

Abstract
Several recent studies have indicated that members of the phylum Planctomycetes are abundantly present at the brine-seawater interface (BSI) above multiple brine pools in the Red Sea. Planctomycetes include bacteria capable of anaerobic ammonium oxidation (anammox). Here, we investigated the possibility of anammox at BSI sites using metagenomic shotgun sequencing of DNA obtained from the BSI above the Discovery Deep brine pool. Analysis of sequencing reads matching the 16S rRNA and hzsA genes confirmed presence of anammox bacteria of the genus Scalindua. Phylogenetic analysis of the 16S rRNA gene indicated that this Scalindua sp. belongs to a distinct group, separate from the anammox bacteria in the seawater column, that contains mostly sequences retrieved from high-salt environments. Using coverage- and composition-based binning, we extracted and assembled the draft genome of the dominant anammox bacterium. Comparative genomic analysis indicated that this Scalindua species uses compatible solutes for osmoadaptation, in contrast to other marine anammox bacteria that likely use a salt-in strategy. We propose the name Candidatus Scalindua rubra for this novel species, alluding to its discovery in the Red Sea.
Electronic supplementary material
The online version of this article (doi:10.1007/s00248-017-0929-7) contains supplementary material, which is available to authorized users.
Keywords: Scalindua, Anammox, Red Sea, Genome binning, Metagenomics, Salt adaptation
Over 25 brine pools have been discovered along the rift through the middle of the Red Sea. These brine pools are characterized by anoxic, salty water, and in some cases geothermal activity [1]. The high salinity of the brine pools prevents mixing with the overlying seawater creating a brine-seawater interface (BSI) featuring steep salt and, in the case of hot brines, temperature gradients. Several studies using 16S rRNA gene amplicon community profiling and shotgun metagenomics have recently revealed the abundant presence of Planctomycetes (5–35%) in the BSI above the Discovery Deep, Atlantis II Deep, and Kebrit Deep brine pools [2–4]. As these are low-oxygen environments, detection of Planctomycetes likely indicates the presence of anammox bacteria. Furthermore, recent studies have shown the presence of ammonia-oxidizing Archaea and nitrite-oxidizing Bacteria in the Atlantis II Deep BSI, indicating an active nitrogen cycle in these systems [5, 6]. To further investigate the presence and nature of anammox bacteria in the Red Sea BSI, we employed genome-resolved shotgun metagenomics of the BSI above the Discovery Deep, where 16S rRNA gene amplicon community profiling indicated that Planctomycetes were more abundant than in other brine pools [2].
Total microbial community DNA (sample DIS-BWI, see [2] for sampling and DNA extraction) was prepared for IonTorrent sequencing as previously described [7]. The resulting library was used for two sequencing runs, resulting in a total of 10.1 million single-end reads. Reads were trimmed on quality (quality limit = 0.05) and length (>100 bp) using CLCgenomics workbench (v8.0.3, CLCbio, Arhus, Denmark). The presence of anammox was investigated by reconstructing full-length sequences of the 16S rRNA and hydrazine synthase alpha (hzsA) genes by mapping and assembly using the CLCgenomics workbench, as described previously [8]. Two 16S rRNA genes matching the Scalindua clade, with coverage 52× and 14×, and two hzsA sequences, with coverage 37× and 16×, could be reconstructed. The 16S sequence obtained from the former, dominant Scalindua species (5.7% of all 16S rRNA gene reads in our dataset), hereafter referred to as Candidatus Scalindua rubra, is only 94% identical to Candidatus Scalindua brodae and clusters with sequences obtained from the Atlantis II Deep BSI, the brine adjacent to the Discovery Deep [4] (Fig. 1a). The latter, low abundant Scalindua species (1.3% of all rRNA gene 16S reads) clusters with sequences from the Arabian Sea oxygen minimum zone and other sequences obtained from the Atlantis II deep (Fig. 1a). Previously sequenced Ca. S. brodae [9] and Candidatus Scalindua profunda [10] formed a third cluster that also includes most sequences obtained from the Eastern Tropical South Pacific oxygen minimum zone (Fig. 1a). Phylogenetic analysis of the hzsA genes corroborates that Ca. S. rubra is distant from Ca. S. profunda and Ca. S. brodae (Fig. 1b). Interestingly, the partial sequences 16S rRNA and hzsA sequences obtained by Borin et al. [11] from the chemocline above Bannock brine in the Mediterranean, cluster with Ca. S. brodae and Ca. S. profunda, rather than with the sequences obtained from the Atlantis II Deep and Discovery Deep BSI.
