should destroy Src42 catalytic function. An in vitro kinase assay confirmed this prediction, whereas the W241R mutation was found to be nearly as active as WT Src42 (Supplementary Figure S4). Given that the D370V mutation impairs Src42 catalytic function, it suggested that catalytic activity per se is not critical for Src42 effect on the pathway and that structural changes imposed by the W241R or D370V mutations are activating other aspects of Src42 function, such as its capacity to associate with specific targets. This possibility is likely considering that even mutations affecting catalytic function can have gain-of-function consequences provided that they disrupt the overall autoinhibited configuration and make available the SH3 and SH2 domains. As the catalytic center of SFK also appears to stabilize the global autoinhibited state (Hubbard, 1999), it is possible that not only the W241R mutation, but also the D370V mutation destabilizes Src42 autoinhibited state thereby increasing the ratio of Src42 molecules bound to some targets, including CNK.
Figure 7.
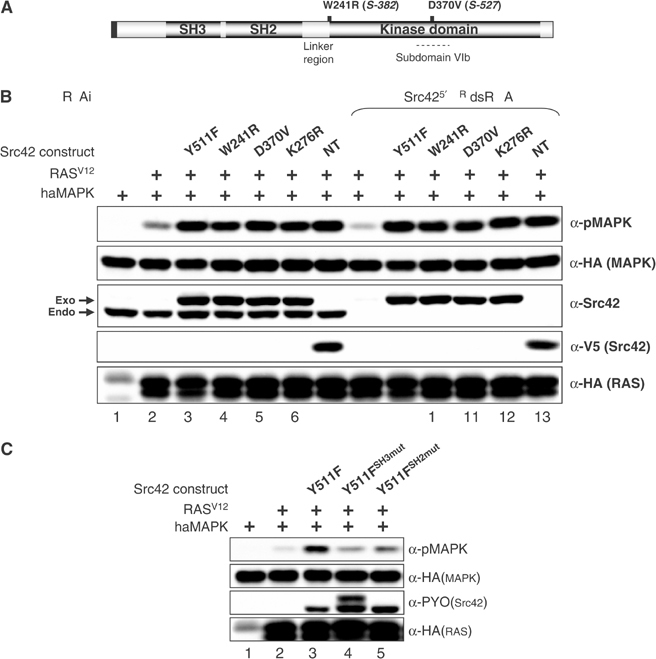
Molecular characterization of Src42 gain-of-function alleles. (A) Schematic structure of Src42. Like mammalian SFKs, the 517-amino-acid-long Src42 contains a myristoylation signal (black box), followed by an SH3, SH2 and kinase domain. Relative positions of amino-acid change found in Src42S-382 and Src42S-527 are shown. (B) S2 cells were first incubated ±the Src42 dsRNA (15 μg/ml) 1 day prior to transfection and then transfected with the HA-MAPK reporter construct (0.15 μg) either alone (lane 1) or with the indicated combinations of HA-RASV12 (0.1 μg) and PYO-Src42 (0.075–0.175 μg) constructs or the truncated V5-tagged NT-Src42 construct (0.075 μg). Endo and exo denote the position of the endogenous and exogenous Src42 proteins, respectively. (C) S2 cells were transfected as in (B). The amount of Src42 constructs used (0.065–0.3 μg) was adjusted to obtain similar protein levels.
