

Abstract
Purpose
A phase Ib study of dasatinib plus ipilimumab in patients with gastrointestinal stromal tumor (GIST) and other sarcomas was performed on the basis of preclinical data demonstrating that combined KIT and CTLA-4 blockade is synergistic.
Experimental Design
A standard 3 + 3 design was used to evaluate the safety, efficacy, and immune correlates of treatment. Dose escalation cohorts received ipilimumab 10 or 3 mg/kg every 3 weeks, followed by maintenance every 12 weeks with escalating doses of dasatinib (70 mg daily, 100 mg daily, or 70 mg twice daily). Response was assessed by RECIST 1.1, Choi, and immune-related RECIST criteria (irRC).
Results
A total of 28 patients (17 male) were enrolled. Histologic subtypes included GISTs (n = 20) and other sarcomas (n = 8.) Dasatinib 70 mg/day with ipilimumab 10 mg/kg or dasatinib 140 mg/day with ipilimumab 3 mg/kg can be safely administered. Dose-limiting toxicities included grade 3 gastric hemorrhage and anemia. No partial or complete responses were noted by RECIST or irRC. There were 7 of 13 partial responses in the GIST patients by Choi criteria, and 3 of 13 patients each had stable and progressive disease, respectively.
Conclusions
Dasatinib and ipilimumab can be safely administered to GIST and sarcoma patients. However, dasatinib was not synergistic with ipilimumab, as there was limited clinical efficacy with the combination. This limited cohort provides prospective data that indoleamine-2,3-dioxygenase (IDO) suppression may potentially correlate with antitumor efficacy in GIST. Given the small cohort, it is only hypothesis generating and additional data would be required. In the era of more modern and effective checkpoint inhibitors, next steps could be consideration of tyrosine kinase inhibitors or IDO inhibitors in combination with anti-PD-1 therapy.
Introduction
Gastrointestinal stromal tumors (GIST) are mesenchymal tumors thought to rise from the interstitial cells of Cajal (1–3). There are approximately 4,000 to 6,000 cases diagnosed in the United States each year (4). Most GISTs contain activating mutations of KIT or PDGFR-α, providing a role for tyrosine kinase inhibitors (TKI), such as imatinib, sunitinib, and regorafenib (5, 6). Imatinib achieves disease control in most patients, with a median progression-free survival (PFS) of about 18 months (7, 8). In addition, approximately 20% of patients with metastatic GIST are alive more than 10 years after initiation of imatinib (7, 8). However, patients who progress on imatinib only have a median PFS of 4 to 6 months on second- and third-line therapy (9, 10). Ultimately, most patients develop resistance to all available TKIs.
It has been demonstrated that TKIs can have “off-target” effects on the immune system, which contribute to the antitumor efficacy of these compounds. TKIs such as sunitinib, imatinib, or dasatinib have been shown to have both suppressive and stimulating effects on multiple subpopulations of immune cells, such as CD4+ and CD8+ T cells (11, 12), natural killer (NK) cells (13), and dendritic cells (14). Imatinib, specifically, can trigger NK cells in mice and humans, promoting its antiproliferative effects (15). These effects have been associated with improved preclinical (12) and clinical outcomes with these agents (13, 16).
It was demonstrated in a genetically engineered KIT-mutant GIST mouse model that the immune system played a major role in the antitumor effects of imatinib (17). Imatinib induced tumor regression correlated with an increase in intratumoral CD8+ cells, a decrease in regulatory T cells, and a decrease in indoleamine-2,3-dioxygenase (IDO) levels, an inhibitor of T-cell proliferation (17). IDO is a heme-containing enzyme that catalyzes the oxidative breakdown of the essential amino acid tryptophan, via the kynurenine pathway (18). IDO has been shown to inhibit T-cell proliferation and blockade of cell-cycle progression by tryptophan depletion (19). In this murine model, imatinib-mediated tumor shrinkage was abrogated by CD8+ depletion. Patients with GIST who develop resistance to TKIs have a decrease in the CD8+-to-T regulatory ratio as well as an increase in IDO expression.
These data suggest that immunotherapy is a logical choice to potentiate responses to TKIs in patients with GIST. In metastatic melanoma, blockade of CTLA-4 with ipilimumab has demonstrated an overall response rate of 10% to 15% and prolonged survival in up to 20% of patients beyond 3 years (20). Using the genetically engineered mouse model of GIST, it was also observed that treatment with either the KIT-targeted TKIs imatinib or dasatinib (Supplementary Fig. S1) for 7 days alone or with chronic anti-CTLA-4 alone decreased tumor volume transiently, followed by invariable regrowth. Mice treated with a KIT-targeted TKI for 7 days plus chronic anti-CTLA-4 blockade, however, showed augmentation and durability of the initial response. The murine GIST model demonstrated that treatment with CTLA-4 blockade synergized with KIT inhibition to prevent regrowth of tumor (17).
These data provided the scientific rationale to launch this phase I study of dasatinib plus ipilimumab in patients with advanced GIST and other sarcomas. As activity has been observed with dasatinib in other sarcoma subtypes, most commonly with high-grade undifferentiated pleomorphic sarcoma, other sarcoma subtypes were included in the dose escalation phase of the trial (21). As the hypothesis driving this study is based upon GIST, we enriched the dose escalation phase of the trial with GIST patients and exclusively enrolled patients with advanced GIST in the expansion cohort.
Materials and Methods
Study design and treatment
NCT01643278 was a single-center study at Memorial Sloan Kettering Cancer Center (New York, NY) to determine the safety and toxicity and identify the dose-limiting toxicity (DLT) and the MTDs of the combination of dasatinib and ipilimumab. The primary objective of this trial was to evaluate the safety profile and identify the MTD of dasatinib + ipilimumab. The secondary objectives included evaluation of the response rate by RECIST version 1.1 (22), immune-related response criteria (irRC; ref. 23), and Choi criteria (24). The exploratory objectives included immunologic analyses of peripheral blood mononuclear cells (PBMC) and evaluation of absolute lymphocyte counts and cytokines. Analyses of tumor tissue included evaluation of IDO expression by Western blotting.
