

Abstract
Intestinal macrophages are uniquely programmed to tolerate exposure to bacteria without mounting potent inflammatory responses. The cytokine IL-10 maintains the macrophage anti-inflammatory response such that loss of IL-10 results in chronic intestinal inflammation. To investigate how IL-10-deficiency alters intestinal macrophage programming and bacterial tolerance, we studied changes in chromatin accessibility in response to bacteria in macrophages from two distinct niches, the intestine and bone-marrow, from both wild-type and IL-10-deficient mice. In both bone-marrow-derived and intestinal macrophages, we identified chromatin accessibility changes associated with bacterial exposure and IL-10-deficiency. Surprisingly, IL-10-deficient intestinal macrophages adopted chromatin and gene expression patterns characteristic of an inflammatory response, even in the absence of bacteria. Further, if IL-10 protein was added to cells that had previously been IL-10-deficient, it could not revert the chromatin landscape to a normal state. Our results demonstrate that IL-10 deficiency results in stable chromatin alterations in macrophages, even in the absence of bacteria. This supports a model where IL-10-deficiency leads to chromatin alterations that contribute to a loss of intestinal macrophage tolerance to bacteria, which is a primary initiating event in chronic intestinal inflammation.
Keywords: Chromatin accessibility, IL-10, macrophages, chronic inflammation, inflammatory bowel disease
INTRODUCTION
Crohn’s Disease is a chronic intestinal inflammatory disorder driven by a combination of genetic and environmental factors, the intestinal bacteria, and the immune system[1]. The lamina propria tissue layer in the intestine is composed of multiple immune cell types including macrophages, B cells, and T cells, which are separated from the intestinal bacteria by a single layer of epithelial cells. Intestinal macrophages have adapted a unique tolerance to the intestinal environment by effectively killing bacteria that breach this epithelial barrier through phagocytosis, but unlike macrophages from other tissues, do not mount a cytokine-mediated inflammatory response thereby preventing damage to surrounding tissue[2]. These mechanisms fail during Crohn’s Disease pathogenesis[3]. In contrast, bacteria-induced changes to histone modifications, transcription factor binding, and transcription reflective of vigorous inflammation have been characterized in bone-marrow-derived macrophages[4–6].
IL-10 maintains the intestinal macrophage anti-inflammatory response, and its expression increases when these cells are exposed to bacteria[7, 8]. Intestinal inflammation is not observed when IL-10-deficient (Il10−/−) mice are raised in a germ-free environment lacking bacteria, but emerges when mice are conventionally-raised with bacteria present or after colonization of germ-free Il10−/− mice with normal intestinal bacteria[9]. More specifically, compared to macrophages from wild-type mice, inflammatory responses to bacterial stimuli are more vigorous in Il10−/− bone marrow macrophages and aberrantly arise in Il10−/− intestinal macrophages[10–12].
In this study, we sought to understand how IL-10-deficiency alters macrophage programming and specifically for intestinal macrophages, bacterial tolerance. We hypothesized that the lack of IL-10 may alter the chromatin landscape in macrophages changing how they respond to bacteria. We thus determined changes in chromatin accessibility in response to bacteria genome-wide in intestinal and bone-marrow-derived macrophages from both wild-type and Il10−/− mice.
RESULTS
To investigate how IL-10-deficiency alters macrophage programming and response to bacteria, we studied changes in chromatin accessibility and gene expression induced by bacteria in two types of macrophages from wild-type (WT) or IL-10-deficient (Il10−/−) mice: bone-marrow-derived (inflammatory in wild-type) and intestinal (non-inflammatory in wild-type). Bone marrow macrophages (BMMs) were treated for 4 hours with either lipopolysaccharide (LPS), a molecule found in the outer membrane of bacteria and known to illicit a strong immune response (stimulated; “+LPS”), or phosphate-buffered saline (unstimulated; “-LPS”). Intestinal macrophages were assayed from mice raised germ-free (no bacteria present; “GF”) or conventionally (bacteria present; “CR”). Due to the small population of cells and expected chromatin effects that are distal from genes[4–6], we measured chromatin accessibility using FAIRE-seq[13], and annotated accessible chromatin using publically available ChIP-seq data from WT BMMs[4–6]. Transcriptional changes were also detected using RNA-seq (Figure 1A).