The observation that one of the Src42 gain-of-function alleles is a kinase-dead variant was intriguing given the correlation that we observed between CNK positive function on the pathway and Src42-mediated CNK tyrosine phosphorylation. To investigate this issue, we first used a simple cotransfection assay that monitored the ability of Src42 constructs to augment RASV12-induced MAPK activation. As shown in Figure 7B, kinase-inactive Src42D370V and Src42K276R variants (lanes 5 and 6) were found to be as competent to collaborate with RASV12 as Src42Y511F and Src42W241R (lanes 3 and 4). Interestingly, even an N-terminal (NT) Src42 protein (including only the SH3 and SH2 domains) was active as the full-length constructs (lane 7). In contrast, a construct corresponding to the catalytic domain alone was inert (data not shown). To ensure that the activity of these mutants was not caused by endogenous Src42, we tested their activity in a context where endogenous Src42 has been depleted by RNAi. As shown in Figure 7B (lanes 9–13), none of the mutants was affected. We showed above that, unlike RAS activity, Src42 activity is integrated by the Y1163 residue of CNK (Figure 5C and D). To verify the relevance of this residue with respect to the ability of kinase-impaired Src42 to collaborate with RASV12, we depleted endogenous CNK and rescued its contribution with CNKY1163. Remarkably, we found that the positive effect of the Src42 mutants strictly depended on the integrity of the Y1163 residue (Supplementary Figure S5). Together, these results are consistent with the idea that Src42 kinase activity is not critical for the positive effect of Src42 on the MAPK pathway and further suggest that Y1163-dependent CNK activity and Src42-mediated CNK tyrosine phosphorylation are not functionally related. In support for these conclusions, we found that kinase-inactive Src42K276R is as competent as WT Src42 to rescue a decrease of SEV-dependent MAPK activation caused by depletion of endogenous Src42 (data not shown).
In addition to their catalytic function, SFKs are known to mediate positive signaling by the binding properties of their SH3 and SH2 domains (Kaplan et al, 1995; Schlaepfer et al, 1997). To verify their respective relevance, we mutagenized them individually in Src42Y511F and tested their ability to augment RAS-mediated MAPK activation. As shown in Figure 7C, inactivation of either domain impaired Src42 activity (lanes 4 and 5). Similar findings were obtained with Src42K276R or NT-Src42 (data not shown), thereby suggesting that Src42 binding was critical for its function through CNK. We then investigated whether the Src42 variants could associate with CNK through its Y1163 region. As shown in Figure 8A, co-immunoprecipitation experiments revealed that NT-Src42 indeed interacted with CNK (lane 1), and this independently of a coexpressed activated RTK. Similar results were obtained with the full-length Src42 constructs (data not shown), thereby possibly explaining their ability to collaborate with RASV12. Nonetheless, as expected, SEV expression greatly increased the interaction (lane 2), which suggested that binding is normally signal-dependent (see below). Using this simple binding assay, we next verified the relevance of the Y1163 region in this interaction. Remarkably, we found that not only the Y1163 residue is critical for binding (lanes 11 and 12), but also the four missense mutations found in this region affected the association (lanes 3–10). Therefore, these results confirm the relevance of this area for Src42 binding and provide a common molecular explanation for the various loss-of-function mutations found in this area. Although it remains to be verified, the correlation between Src42 binding to CNK and its positive effect on the pathway is consistent with the type of mutations found in the two putative Src42 gain-of-function alleles.
Figure 8.