Eligible patients included those with advanced/unresectable GIST who were refractory or intolerant of imatinib and sunitinib. For the dose escalation, metastatic sarcoma patients who have received at least 1 prior therapy were also eligible. All patients needed adequate performance status (ECOG greater than or equal to 1; Karnofsky performance status greater than or equal to 70%) and measurable disease per RECIST. Those with brain metastases, autoimmune diseases, and any therapy within 4 weeks were excluded.
The design of this phase Ib clinical trial included ipilimumab at 10 mg/kg, with escalating doses of dasatinib starting at 70 mg daily up to 140 mg daily. A standard 3 + 3 dose escalation was used. Once the MTD of dasatinib in combination with ipilimumab 10 mg/kg was determined, a dose expansion cohort enrolled 6 additional patients. Ipilimumab was ultimately FDA approved at 3 mg/kg for metastatic melanoma; thus, we conducted a second dose escalation study of dasatinib in combination with ipilimumab 3 mg/kg.
There was a 1-week dasatinib lead in, followed by initiation of ipilimumab. Ipilimumab was administered four times every 3 weeks until week 12. In the absence of DLT or overt clinical progression, dasatinib was administered continuously and ipilimumab was given every 12 weeks thereafter.
DLT
Toxicity was graded according to NCI Common Terminology Criteria for Adverse Events (CTCAE v 4.0). Any adverse event (AE) that met criteria as defined in the protocol that occurred prior to the third dose of ipilimumab (week 7) was considered a DLT for the purposes of dose escalation to the next dose level. The DLT period was established to allow for the potentially delayed toxicities observed with immunologic checkpoint blockade. Patients that did not complete the 7-week DLT period were replaceable and considered inevaluable for the purpose of safety and efficacy. The highest tolerated dose was defined as the maximum dose of ipilimumab administered in combination with dasatinib at which no more than 1 of 6 patients experienced a DLT.
Evaluation of clinical activity
Baseline imaging was obtained within 4 weeks of starting the trial. Tumor assessments continued at weeks 12, 18, 24, and every 12 weeks thereafter. Response was assessed using RECIST 1.1, the irRC, and Choi criteria. Response and progression as defined by modified irRC was used for all clinical decision making in this trial. Patients were allowed to remain on the study until the time of confirmed progressive disease as defined by the modified irRC (≥25% increase in tumor burden) or the development of unacceptable toxicity.
Correlative studies
All patients had serum and PBMCs collected at baseline, after 1 week of therapy with dasatinib alone, and then prior to each dose of ipilimumab. Patients enrolled at the MTD extension underwent a biopsy prior to initiation of therapy, after the 1 week of dasatinib alone, and after the second induction dose of ipilimumab that was administered concurrently with dasatinib. All patients had tumor KIT mutational status identified using a CLIA-certified assay.
Immunologic analysis in blood
Pharmacodynamic effects were assessed for measures of absolute lymphocyte count (ALC) and serum cytokines. Human plasma cytokines were quantitated using the Meso Scale Discovery (MSD) electrochemiluminescence multiplex immunoassay platform. IFNγ, IL10, IL12p70, IL13, IL1β, IL2, IL4, IL5, IL8, and TNFα were measured using the MSD 10-plex human Th1-Th2 ultrasensitive assay plates, whereas human IL17A and VEGF were measured using custom MSD 2-plex V-Plex human cytokine panel assay plates. Briefly, 50 μL of 2-fold diluted sample was added per well into the MSD assay plates precoated with capture antibodies to the various analytes and allowed to incubate at room temperature with shaking for 2 hours. Plates were then washed 3 times with 150 μL/well of wash buffer, and 25 μL of the appropriate cytokine detection antibody was added to each well for additional 2-hour incubation at room temperature with shaking. After washing plates again 3 times with 150 μL/well wash buffer, 150 μL/well of read buffer was added to each well, and plates were read on the MSD imager. Final cytokine concentrations were interpolated from a standard curve generated using a 4-parameter logistic model fit through the calibrator results using the MSD Discovery Workbench software.
Western blot analysis
Whole protein was extracted from frozen GIST biopsy cores using lysis buffer with complete protease inhibitor cocktail (Roche Diagnostics). Cores were obtained from 6 patients with GIST who consented to tissue analysis under an Institutional Review Board protocol. Protein concentration was determined using the Bradford method, and Western blotting was performed using standard methods. Antibodies were used against total and phosphorylated KIT (Tyr719) and GAPDH (Cell Signaling Technology). Anti-IDO antibody (clone 10.1) was purchased from EMD Millipore.
Results
Patient characteristics
Between July 2012 and October 2015, a total of 28 patients (17 male) were enrolled. The median age was 56 (range, 40–75). Histologic subtypes included GIST (n = 20), high-grade sarcoma (n = 2), leiomyosarcoma (n = 1), epithelioid sarcoma (n = 1), uterine smooth muscle tumor of uncertain malignant potential (n = 1), malignant peripheral nerve sheath tumor (n = 1), chordoma (n = 1), and clear cell sarcoma (n = 1.) The median numbers of prior treatments were 3 (1–8; Table 1A).
Table 1A. Demographics, all patients.
| N = 28 | |
|---|---|
| Gender, M:F | 17:11 |
| Age, median (range) | 56 (40–75) |
| ECOG, median (range) | 0 (0–1) |
| Prior Tx, median (range) | 3 (1–8) |
| Histologies | |
| Gastrointestinal stromal tumor | 20 |
| HG sarcoma, NOS | 2 |
| MPNST | 1 |
| Leiomyosarcoma | 1 |
| Chordoma | 1 |
| Clear cell | 1 |
| Uterine smooth muscle tumor of uncertain malignant potential | 1 |
| Epithelioid sarcoma | 1 |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; F, female; HG sarcoma NOS, high-grade sarcoma not otherwise specified; M, male; MPNST, malignant peripheral nerve sheath tumor; Tx, treatment.