Fig. 1.
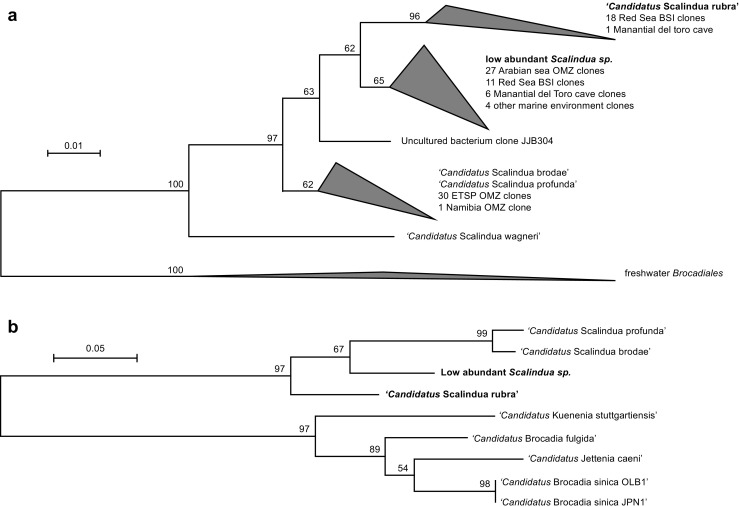
Maximum likelihood trees of anammox 16S rRNA and hzsA genes. a Maximum likelihood tree of 109 near full-length Brocadiales 16S rRNA genes matching >90% of the length the Ca. S. rubra sequence, originating from enrichment cultures, draft genomes, and clone libraries of marine environments. b Maximum likelihood tree of all available full-length hzsA gene sequences obtained from draft genomes. Sequences obtained in this study are indicated in bold. Trees were constructed using MEGA5 [36], bootstrapped with 1000 replicates, and visualized using the interactive tree of life (iTOL) v3 webserver [37]. Wedge height was scaled proportional to number of sequences. OMZ oxygen minimum zone, BSI brine-seawater interface, ETSP Eastern Tropical South Pacific
To obtain a draft genome of Ca. S. rubra, we assembled the metagenome de novo using the CLCgenomics workbench with word size 31 and bubble size 5000. Contigs were assigned to Ca. S. rubra using emergent self-organizing maps [12, 13], coverage, and GC content [14]. Scripts used for binning are available at www.github.com/dspeth. The resulting 1020 Ca. S. rubra contigs were used for iterative reassembly using SPAdes (version 3.5.0) [15] and Bowtie2 [16], resulting in 443 contigs assigned to Ca. S. rubra (Table 1). Contigs were error corrected to account for persistent IonTorrent-specific errors as described previously [7] and annotated using Prokka (version 1.10) [17] using a custom database containing the six Brocadiales draft genomes in Genbank [7, 9, 18–21]. Coverage of the contigs representing the low-abundance Scalindua species was too low (∼15×) to enable extraction of a good quality draft genome of this organism.
Table 1.
Metrics of the available Scalindua spp. draft genomes
| Species | Genome size (Mbp) | GC content | Completeness (%) | Contamination (%) | # of contigs | Reference |
|---|---|---|---|---|---|---|
| Candidatus Scalindua profunda | 5.14 | 39.1 | 95 | 3 | 1580 | [10] |
| Candidatus Scalindua brodae | 4.08 | 39.6 | 92 | 2.3 | 282 | [9] |
| Candidatus Scalindua rubra | 5.19 | 37.3 | 92 | 5.1 | 443 | This study |
Completeness and contamination of the Scalindua spp. draft genomes were estimated using checkM [35]
The Ca. S. rubra draft genome encoded the genes required for hydrazine metabolism, hydrazine synthase [22] (SCARUB_01028–SCARUB_01030), and hydrazine dehydrogenase [23] (SCARUB_00654). The genes encoding hydrazine synthase subunits B and C are not fused in Ca. S. rubra, suggesting that the fusion of these genes in Ca. S. profunda and Ca. S. brodae is a recent event. Like the other Scalindua species, Ca. S. rubra encodes a heme-cd 1 type nitrite reductase (nirS) (SCARUB_03231). In contrast to Ca. S. profunda, neither Ca. S. brodae nor Ca. S. rubra encode a cyanase. Another interesting feature in the Ca. S. rubra genome is the apparent capability to synthesize gas vesicles, as 11 gas vesicle synthesis proteins are present. Although gas vesicles are often regulated by light intensity, gas vesicle formation is induced by high salinity in halophilic Archaeon Haloferax mediterranei [24]. It is possible that Ca. S. rubra uses gas vesicles to stabilize its position within the BSI and prevent osmotic and/or heat shock as a result of the steep gradients in the BSI. The cellular location of gas vesicles in the already complicated cell architecture of an anammox bacterium is an interesting topic for further investigation.