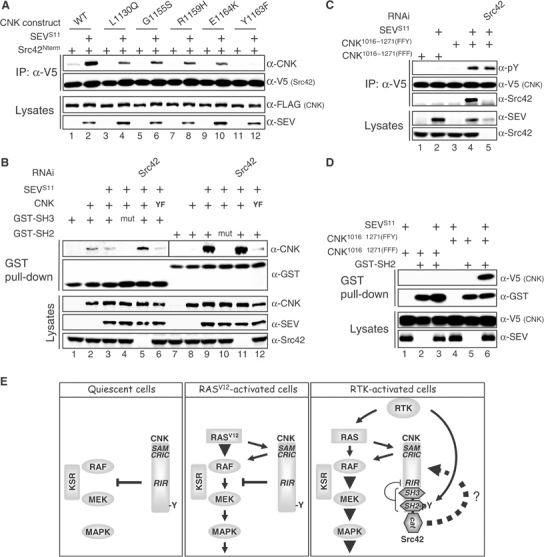
Characterization of the Src42 binding function on CNK. (A) S2 cells were transfected with the indicated FL-CNK variants (0.55–0.8 μg) and the NT-Src42 construct (0.09–0.25 μg) ±SEVS11 (0.6 μg). The CNK variants were expressed for a total of 36 h under the control of the constitutive cnk promoter, whereas NT-Src42 and SEVS11 expressions were induced 16 and 3 h, respectively, prior to harvesting the cells. (B) S2 cells were first incubated ±the Src42 dsRNA (15 μg/ml) 1 day prior to transfection and then transfected with the indicated combinations of CNK (0.5 μg), SEVS11 (0.6 μg) or GST constructs (0.1–0.3 μg). CNK and SEVS11 expression time was as in (A), whereas GST constructs were expressed for 16 h prior to cell lysis. GST-fused proteins were collected using glutathione-Sepharose beads. YF denotes the CNKY1163F variant. Interestingly, depletion of endogenous Src42 slightly increased GST-SH3 binding to CNK (lane 5). (C) The short CNK mutant variants (0.35–0.8 μg) were cotransfected ±SEVS11 (0.5 μg) as indicated and expressed for 16 h prior to cell lysis. RNAi treatment was as in (B). (D) Transfections, including the GST-SH2 construct (0.15–0.8 μg), were conducted as in (C). (E) Model depicting the integration of RAS and Src42 signals by CNK. See text for details.
The ability of full-length or NT-Src42 variants to bind CNK without SEV expression was intriguing as it depended on the Y1163 residue (Figure 8B, compare lanes 1 and 11, and data not shown) as well as on the SH3 and SH2 domains (data not shown). Moreover, it occurred independently of endogenous Src42 (data not shown). These observations suggested that both domains are engaged in binding CNK and that either the Y1163 residue is phosphorylated at an undetectable level or binding of the SH3 domain somehow stabilizes the SH2 domain to its unphosphorylated binding site. To eliminate a possible interdependency between the two domains with respect to CNK binding, we separately fused them to GST and directly assessed their respective binding ability. As shown in Figure 8B, the SH3 domain associated with CNK independently of SEV (compare lanes 2 and 3) and of the Y1163 residue (lane 6). In sharp contrast, the SH2 domain associated with CNK in a strict SEV-dependent manner (compare lanes 8 and 9) that required the integrity of the Y1163 residue (lane 12). Therefore, given that the SH2 domain cannot bind CNK when tested alone in the absence of SEV, but is involved when the SH3 domain is present, it suggested that binding of the SH3 domain to CNK stabilizes the SH2 domain to its binding site thereby allowing it to participate in CNK binding in the absence of Y1163 phosphorylation. This phenomenon, possibly magnified by the artificial setup used to test the constructs, might explain why the collaboration between Src42 and RASV12 depends on the SH3 and SH2 domains of Src42 as well as on the Y1163 residue of CNK.
Src42 does not phosphorylate the Y1163 residue of CNK
The strong SEV dependency of Src42 SH2 domain binding to CNK suggests that in normal conditions, binding of this domain requires the phosphorylation of at least one tyrosine residue in CNK. Because we found that endogenous Src42 is mediating SEV-induced CNK tyrosine phosphorylation, it is conceivable that upon activation by SEV, Src42 phosphorylates its own binding site on CNK thereby inducing normal binding. However, this would be inconsistent with the functional data that suggest that Src42 kinase activity is not involved in the regulation of MAPK activation through CNK. To verify this, we eliminated endogenous Src42 by RNAi and tested the impact on SEV-dependent GST-SH2 domain binding to CNK. As shown in Figure 8B (compare lanes 9 and 11), Src42 depletion did not reduce the interaction. Identical results were obtained with NT-Src42 (data not shown). Therefore, in agreement with our functional results, these findings indicate that the kinase activity of Src42 is not involved in generating a high-affinity binding site on CNK for its SH2 domain.