Of the 20 GIST patients enrolled (13 males), the median age was 54 (range, 40–75), and patients were extensively pretreated, with a median of 3 prior treatments (range, 1–8). There were 5 patients who had tumors with mutations in exon 11 or 9, 10 patients with tumors that harbored secondary mutations in exon 13, 14, or 17, 2 patients whose tumors had mutations in PDGFR exon 18 D842V, and 1 patient with an NF1 mutation and SDH deficiency, respectively. Mutational status was unknown in 1 patient (Table 1B).
Among the enrolled 28 patients, 24 were evaluable for DLT. Four patients were considered inevaluable for the primary endpoint of DLT assessment due to rapid disease progression prior to the end of the DLT period.
Dose escalation and MTD
In dose level 1 (70 mg dasatinib and 10 mg/kg ipilimumab), there was one patient that had a grade 3 gastric hemorrhage. The patient was admitted with melena, failure to thrive, and ultimately noted to have progression of disease. In dose level 2 (100 mg dasatinib and 10 mg/kg ipilimumab), there were 2 patients that experienced grade 3 anemia considered possibly related to therapy. Work-up excluded an autoimmune-mediated anemia. As a result, the MTD was established with dasatinib at 70 mg daily and ipilimumab at 10 mg/kg, dose level 1. There was a dose expansion of 6 patients at this dose level, all with GIST.
Because of the concern of inadequate dasatinib exposure as well as FDA approval of ipilimumab at 3 mg/kg, ipilimumab was reduced to 3 mg/kg and the dasatinib was reescalated. In this dose escalation, there were no DLTs identified at dose level 1b (100 mg dasatinib and 3 mg/kg ipilimumab) or dose level 2b (140 mg dasatinib and 3 mg/kg ipilimumab), and the MTD was identified to be dasatinib 140 mg daily with ipilimumab 3 mg/kg (Table 2). There were no further expansions planned at this dose level.
Table 2. DLTs.
| Dose level | Number of patients | Doses | DLTs |
|---|---|---|---|
| 1 | 6 | 70 mg dasatinib and 10 mg/kg ipilimumab | Grade 3 gastric hemorrhage |
| 2 | 6 | 100 mg dasatinib and 10 mg/kg ipilimumab | Grade 3 gastric hemorrhage Grade 3 anemia |
| 1B | 3 | 100 mg dasatinib and 3 mg/kg ipilimumab | None |
| 2B | 3 | 140 mg dasatinib and 3 mg/kg ipilimumab | None |
Safety
Of the 24 evaluable patients, 23 of 24 (96%) experienced treatment-related AEs of any grade. The most common treatment-related AEs were anemia (92%), fatigue (58%), diarrhea (38%), edema (29%), hyperglycemia (29%), elevated ALT/AST (21%), elevated lipase (17%), dyspnea (17%), and lymphopenia (13%; Table 3).
Table 3. All grade toxicity possibly, probably, or definitely related to study therapy by dose level.
| Grade | Dose level 1 (n = 6) | Dose level 2 (n = 6) | Dose expansion (n = 6) | Dose level 1B (n = 3) | Dose level 2B (n = 3) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
||||||
| 70 mg dasatinib, 10 mg/kg ipilimumab | 100 mg dasatinib, 10 mg/kg ipilimumab | 70 mg dasatinib, 10 mg/kg ipilimumab | 100 mg dasatinib, 3 mg/kg ipilimumab | 140 mg dasatinib, 3 mg/kg ipilimumab | ||||||
|
|
|
|
|
|
||||||
| 1/2 | 3/4 | 1/2 | 3/4 | 1/2 | 3/4 | 1/2 | 3/4 | 1/2 | 3/4 | |
| Acute kidney injury | 1 (17%) | |||||||||
| ALT increased | 1 (17%) | 2 (67%) | 2 (67%) | |||||||
| AST increased | 2 (67%) | 3 (100%) | ||||||||
| ALK P increased | 1 (17%) | 2(67%) | ||||||||
| Anemia | 3 (50%) | 3 (50%) | 3 (50%) | 2 (33%) | 5 (83%) | 1 (17%) | 2 (67%) | 3 (100%) | ||
| Anorexia | 1 (17%) | 1 (17%) | ||||||||
| Ascites | 1 (17%) | |||||||||
| Chills | 1 (33%) | |||||||||
| Constipation | 1 (33%) | |||||||||
| Cough | 2 (33%) | |||||||||
| Diarrhea | 2 (33%) | 2 (33%) | 2 (33%) | 1 (17%) | 1 (33%) | 1 (33%) | ||||
| Dry mouth | 1 (17%) | |||||||||
| Dysgeusia | 1 (17%) | |||||||||
| Dyspnea | 1 (17%) | 1 (17%) | 1 (17%) | 1(33%) | 1 (33%) | |||||
| Edema | 1 (17%) | 1 (17%) | 3(50%) | 1 (17%) | 1 (33%) | |||||