To assess further adaptations to life in the BSI we searched the draft genome of Ca. S. rubra for mechanisms of osmoadaptation. Based on the recent work of Ngugi and colleagues [5], we used protein isoelectric point (IEP) distributions as indicator for a charged cytoplasm resulting from a “salt-in” osmoadaptation strategy [25]. We calculated the IEP of all predicted proteins in the eight available genomes of anammox bacteria using the “iep” script from the EMBOSS package (v6.5.7) [26]. Surprisingly, the median protein IEP of Ca. S. rubra is more basic than the median protein IEP of Ca. S. brodae and Ca. S. profunda and comparable to that of the freshwater species (Fig. 2). The acid-shifted distribution of protein IEP indicates that both previously sequenced Scalindua species have adapted to seawater salinity using a “salt-in” strategy, adding acidic residues to prevent protein denaturation in high-ion concentrations [27]. The observations that, in contrast to freshwater species, salt in the growth medium was required to enrich Ca. S. profunda and that 90% of dry weight of this organism consisted of salt further support this interpretation [28, 29]. As expected, the acid shift is more pronounced if only cytoplasmic proteins are considered, and absent from membrane proteins (Supplemental Figure S1). In contrast, the more basic IEP of Ca. S. rubra proteins suggests that it relies on compatible solutes to cope with the salinity at the BSI. Synthesis of compatible solutes is energetically more costly than coping with salinity using a salt-in strategy [30]. Although some halophiles use a salt-in strategy at higher salinity than observed at the Discovery Deep BSI [31, 32], it is possible that Ca. S. rubra uses compatible solutes to adapt to the range of salt concentrations resulting from the steep salt gradient in the Discovery Deep BSI [2]. In line with this hypothesis, the recently published genomes of ammonia-oxidizing Archaea and nitrite-oxidizing Bacteria from the BSI above the Atlantis II Deep, which is adjacent to the Discovery Deep, also indicate that these organisms employ compatible solutes [5, 6].
Fig. 2.
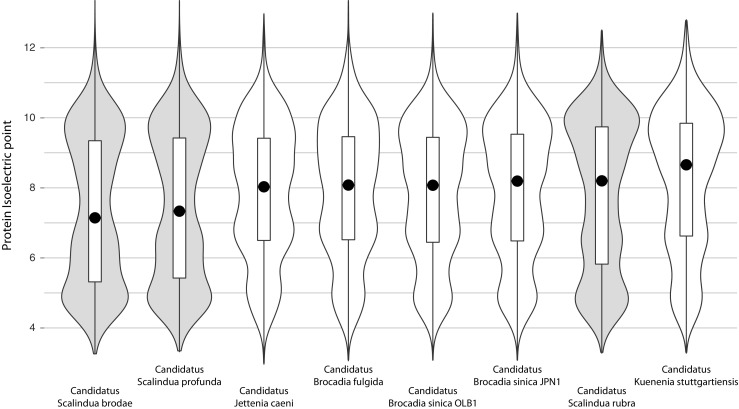
Protein isoelectric point distribution in eight genomes of anammox bacteria. Violin plots indicating the isoelectric point distribution of total protein set of all eight available anammox genomes, ordered from lowest to highest median value. Box plots (white bars) indicate 50% of the values around the median, indicated by a black circle. The three available genomes of Scalindua sp. are indicated by gray shading
We searched the Ca. S. rubra draft genome for proteins required for biosynthesis and transport of common compatible solutes. Many organisms use the amino acids glutamate, glutamine, or proline as compatible solutes [33]. All anammox bacteria can synthesize these amino acids, and thus, it is possible that Ca. S. rubra utilizes any or all three of these amino acids. This could also provide an explanation for the adaptation of freshwater anammox species Ca. K. stuttgartiensis to marine salt concentrations [34]. None of the Scalindua species is capable of synthesizing amino acid-derived compatible solutes glycine-betaine or (hydroxy)ectoine, but all three encode a glycine-betaine transporter. Furthermore, none of the Scalindua genomes encode the potential for biosynthesis of glycerate-derived compatible solutes or mannitol or sorbitol [33]. Conclusive evidence on the presence, and nature, of compatible solutes in Ca. S. rubra will require biomass for experimental verification of the amino acid content.