The binding data presented thus far strongly suggest that the Y1163 residue is part of an Src42 SH2 domain binding site. Furthermore, this residue is probably specifically phosphorylated in response to SEV expression, but this event should not be mediated by Src42. To monitor specifically the phosphorylation status of the Y1163 residue, we generated a short V5-tagged CNK construct (positions 1016–1271) that encompassed the Y1163 region and included only three tyrosine residues (Y1085, Y1136 and Y1163; Figure 2). We tested two versions of it: one had the three tyrosine residues changed to phenylalanine (FFF), while the other (FFY) only kept intact the Y1163 residue. As shown in Figure 8C, we found that the FFY protein, but not the FFF variant, was tyrosine phosphorylated upon SEVS11 expression and associated with endogenous Src42, therefore demonstrating that the Y1163 residue is genuinely phosphorylated upon SEV expression and that this event is critical for endogenous Src42 association. Consistent with the results shown above, depletion of endogenous Src42 did not affect the tyrosine phosphorylation of the FFY protein, thus providing strong evidence that a tyrosine kinase other than Src42 is phosphorylating the Y1163 residue. Finally, to show that the SH2 domain of Src42 is most likely interacting with the phosphorylated Y1163 residue, we tested the ability of the GST-SH2 domain protein to associate with the short CNK variants in a SEV-dependent manner. As shown in Figure 8D, the SH2 domain specifically associated with the FFY variant and this occurred only upon SEV coexpression (lane 6). Therefore, these results provide compelling evidence that the Y1163 residue is indeed phosphorylated in a SEV-dependent but Src42-independent manner, and serves as a binding site for the SH2 domain of Src42. Given that Src42 is apparently not phosphorylating the Y1163 residue, it remains possible that the receptor itself is performing this event and that inappropriate in vitro enzymatic condition explains our failure to detect it. Alternatively, another tyrosine kinase could be involved.
Concluding remarks
In this study, we showed that CNK integrates RAS and Src42 signals as a binary input, thereby allowing RAF to send signals to MEK (Figure 8E). The RAS signal is received by the SAM and CRIC domains of CNK, which appears to enhance RAF catalytic function (Douziech et al, 2003), whereas Src42 activity is integrated by the Y1163 region of CNK and seems to relieve the inhibitory effect that the RIR imposes on RAF's ability to phosphorylate MEK (Douziech et al, 2003). Why would RAF activation depend on two distinct but corequired signals emitted by the same RTK? One possibility is that this requirement generates specificity downstream of an RTK. For example, only receptors that activate both RAS and Src42 would lead to activation of the MAPK module within discretely localized CNK complexes. Consequently, the combinatorial use of multifunctional signals might be a means to produce a specific output from generic signals.
Intriguingly, despite the fact that the second Drosophila SFK, Src64, is naturally expressed in S2 cells, it did not act like Src42 in response to SEV, EGFR and InsR activation. Although the reason for this difference is not immediately clear, we found that, unlike Tec29, overexpression of an Src64YF variant is nonetheless capable of associating with CNK and inducing its tyrosine phosphorylation (data not shown). It is thus possible that Src64 fulfills a similar role to Src42, but downstream of other RTKs or in response to other types of stimuli and that difference in their subcellular localization, requirement for specific cofactors or additional regulatory events account for their distinct behavior.
The mechanism by which the binding of Src42 to CNK deactivates the RIR is currently unknown and a number of scenarios can be envisioned. For example, it might induce a conformational change that suppresses the inhibitory effect that the RIR imposes on RAF catalytic activity. Alternatively, it is possible that Src42 binding displaces an inhibitory protein interacting with CNK or facilitates the relocalization of a CNK/RAF complex to a subcellular compartment that is required for RAS-dependent RAF activation. However, we do not believe that this mechanism involves displacing CNK away from RAF as neither SEV expression nor Src42 depletion altered the CNK/RAF interaction (Figure 4B, and data not shown).