| Fatigue | 6 (100%) | 1 (17%) | 4 (67%) | 2 (67%) | 1 (33%) | |||||
| Flatulence | 1 (33%) | |||||||||
| Gastric hemorrhage | 1 (17%) | 1 (17%) | 1 (17%) | |||||||
| Generalized weakness | 1 (17%) | 1 (17%) | 1 (17%) | |||||||
| GERD | 1 (17%) | |||||||||
| Headache | 1 (17%) | 1 (17%) | ||||||||
| Hypoalbuminemia | 1 (17%) | 1 (33%) | 1 (33%) | |||||||
| Hypocalcemia | 1 (17%) | 1 (33%) | ||||||||
| Hypoglycemia | 1 (17%) | |||||||||
| Hyponatremia | 1 (33%) | 2 (67%) | ||||||||
| Hypophosphatemia | 2 (67%) | |||||||||
| Hyperglycemia | 1 (17%) | 3 (100%) | 3 (100%) | |||||||
| Hyperkalemia | 1 (33%) | |||||||||
| Hypernatremia | 1 (33%) | |||||||||
| Infection, anorectal | 1 (17%) | |||||||||
| Intestinal stoma site bleeding | 1 (17%) | |||||||||
| Lipase elevated | 1 (17%) | 1 (17%) | 1 (17%) | 1 (33%) | ||||||
| Lymphopenia | 1 (17%) | 1 (17%) | 1 (17%) | |||||||
| Mucositis | 1 (17%) | 1 (33%) | ||||||||
| Myalgias | 1 (33%) | |||||||||
| Nausea | 1 (17%) | 1 (17%) | 3 (50%) | 2 (33%) | ||||||
| Neutropenia | 1 (33%) | |||||||||
| Pain | 3 (33%) | 1 (17%) | ||||||||
| Paresthesia | 1 (33%) | |||||||||
| Pericardial effusion | 1 (33%) | |||||||||
| Peripheral neuropathy | 2 (33%) | 1 (17%) | ||||||||
| Pleural effusion | 2 (33%) | 1 (17%) | 1 (33%) | |||||||
| Pruritus | 1 (17%) | 1 (17%) | ||||||||
| Rash | 2 (33%) | 2 (33%) | 1 (17%) | 1 (33%) | 1 (33%) | |||||
| Thrombocytopenia | 1 (17%) | 1 (17%) | 1 (17%) | 1 (33%) | 1 (33%) | |||||
| Urinary urgency | 1 (17%) | |||||||||
| Vomiting | 1 (17%) | 2 (33%) | 1 (17%) | |||||||
| Watery eyes | 1 (33%) | |||||||||
| WBC decreased | 2 (33%) | 1 (33%) | 1 (33%) | |||||||
Abbreviations: ALK P, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GERD, gastroesophageal reflux disease; WBC, white blood cells.
Grade 3 AEs included anemia in 6 patients (21%), lymphopenia in 3 patients (13%), and diarrhea, edema, infection, nausea, pericardial effusion, and vomiting each in 1 patient (4%). A patient treated at dose level 2b after about 10 weeks on treatment had a pericardial effusion. She had her pericardial effusion drained, and her dasatinib was resumed at a lower treatment dose. There were no grade 4 AEs (Table 3).
Evaluation of clinical activity
Of the 24 patients evaluable for toxicity, 18 patients (13 with GIST) made it to their first disease assessment and were evaluable for radiographic response. Six patients were taken off the study therapy prior to this first imaging time point due to clinical decline. The median follow-up on these patients was 17.1 months. The median PFS was 2.8 months [95% confidence interval (CI), 2.7–3.0]. The median overall survival (OS) was 13.5 months (95% CI, 11.4–NR; Fig. 1A and B).
Figure 1.
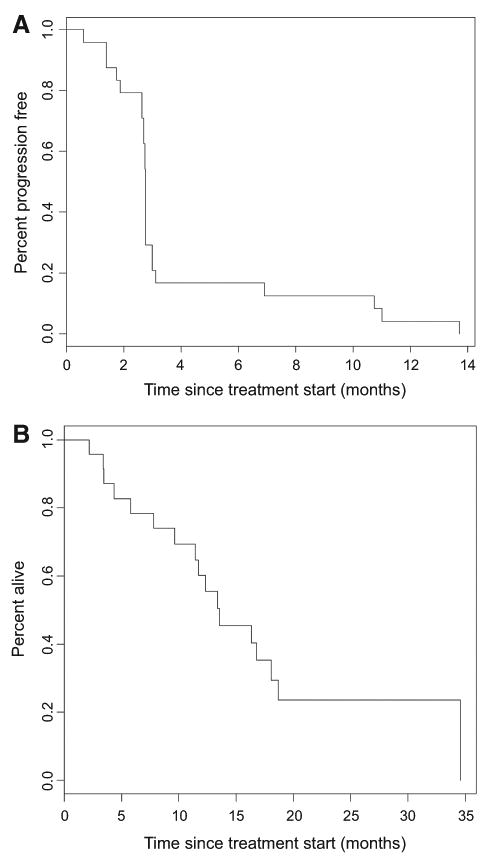
A, Kaplan–Meier curve of PFS. The median follow-up on these 18 patients was 17.1 months. The median PFS was 2.8 months (95% CI, 2.7–3.0). B, Kaplan–Meier curve of OS. The median follow-up on these 18 patients was 17.1 months. The median OS was 13.5 months (95% CI, 11.4–NR).
There were no partial or complete responses noted by RECIST or irRC. In the non-GIST sarcoma patients (n = 5), there was 1 patient with a high-grade sarcoma with rapidly progressive disease before the start of treatment, who achieved stable disease for 47 weeks. Furthermore, 2 patients with clear cell sarcoma and epithelioid sarcoma both had stable disease for 20 and 16 weeks, respectively.
By Choi criteria, 7 of 13 evaluable patients with GIST had partial responses, whereas 3 of 13 patients each had stable and progressive disease, respectively (Table 4). The duration of clinical responses was a median of 82 days with a range of 0–146 days. There were 2 patients who showed evidence of Choi partial responses but had RECIST and clinical progression and therefore were taken off study.