In conclusion, we have presented the draft genome of a moderately halophilic anammox bacterium, Ca. S. rubra. Our analysis of the adaptations to salt stress in this genome has shed new light on previous results of salt adaptation in anammox bacteria.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(PDF 779 kb)
Acknowledgements
Daan R. Speth was supported by BE-Basic FP 07.002.01. Bas E. Dutilh was supported by the Netherlands Organization for Scientific Research (NWO) Vidi grant 864.14.004. Mike S. M. Jetten was supported by the European Research Council advanced grants 232937 and 339880 and the NWO gravitation SIAM 024002002.
Accession Numbers
The raw sequencing reads described in this paper has been deposited to Genbank/EBI/DDBJ under SRA accession number SRX1894129. The assembled, annotated draft genome has been deposited at DDBJ/ENA/GenBank under the accession MAYW00000000. The version described in this paper is version MAYW01000000.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s00248-017-0929-7) contains supplementary material, which is available to authorized users.
References
- 1.Bischoff JL. Red Sea geothermal brine deposits: their mineralogy, chemistry, and genesis. In: Degens ET, Ross DA, editors. Hot brines and recent heavy metal deposits in the Red Sea. Berlin Heidelberg, Berlin, Heidelberg: Springer; 1969. pp. 368–401. [Google Scholar]
- 2.Bougouffa S, Yang JK, Lee OO, et al. Distinctive microbial community structure in highly stratified deep-sea brine water columns. Appl Environ Microbiol. 2013;79:3425–3437. doi: 10.1128/AEM.00254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdallah RZ, Adel M, Ouf A, et al. Aerobic methanotrophic communities at the Red Sea brine-seawater interface. Front Microbiol. 2014;5:487. doi: 10.3389/fmicb.2014.00487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan Y, Hikmawan T, Antunes A, et al. Diversity of methanogens and sulfate-reducing bacteria in the interfaces of five deep-sea anoxic brines of the Red Sea. Res Microbiol. 2015 doi: 10.1016/j.resmic.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Ngugi DK, Blom J, Alam I, et al. Comparative genomics reveals adaptations of a halotolerant thaumarchaeon in the interfaces of brine pools in the Red Sea. ISME J. 2015;9:396–411. doi: 10.1038/ismej.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ngugi DK, Blom J, Stepanauskas R, Stingl U. Diversification and niche adaptations of Nitrospina-like bacteria in the polyextreme interfaces of Red Sea brines. ISME J. 2016;10:1383–1399. doi: 10.1038/ismej.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Speth DR, In ‘t Zandt MH, Guerrero-Cruz S, et al. Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system. Nat Comms. 2016;7:11172. doi: 10.1038/ncomms11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lüke C, Speth DR, Kox MAR, et al. Metagenomic analysis of nitrogen and methane cycling in the Arabian Sea oxygen minimum zone. Peer J. 2016;4:e1924. doi: 10.7717/peerj.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speth DR, Russ L, Kartal B, et al. Draft genome sequence of anammox bacterium “Candidatus Scalindua brodae,” obtained using differential coverage binning of sequencing data from two reactor enrichments. Genome Announc. 2015 doi: 10.1128/genomeA.01415-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van de Vossenberg J, Woebken D, Maalcke WJ, et al. The metagenome of the marine anammox bacterium “Candidatus Scalindua profunda” illustrates the versatility of this globally important nitrogen cycle bacterium. Environ Microbiol. 2013;15:1275–1289. doi: 10.1111/j.1462-2920.2012.02774.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borin S, Mapelli F, Rolli E, et al. Anammox bacterial populations in deep marine hypersaline gradient systems. Extremophiles. 2013;17:289–299. doi: 10.1007/s00792-013-0516-x. [DOI] [PubMed] [Google Scholar]
- 12.Ultsch A, Mörchen F (2005) ESOM-Maps: tools for clustering, visualization, and classification with Emergent SOM. https://www.uni-marburg.de/fb12/datenbionik/pdf/pubs/2005/ultsch05esom