Although several questions are left unanswered regarding the Src42/CNK association, collectively, our data suggest a subtle binding mode reminiscent of the mammalian Src/FAK interaction (Thomas et al, 1998). Indeed, it appears that CNK is phosphorylated on the Y1163 residue not by Src42 itself, but either by the receptor or by another kinase (Figure 8E) and that this step generates a high-affinity binding site for the SH2 domain of Src42 thereby triggering its recruitment. This event is presumably not sufficient for a stable association and/or derepression of the RIR, but also requires the binding of the SH3 domain to an unidentified sequence element within CNK. The engagement of the SH3 and SH2 domains of Src42 on CNK would not only relieve the RIR's inhibitory effect, but would also derepress Src42 autoinhibited configuration and possibly orient favorably Src42 to phosphorylate one or a few specific tyrosine residues on CNK (dotted arrow in Figure 8E). This scenario is certainly plausible considering that CNK has a total of 39 tyrosine residues. This would explain why depletion of endogenous Src42 led to a reduction, but not a complete elimination, of SEV-induced CNK tyrosine phosphorylation (Figure 4) or why the Y1163F mutation impaired CNK phosphorylation mediated by Src42Y511F (Supplementary Figure S3) as a disruption of the Src42/CNK association would prevent Src42 from phosphorylating the other sites. Although these Src42-dependent phosphorylated residues do not appear to play a role in activating the MAPK module, their concerted regulation suggests that CNK is coordinating signaling between the MAPK module and at least another pathway.
Materials and methods
Plasmids
Copper-inducible Flag-tagged FL-CNK, NT-CNK and CT-CNK constructs have been described by Therrien et al (1998). Amino-acid positions corresponding to additional CNK constructs are indicated in the text and were generated by standard procedures. CNKY1163F cDNA was introduced in the psE P-element vector to create transgenic lines similar to P[sE-CNK] (Therrien et al, 1998). For genomic rescue experiments, we assembled in pBlueScript an ∼5.5 kb cnk genomic fragment extending 875 bp upstream of the first methionine (corresponds to sequences between cnk ORF and the first upstream gene) and ending immediately after the STOP codon. ADH 3′UTR sequences were introduced at the 3′-end and a 3XFlag tag was inserted at the first ATG. The engineered cnk genomic fragment (pcnk-cnk) was then mutagenized to generate desired point mutations. The genomic constructs were either used for expression in S2 cells or the inserts were moved to a P-element vector.
For Src42 constructs, a PCR-generated cDNA was introduced in a copper-inducible vector. A PYO epitope tag was introduced at the C-terminus and then was mutagenized to produce the different mutants used in this study. N- and C-terminal Src42 constructs correspond to amino-acid positions 2–240 and 224–517, respectively. A V5 epitope was introduced at the N-terminus for these two constructs. SH3m and SH2m variants had a W100K and R157K replacement, respectively. The SH3 and SH2 domains (WT and mutant versions as above) fused to GST encompassed amino-acid positions 59–128 and 120–231, respectively, and were expressed under a copper-inducible promoter.
The SevS11 constructs have been described by Therrien et al (1998), whereas the RASV12 and MAPK constructs have been described by Douziech et al (2003).
Genetics, molecular analysis and histology
Fly culture, crosses and P-element-mediated germline transformations were conducted according to standard procedures. The sE-CNKWT and RASV12 transgenic flies have been described by Therrien et al (1998). At least two independent lines were characterized for each construct (sE-cnkY1163F and cnk genomic rescue constructs). The Src42l(2)k10108 allele has a P-element inserted in the 5′UTR of the Src42 locus, which obliterates expression (data not shown).
Molecular characterization of cnk and Src42 alleles (Therrien et al, 2000) was performed essentially as described (Therrien et al, 1998).
Scanning electron microscopy and sectioning of adult Drosophila eyes were conducted as described by Wassarman et al (2000) and Tomlinson and Ready (1987), respectively. At least three independent eyes were analyzed per genotype.