Table 4. Clinical activity.
| Parameter (n = 24) | Dose escalation (n = 8/12 evaluable) | Dose expansion (n = 4/6 evaluable) | “B” dose escalation (n = 6/6 evaluable) |
|---|---|---|---|
| Best ORR by RECIST | |||
| CR/PR | 0 | 0 | 0 |
| SD | 3 | 2 | 4 |
| PD | 5 | 2 | 2 |
| Best ORR by irRC | |||
| CR/PR | 0 | 0 | 0 |
| SD | 6 | 2 | 3 |
| PD | 2 | 2 | 3 |
| GIST patients only (n = 17) | n = 5/7 evaluable | 4/6 evaluable | 4/4 evaluable |
| Best ORR by Choi | |||
| CR/PR | 3 | 1 | 3 |
| SD | 1 | 2 | 0 |
| PD | 1 | 1 | 1 |
Abbreviations: ORR, overall response rate; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease.
In the dose escalation, the median treatment time was 5.0 months, ranging from 1.1 to 16.5 months. In the expansion cohort, the median treatment time was 4.2 months (range, 0.6–11 months). There is one patient that continues to receive treatment at the second dose escalation cohort. As of the last date of follow-up for this patient, the median treatment time was 5.7 months (range, 2.7–13.9 months).
Correlative data
A panel of cytokines was analyzed in 8 patients in cohorts utilizing ipilimumab 10 mg/kg, including IL2, IL4, IL5, IL8, IL10, IL12, IL13, IL17, IFNγ, TNFα, and VEGF. Although most cytokines were not detectable, the two consistently detectable in GIST samples were VEGF and IL8. There was a nonsignificant trend toward decrease in plasma VEGF levels with dasatinib and dasatinib plus ipilimumab (medians at baseline, week 1, week 7 were 67, 30, and 32 pg/mL, respectively; paired t test for baseline to week 1, P = 0.33; baseline to week 7, P = 0.23). Plasma IL8 levels were widely variable at baseline in GIST (range, 10–5,398 pg/mL). Median IL8 did not change significantly with dasatinib or dasatinib + ipilimumab therapy (51 vs. 39 vs. 59 pg/mL). (Fig. 2A and B)
Figure 2.
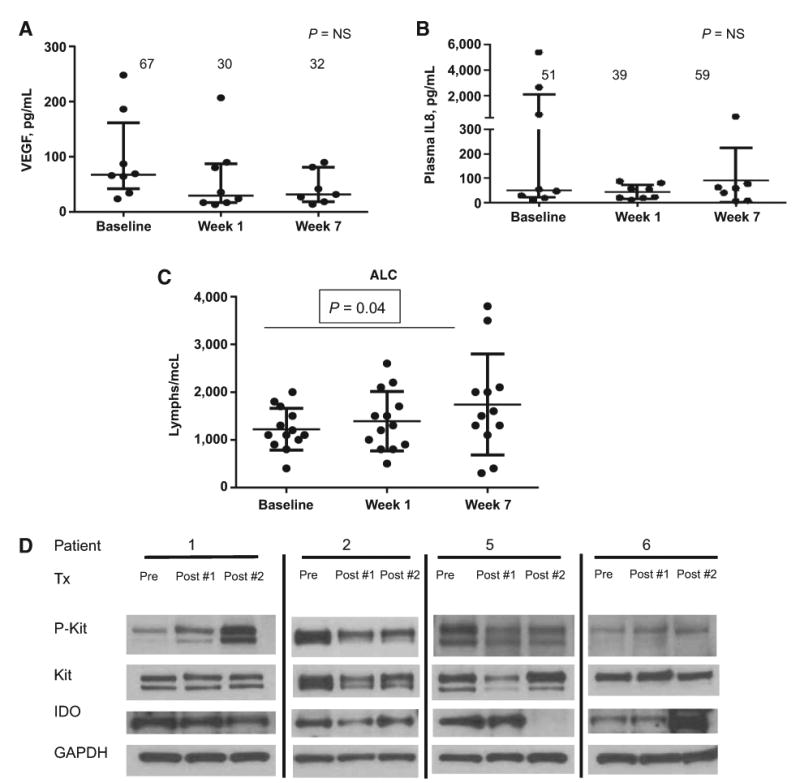
A, For GIST patients (n = 8), VEGF is detectable in plasma, and there are nonsignificant trends toward decrease with therapy. B, IL8 is widely variable at baseline without any changes seen with therapy. C, For patients (n =13), median ALC at baseline =1,100; after 1 week of dasatinib = 1,300 (P = NS); dasatinib and two doses of ipilimumab = 1,550 (P = 0.04, paired t test). No relationship with clinical benefit was observed. D, Western blot analysis: Pre represents prior to treatment, post #1 is after dasatinib alone, and post #2 is after dasatinib + two doses of ipilimumab. There were 4 of 6 patients with adequate tissue for analysis. All patients had baseline expression of KIT, p-KIT, and IDO. Patients 1 and 2 had no changes after treatment (Tx) and evidence of progression. Patient 5 (duodenal GIST, exon 13 mutation) had loss of IDO expression at the second posttreatment biopsy with stable disease per RECIST, irRC, and Choi lasting 19 weeks. Patient 6 had SDH-deficient GIST with evidence of stable disease for 48 weeks without loss of IDO expression.
Absolute lymphocyte count was increased after treatment with dasatinib/ipilimumab. For all 13 GIST patients, median ALC at baseline = 1,100, after 1 week of dasatinib = 1,300 (P = NS), dasatinib and 2 doses of ipilimumab = 1,550 (P = 0.04, paired t test). No relationship with clinical benefit was observed (Fig. 2C).