- 13.Dick GJ, Andersson AF, Baker BJ, et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009;10:R85. doi: 10.1186/gb-2009-10-8-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tyson GW, Chapman J, Hugenholtz P, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 15.Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 18.Strous M, Pelletier E, Mangenot S, et al. Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature. 2006;440:790–794. doi: 10.1038/nature04647. [DOI] [PubMed] [Google Scholar]
- 19.Hira D, Toh H, Migita CT, et al. Anammox organism KSU-1 expresses a NirK-type copper-containing nitrite reductase instead of a NirS-type with cytochrome cd1. FEBS Lett. 2012;586:1658–1663. doi: 10.1016/j.febslet.2012.04.041. [DOI] [PubMed] [Google Scholar]
- 20.Ferousi C, Speth DR, Reimann J, et al. Identification of the type II cytochrome c maturation pathway in anammox bacteria by comparative genomics. BMC Microbiol. 2013;13:265. doi: 10.1186/1471-2180-13-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oshiki M, Shinyako-Hata K, Satoh H, Okabe S. Draft genome sequence of an anaerobic ammonium-oxidizing bacterium, “Candidatus Brocadia sinica”. Genome Announc. 2015 doi: 10.1128/genomeA.00267-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dietl A, Ferousi C, Maalcke WJ, et al. The inner workings of the hydrazine synthase multiprotein complex. Nature. 2015;527:394–397. doi: 10.1038/nature15517. [DOI] [PubMed] [Google Scholar]
- 23.Maalcke WJ, Reimann J, de Vries S et al (2016) Characterization of anammox hydrazine dehydrogenase, a key N 2-producing enzyme in the global nitrogen cycle. J Biol Chem jbc.M116.735530. doi: 10.1074/jbc.M116.735530 [DOI] [PMC free article] [PubMed]
- 24.Englert C, Horne M, Pfeifer F. Expression of the major gas vesicle protein gene in the halophilic archaebacterium Haloferax mediterranei is modulated by salt. Mol Gen Genet. 1990;222:225–232. doi: 10.1007/BF00633822. [DOI] [PubMed] [Google Scholar]
- 25.Sleator RD, Hill C. Bacterial osmoadaptation: the role of osmolytes in bacterial stress and virulence. FEMS Microbiol Rev. 2002;26:49–71. doi: 10.1111/j.1574-6976.2002.tb00598.x. [DOI] [PubMed] [Google Scholar]
- 26.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 27.Zaccai G, Cendrin F, Haik Y, Borochov N, Eisenberg H. Stabilization of halophilic malate dehydrogenase. J Mol Biol. 1989;208:491–500. doi: 10.1016/0022-2836(89)90512-3. [DOI] [PubMed] [Google Scholar]
- 28.van de Vossenberg J, Rattray JE, Geerts W, et al. Enrichment and characterization of marine anammox bacteria associated with global nitrogen gas production. Environ Microbiol. 2008;10:3120–3129. doi: 10.1111/j.1462-2920.2008.01643.x. [DOI] [PubMed] [Google Scholar]
- 29.Rattray JE, van de Vossenberg J, Hopmans EC, et al. Ladderane lipid distribution in four genera of anammox bacteria. Arch Microbiol. 2008;190:51–66. doi: 10.1007/s00203-008-0364-8. [DOI] [PubMed] [Google Scholar]
- 30.Oren A. Bioenergetic aspects of halophilism. Microbiol Mol Biol Rev. 1999;63:334–348. doi: 10.1128/mmbr.63.2.334-348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowers KJ, Mesbah NM, Wiegel J. Biodiversity of poly-extremophilic bacteria: does combining the extremes of high salt, alkaline pH and elevated temperature approach a physico-chemical boundary for life? Saline Syst. 2009;5:9. doi: 10.1186/1746-1448-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowers KJ, Wiegel J. Temperature and pH optima of extremely halophilic archaea: a mini-review. Extremophiles. 2011;15:119–128. doi: 10.1007/s00792-010-0347-y. [DOI] [PubMed] [Google Scholar]
- 33.Empadinhas N, da Costa M. Osmoadaptation mechanisms in prokaryotes: distribution of compatible solutes. Int Microbiol. 2008;11:151–161. [PubMed] [Google Scholar]
- 34.Kartal B, Koleva M, Arsov R, et al. Adaptation of a freshwater anammox population to high salinity wastewater. J Biotechnol. 2006;126:546–553. doi: 10.1016/j.jbiotec.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 35.Parks DH, Imelfort M, Skennerton CT, et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura K, Peterson D, Peterson N, et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 779 kb)