Cell culture and transfections
Plain and stable S2 cell lines were maintained and transfected as described by Roy et al (2002). Heat-inducible SevS11- and copper-inducible RASV12-expressing stable cell lines have been described previously (Therrien et al, 1998; Roy et al, 2002). Unless specified otherwise, SevS11 expression (in stable cells or following hs-SevS11 transfection) was induced by a 30 min heat treatment at 37°C and cells were harvested 2 h later. For Egfr activation, an Egfr-expressing S2 cell line was stimulated with the supernatant from Spitz-secreting cells (Schweitzer et al, 1995). RASV12 expression or other copper-inducible expression constructs were induced 48 h post-transfection by adding 0.7 mM CuSO4 to the cell medium and cells were harvested 16 h later. The amount of transfected plasmids was always adjusted among related constructs to obtain similar protein levels. RNAi experiments were conducted essentially as described by Douziech et al (2003).
Protein analysis and antibodies
NP-40 cell lysates were prepared and analyzed either directly or following immunoprecipitation as described by Roy et al (2002). Sources for α-CNK, α-SEV, α-RAS1, α-pMAPK, α-FLAG, α-PYO, α-MYC and α-HA antibodies (Ab) have been mentioned by Douziech et al (2003). α-MAPK and α-pY (clone 4G10) Ab were from Chemicon and USB, respectively, whereas the α-V5 and α-GST Ab were from Invitrogen and Oncogene Research Products, respectively. α-Src42 and α-Src64 polyclonal Ab were generously provided by J Dixon, and the α-Tec29 monoclonal Ab (clone Il9) was a kind gift of S Beckendorf. Glutathione-Sepharose beads were from Amersham Biosciences.
For in vitro kinase assays, NP-40 cell lysates were immunoprecipitated using α-CNK or α-PYO (Src42) and immunoprecipitates were washed three times in NP-40 lysis buffer and once in kinase buffer (25 mM Hepes pH 7.2, 150 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, 1 mM DTT and 1 mM Na3VO4). Kinase reactions were initiated by adding 10 μCi of [γ-32P]ATP, 5 μM ATP and 2 μg of acid-treated enolase (Roche) and incubated for 20 min at 30°C.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Acknowledgments
We are grateful to David Wassarman and Caroline Baril for critical reading of the manuscript and Frank Sicheri for helpful discussions. We thank S Beckendorf, J Dixon, S Katzav and B Shilo for providing reagents. We also thank Allan Wong for generating a number of Src42 constructs, Alexandre Viau for assembling the initial pcnk-cnk construct and François Roy for help with the RNAi technique. MT is recipient of a Canadian Research Chair. This work was supported in part by a CIHR and a NCIC grant to MT.