Western blot analysis
Serial tumor biopsies were obtained on 6 patients. For each patient, three biopsies were performed: 1 pretreatment before initiating dasatinib and ipilimumab, a second biopsy after 1 week of treatment with dasatinib, and a third biopsy after ongoing dasatinib and two doses of ipilimumab. Tissue was adequate for analysis in 4 of 6 patients. All patients had baseline expression of KIT, p-KIT, and IDO. There were no changes in the expression of KIT, p-KIT, and IDO noted after 1 week of dasatinib as well as two doses of ipilimumab for patients 1 and 2. Both of these patients had progressive disease at the first imaging study. Of note, patient 5 had loss of IDO expression at the second posttreatment biopsy. Interestingly, this patient had a duodenal primary GIST with an exon 13 KIT mutation and had evidence of stable disease per RECIST, irRC, and Choi lasting 19 weeks. Patient 6 had succinate dehydrogenase–deficient (SDH) GIST with evidence of stable disease for 48 weeks despite not having evidence of IDO suppression. On the contrary, there appears to be an incremental increase in IDO expression; the significance of this remains unknown (Fig. 2D).
Discussion
After progression on standard TKIs, patients with metastatic GIST have limited therapeutic options. Thus, there is an unmet clinical need for more effective and durable systemic therapeutic options. There is increasing evidence that TKIs can potentially enhance immunologic effects. Data from our group in GIST mouse models have suggested that when TKIs, such as imatinib, lead to tumor regression, there are favorable changes in the immune milieu, including an increase in intratumoral CD8+ T cells, a decrease in regulatory T cells, and a decrease in IDO levels. Our hypothesis suggested that if a TKI is effective in altering the immune milieu and decreasing IDO expression, anti-CTLA-4 would enhance these antitumor effects by further enhancing T-cell activation.
We found that dasatinib 70 mg/day with ipilimumab 10 mg/kg can be safely administered in patients with advanced GIST and other sarcomas. The DLTs included grade 3 gastric hemorrhage at dose level 1 and grade 3 anemia at dose level 2.There was concern of inadequate dasatinib exposure, and further, ipilimumab was ultimately FDA approved at 3 mg/kg for metastatic melanoma. Higher doses of ipilimumab can lead to higher response rates but with significantly more toxicity (25). Therefore, a second dose escalation was performed. Ipilimumab was reduced to 3 mg/kg and dasatinib was reescalated. There were no DLTs identified; we determined that dasatinib 140 mg/day + ipilimumab 3 mg/kg was the MTD.
Almost all patients experienced a treatment-related AE, but mostly of grade 1 or 2. Interestingly, the incidence of grade 3 or 4 AEs on the study was not very high. Generally, the lower dose of ipilimumab was better tolerated, and in those cohorts, there were no DLTs identified. This is consistent with published literature, noting that higher doses of ipilimumab are generally less tolerated (25).
Of the 8 patients with sarcoma, most had rapid disease progression. Interestingly, there were 3 patients each with high-grade sarcoma, clear cell sarcoma, and epithelioid sarcoma who were on the study for 47, 20, and 16 weeks, respectively. Unsurprisingly, there was one patient with leiomyosarcoma, who was only on study for less than 4 weeks. Recent data have demonstrated the lack of responses of single-agent pembrolizumab, which targets PD-1, in patients with leiomyosarcoma but promising responses in selected sarcomas, such as high-grade undifferentiated pleomorphic sarcomas (26). Data are not yet available from the Alliance Clinical Trials in Oncology study, A091401, evaluating the efficacy of nivolumab with or without ipilimumab in all sarcoma subtypes (NCT02500797). This study will evaluate the efficacy of potential combinatorial strategies and further allowed enrolment of other more rare sarcoma histologies. The challenge for this heterogeneous disease is to identify the most effective way to modulate the microenvironment favorably. There is a general lack of preclinical data in other sarcoma subtypes compared with GIST, where the immune microenvironment has been well characterized by our group.
Enrolled GIST patients were extensively pretreated, and most patients had TKI-resistant disease with secondary resistant mutations, such as exon 13 and 17. Generally, these mutations tend to be resistant to TKIs, such as imatinib, sunitinib, as well as dasatinib. Many patients had rapid disease progression, rendering them inevaluable for response, as they did not make it to the first disease assessment at week 12. There were no complete or partial responses noted per RECIST. There were a few patients with Choi responses, and the median PFS was less than 3 months.
This is likely reflective of the aggressive nature of TKI-resistant disease as well as the presumed ineffectiveness of dasatinib for GIST patients. In a phase II trial of dasatinib for patients with metastatic GIST refractory to imatinib and sunitinib, the partial response rate per Choi was 32%, but the median PFS was only 2 months and only 21% were progression free for >2 months (27). Interestingly, our reported median PFS was 2.8 months, which is similar to the reported literature. In that study, no statistically significant differences in Choi responses by molecular status were seen, but a patient whose tumor harbored a PDGFR exon 18 D842V had a PFS of 9.67 months. In our trial, there is one patient with a tumor that also had a PDGFR exon 18 D842V mutation that continues to remain on trial for greater than 13.9 months. This highlights that dasatinib may potentially have some activity in tumors with PDGFR D842V mutations, but beyond this variant, there is no clear role for dasatinib in GIST.
In this cohort of multiple refractory GISTs, one patient with suppression of IDO by Western blot analysis had evidence of stable disease for 4.75 months. The biopsy specimens of 2 other patients did not demonstrate IDO suppression, and both of these patients had disease progression. Despite lack of IDO suppression, there was 1 patient with SDH-B deficiency that was on study for nearly 47 weeks with a Choi partial response and stable disease per RECIST. This may be reflective of the rather indolent course of SDH-deficient GIST, but the value of the KIT-mutant GIST mouse model may be limited in this variant. Although this is a limited cohort, one can hypothesize that, in the setting of ongoing IDO expression, T-cell proliferation was hindered and ipilimumab was not adequate to overcome this negative regulation. Perhaps these multiple refractory tumors were too heterogeneous on a molecular level to be able to suppress IDO after treatment with dasatinib. Additional data would be needed to better evaluate this hypothesis.
Beyond tumor tissue correlatives, multiple cytokines were explored in the peripheral blood. Most were not detectable except IL8 and VEGF. These levels did not change as a result of treatment. In addition, ALCs were explored. In patients with metastatic melanoma treated with ipilimumab, ALC significantly increases over time, and the degree of increase is associated with OS (28–30). In our study, there was a trend toward an increase in ALC levels after treatment with ipilimumab and dasatinib; however, this change did not correlate with clinical efficacy.
We have found that dasatinib does not alter the immune microenvironment favorably to enhance responses with the checkpoint inhibitor ipilimumab. At the time this study was designed, we were unable to secure drug supply for an alternate TKI. Nonetheless, this was the first study combining a checkpoint inhibitor with a TKI specifically for patients with GIST. Future efforts should focus on earlier line therapy when tumors are expected to be more molecularly homogeneous and responsive to therapy. This limited cohort provides useful prospective data that IDO suppression may potentially correlate with antitumor efficacy in GIST. Furthermore, we have recently found that intratumoral CD8+ T cells inhuman GISTs have high PD-1 expression, and in a murine model of GIST, anti-PD-1 and anti-PD-L1 increase the effects of imatinib (31). Moving forward, in the era of more modern and effective checkpoint inhibitors, consideration of TKIs or IDO inhibitors in combination with anti-PD-1 therapy may offer more promising results.
Supplementary Material
Table 1B. Demographics, GIST patients.
| n = 20 | |
|---|---|
| Gender (M:F) | 13:7 |
| Age (median, range) | 54 (40–75) |
| ECOG (median, range) | 0 (0–1) |
| Prior treatments (median, range) | 3 (1–8) |
| Primary site | |
| Gastric | 8 |
| Small bowel | 12 |
| Mutationsa | |
| KIT exon 9 (502_503 duplication) | |
| KIT exon 9 (S501) | |
| KIT exon 9 (6bp deletion) and KIT exon 17 (D820Y) | |
| KIT exon 9 (6bp insertion) and KIT exon 14 (N680K) | |
| KIT exon 11 (9bp insertion/deletion) | |
| KIT exon 11 (L575P) and KIT exon 9 (6bp insertion) | |
| KIT exon 11 (point mutation) and exon 11 (21 bp in frame deletion) | |
| KIT exon 11 (W557R) and KIT exon 11 (G565V) | |
| KIT exon 11 (6bp deletion) and KIT exon 13 (V654A) | |
| KIT exon 11 (L576P) and KIT exon 13 (V654A) | |
| KIT exon 11 (WK557) and KIT exon 13 (V654A) | |
| KIT exon 11 (6bp deletion) and KIT exon 17 (Y823D) | |
| KIT exon 11 (6bp deletion) and KIT exon 17 (D820Y) | |
| KIT exon 13 (V654A) | |
| KIT exon 13 (V654A) | |
| PDGFR exon 18 D842V | |
| PDGFR exon 18 D842V | |
| mTOR exon 30 and SDHB exon 2 | |
| NF1 (deletion), TERT (amplification), and SDHA (amplification) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; F, female; M, male.
Missing for 1 patient.
Translational Relevance.
The immune system has been shown to play a major role in cancer control and progression. There have been tremendous breakthroughs in other malignancies through manipulation of the immune system with checkpoint inhibitors, but limited data exist in sarcoma. Targeted therapies, such as tyrosine kinase inhibitors (TKI), can stimulate immune cells providing the rationale to evaluate TKIs with immunotherapy. Furthermore, combined blockade of CTLA-4 plus KIT, via imatinib or dasatinib, led to improved antitumor effect in a murine model of GIST. In this phase I study, we demonstrated the safety of dasatinib plus ipilimumab in patients with advanced GIST and other sarcomas. Our correlative studies, albeit in a limited sample size, provided interesting data that indoleamine-2,3-dioxygenase (IDO) suppression may potentially correlate with antitumor efficacy in GIST. In the era of more modern and effective checkpoint inhibitors, consideration of TKIs or IDO inhibitors with anti-PD-1 therapy may offer more promising results.
Acknowledgments
Grant Support: This work was supported by MSK Cancer Center Support Grant/Core Grant (P30 CA008748; to Craig Thompson) and NIH grants (R01 CA102613; to R.P. DeMatteo).
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
ClinicalTrials.gov identifier: NCT01643278
Authors' Contributions: Conception and design: S.P. D'Angelo, A.N. Shoushtari, M.L. Keohan, M.A. Dickson, H. Streicher, N. Takebe, L.-X. Qin, R.P. DeMatteo, R.D. Carvajal Development of methodology: S.P. D'Angelo, A.N. Shoushtari, M.L. Keohan, M.A. Dickson, N. Takebe, R.P. DeMatteo, R.D. Carvajal
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S.P. D'Angelo, A.N. Shoushtari, M.L. Keohan, M.A. Dickson, M.M. Gounder, P. Chi, J.K. Loo, L. Schneider, J.P. Erinjeri, A. Sjoberg, N. Takebe, R.P. DeMatteo, R.D. Carvajal, W.D. Tap
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S.P. D'Angelo, A.N. Shoushtari, M.A. Dickson, M.M. Gounder, P. Chi, L.-X. Qin, C. Antonescu, R.P. DeMatteo, R.D. Carvajal, W.D. Tap
Writing, review, and/or revision of the manuscript: S.P. D'Angelo, A.N. Shoushtari, M.L. Keohan, M.A. Dickson, M.M. Gounder, P. Chi, J.K. Loo, J.P. Erinjeri, N. Takebe, L.-X. Qin, C. Antonescu, R.P. DeMatteo, R.D. Carvajal, W.D. Tap
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S.P. D'Angelo, J.K. Loo, L. Gaffney, L. Schneider, Z. Patel, N. Takebe, W.D. Tap
Study supervision: S.P. D'Angelo, L. Gaffney, H. Streicher, N. Takebe, R.D. Carvajal, W.D. Tap
Other (provided recist reads): M. Bluth
Reprints and Subscriptions: To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at pubs@aacr.org.