References
- Anselmo AN, Bumeister R, Thomas JM, White MA (2002) Critical contribution of linker proteins to Raf kinase activation. J Biol Chem 277: 5940–5943 [DOI] [PubMed] [Google Scholar]
- Bumeister R, Rosse C, Anselmo A, Camonis J, White MA (2004) CNK2 couples NGF signal propagation to multiple regulatory cascades driving cell differentiation. Curr Biol 14: 439–445 [DOI] [PubMed] [Google Scholar]
- Chong H, Vikis HG, Guan KL (2003) Mechanisms of regulating the Raf kinase family. Cell Signal 15: 463–469 [DOI] [PubMed] [Google Scholar]
- Dhillon AS, Kolch W (2002) Untying the regulation of the Raf-1 kinase. Arch Biochem Biophys 404: 3–9 [DOI] [PubMed] [Google Scholar]
- Douziech M, Roy F, Laberge G, Lefrancois M, Armengod AV, Therrien M (2003) Bimodal regulation of RAF by CNK in Drosophila. EMBO J 22: 5068–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou XS, Chou TB, Melnick MB, Perrimon N (1995) The torso receptor tyrosine kinase can activate Raf in a Ras-independent pathway. Cell 81: 63–71 [DOI] [PubMed] [Google Scholar]
- Hubbard SR (1999) Src autoinhibition: let us count the ways. Nat Struct Biol 6: 711–714 [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Aspenstrom P, Hall A (2004) Human CNK1 acts as a scaffold protein, linking Rho and Ras signal transduction pathways. Mol Cell Biol 24: 1736–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Swedlow JR, Morgan DO, Varmus HE (1995) c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev 9: 1505–1517 [DOI] [PubMed] [Google Scholar]
- Lanigan TM, Liu A, Huang YZ, Mei L, Margolis B, Guan KL (2003) Human homologue of Drosophila CNK interacts with Ras effector proteins Raf and Rlf. FASEB J 17: 2048–2060 [DOI] [PubMed] [Google Scholar]
- Li W, Noll E, Perrimon N (2000) Identification of autosomal regions involved in Drosophila Raf function. Genetics 156: 763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Li Y (1999) Drosophila Src42A is a negative regulator of RTK signaling. Dev Biol 208: 233–243 [DOI] [PubMed] [Google Scholar]
- Rabizadeh S, Xavier RJ, Ishiguro K, Bernabeortiz J, Lopez-Ilasaca M, Khokhlatchev A, Mollahan P, Pfeifer GP, Avruch J, Seed B (2004) The scaffold protein CNK1 interacts with the tumor suppressor RASSF1A and augments RASSF1A-induced cell death. J Biol Chem 279: 29247–29254 [DOI] [PubMed] [Google Scholar]
- Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M (2002) KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev 16: 427–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Broome MA, Hunter T (1997) Fibronectin-stimulated signaling from a focal adhesion kinase–c-Src complex: involvement of the Grb2, p130cas, and Nck adaptor proteins. Mol Cell Biol 17: 1702–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer R, Shaharabany M, Seger R, Shilo BZ (1995) Secreted spitz triggers the DER signaling pathway and is a limiting component in embryonic ventral ectoderm determination. Genes Dev 9: 1518–1529 [DOI] [PubMed] [Google Scholar]
- Simon MA, Drees B, Kornberg T, Bishop JM (1985) The nucleotide sequence and the tissue-specific expression of Drosophila c-src. Cell 42: 831–840 [DOI] [PubMed] [Google Scholar]
- Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M (2001) The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. BioEssays 23: 436–446 [DOI] [PubMed] [Google Scholar]
- Takahashi F, Endo S, Kojima T, Saigo K (1996) Regulation of cell–cell contacts in developing Drosophila eyes by Dsrc41, a new, close relative of vertebrate c-src. Genes Dev 10: 1645–1656 [DOI] [PubMed] [Google Scholar]
- Therrien M, Morrison DK, Wong AM, Rubin GM (2000) A genetic screen for modifiers of a kinase suppressor of Ras-dependent rough eye phenotype in Drosophila. Genetics 156: 1231–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Therrien M, Wong AM, Rubin GM (1998) CNK, a RAF-binding multidomain protein required for RAS signaling. Cell 95: 343–353 [DOI] [PubMed] [Google Scholar]
- Thomas JW, Ellis B, Boerner RJ, Knight WB, White GC II, Schaller MD (1998) SH2- and SH3-mediated interactions between focal adhesion kinase and Src. J Biol Chem 273: 577–583 [DOI] [PubMed] [Google Scholar]
- Tomlinson A, Ready DF (1987) Neuronal differentiation in the Drosophila ommatidium. Dev Biol 120: 366–376 [DOI] [PubMed] [Google Scholar]
- Wassarman DA, Aoyagi N, Pile LA, Schlag EM (2000) TAF250 is required for multiple developmental events in Drosophila. Proc Natl Acad Sci USA 97: 1154–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL (1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev 79: 143–180 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1