Disclosure of Potential Conflicts of Interest: A.N. Shoushtari is a consultant/advisory board member for Immunocore and Vaccinex and reports receiving commercial research support from Bristol-Myers Squibb and Immunocore. R.D. Carvajal is a consultant/advisory board member for AstraZeneca, Aura Biosciences, Iconic Therapeutics, Janssen, Merck, and Novartis. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Chan JK. Mesenchymal tumors of the gastrointestinal tract: a paradise for acronyms (STUMP, GIST, GANT, and now GIPACT), implication of c-kit in genesis, and yet another of the many emerging roles of the interstitial cell of Cajal in the pathogenesis of gastrointestinal diseases? Adv Anat Pathol. 1999;6:19–40. doi: 10.1097/00125480-199901000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Sakurai S, Fukasawa T, Chong JM, Tanaka A, Fukayama M. C-kit gene abnormalities in gastrointestinal stromal tumors (tumors of interstitial cells of Cajal. Jpn J Cancer Res. 1999;90:1321–8. doi: 10.1111/j.1349-7006.1999.tb00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seidal T, Edvardsson H. Expression of c-kit (CD117) and Ki67 provides information about the possible cell of origin and clinical course of gastrointestinal stromal tumours. Histopathology. 1999;34:416–24. doi: 10.1046/j.1365-2559.1999.00643.x. [DOI] [PubMed] [Google Scholar]
- 4.Brennan MF, Antonescu CR, Maki RG. Management of soft tissue sarcoma. New York, NY: Springer; 2012. [Google Scholar]
- 5.Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9. doi: 10.1200/JCO.2003.04.190. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10. doi: 10.1126/science.1079666. [DOI] [PubMed] [Google Scholar]
- 7.Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–32. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 8.Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–34. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 9.Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 11.Lee KC, Ouwehand I, Giannini AL, Thomas NS, Dibb NJ, Bijlmakers MJ. Lck is a key target of imatinib and dasatinib in T-cell activation. Leukemia. 2010;24:896–900. doi: 10.1038/leu.2010.11. [DOI] [PubMed] [Google Scholar]
- 12.Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–22. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kreutzman A, Juvonen V, Kairisto V, Ekblom M, Stenke L, Seggewiss R, et al. Mono/oligoclonal T- and NK-cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood. 2010;116:772–82. doi: 10.1182/blood-2009-12-256800. [DOI] [PubMed] [Google Scholar]
- 14.Ray P, Krishnamoorthy N, Oriss TB, Ray A. Signaling of c-kit in dendritic cells influences adaptive immunity. Ann N Y Acad Sci. 2010;1183:104–22. doi: 10.1111/j.1749-6632.2009.05122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borg C, Terme M, Taieb J, Ménard C, Flament C, Robert C, et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J Clin Invest. 2004;114:379–88. doi: 10.1172/JCI21102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mustjoki S, Ekblom M, Arstila TP, Dybedal I, Epling-Burnette PK, Guilhot F, et al. Clonal expansion of T/NK-cells during tyrosine kinase inhibitor dasatinib therapy. Leukemia. 2009;23:1398–405. doi: 10.1038/leu.2009.46. [DOI] [PubMed] [Google Scholar]
- 17.Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094–100. doi: 10.1038/nm.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–96. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mellor AL, Munn DH. Immunology at the maternal-fetal interface: lessons for T cell tolerance and suppression. Annu Rev Immunol. 2000;18:367–91. doi: 10.1146/annurev.immunol.18.1.367. [DOI] [PubMed] [Google Scholar]
- 20.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33:1889–94. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuetze SM, Wathen JK, Lucas DR, Choy E, Samuels BL, Staddon AP, et al. SARC009: Phase 2 study of dasatinib in patients with previously treated, high-grade, advanced sarcoma. Cancer. 2016;122:868–74. doi: 10.1002/cncr.29858. [DOI] [PubMed] [Google Scholar]
- 22.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 23.Wolchok JD, Hoos A, O'Day S, Weber JS, Hamid O, Lebbé C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 24.Choi H. Response evaluation of gastrointestinal stromal tumors. Oncologist. 2008;13:4–7. doi: 10.1634/theoncologist.13-S2-4. [DOI] [PubMed] [Google Scholar]
- 25.Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11:155–64. doi: 10.1016/S1470-2045(09)70334-1. [DOI] [PubMed] [Google Scholar]
- 26.Tawbi HAH, Burgess MA, Crowley J, Van Tine BA, Hu J, Schuetze S, et al. Safety and efficacy of PD-1 blockade using pembrolizumab in patients with advanced soft tissue (STS) and bone sarcomas (BS): results of SARC028, a multicenter phase II study. J Clin Oncol. 2016;34 (suppl; abstr 11006) [Google Scholar]
- 27.Trent JC, Wathen K, von Mehren M, Samuels BL, Staddon AP, Choy E, Butrynski JE, et al. A phase II study of dasatinib for patients with imatinib-resistant gastrointestinal stromal tumor (GIST) J Clin Oncol. 2011;29 (suppl; abstr 10006) [Google Scholar]
- 28.Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, et al. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013;24:1697–703. doi: 10.1093/annonc/mdt027. [DOI] [PubMed] [Google Scholar]
- 29.Postow MA, Chasalow SD, Yuan J, Kuk D, Panageas KS, Cheng M, et al. Pharmacodynamic effect of ipilimumab on absolute lymphocyte count (ALC) and association with overall survival in patients with advanced melanoma. J Clin Oncol. 2013;31 doi: 10.1097/CMR.0000000000000633. (suppl; abstr 9052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63:675–83. doi: 10.1007/s00262-014-1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seifert AM, Zeng S, Zhang JQ, Kim TS, Cohen NA, Beckman MJ, et al. PD-1/PD-L1 blockade enhances T cell activity and antitumor efficacy of imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2017;23:454–65. doi: 10.1158/1078-0432.CCR-16-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.