Fig. 1. Three regulatory element classes in bone marrow macrophages driven by IL-10 deficiency and bacterial stimulation.
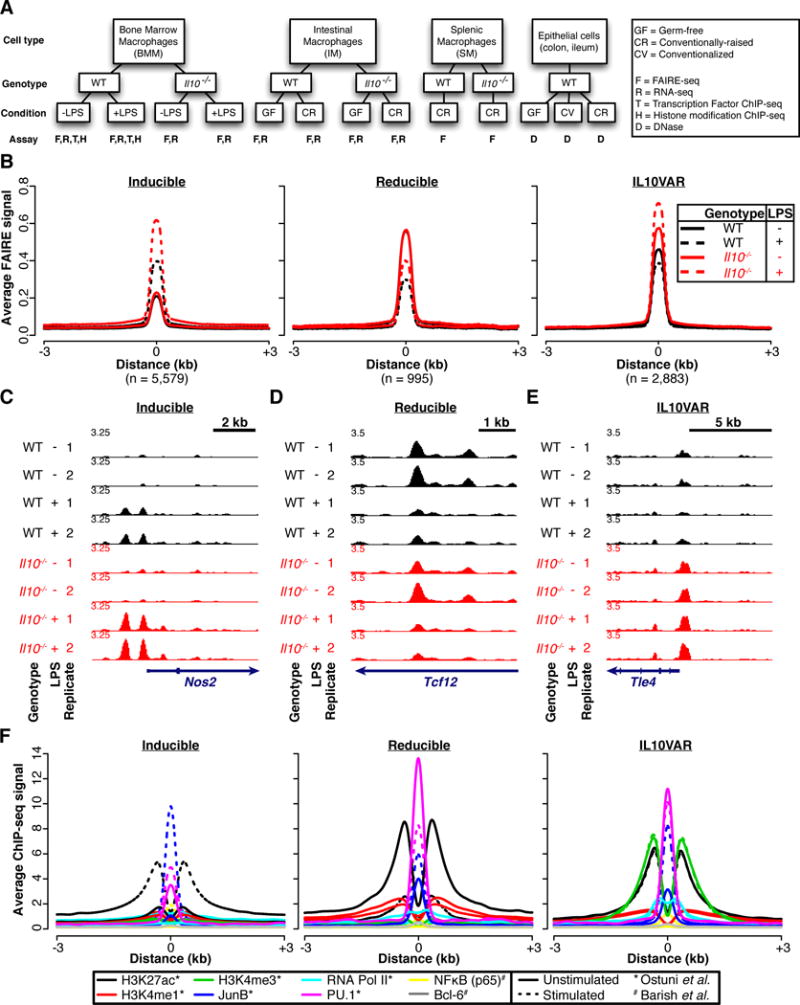
A. Experimental schema. Bone Marrow Macrophages (BMMs) were isolated from wild-type (WT) and IL-10-deficient (Il10−/−) mice. BMMs stimulated with lipopolysaccharide (LPS) were compared against control (-LPS). Intestinal macrophages (IMs) were isolated from the lamina propria intestinal layer of wild-type (WT) and IL-10-deficient (Il10−/−) mice. Intestinal macrophages isolated from germ-free (GF) mice were compared against conventionally-raised (CR) mice. Data were also generated on WT and Il10−/− conventionally-raised splenic macrophages (SM). B. Average normalized FAIRE-seq signal across two biological replicates for WT (black) and Il10−/− (red) for both LPS-stimulated (dotted) and control (solid) BMMs is plotted around regions (±3 kb) classified as inducible (left), reducible (middle), or IL10VAR (right). C–E Representative loci for inducible (C), reducible (D), and IL10VARs (E). F. Annotation with published ChIP-seq signal[4–6].
LPS and IL-10 regulate three distinct classes of differentially accessible chromatin
We first studied bone-marrow-derived macrophages (BMMs), where changes in histone modifications and transcription factor binding reflective of the inflammatory response have been studied in wild-type cells[4–6]. The effect of IL-10-deficiency on chromatin and gene expression in BMMs is unknown, though they exhibit an exaggerated inflammatory response to bacteria[11, 14].
We distinguished genomic regions with accessible chromatin selective to IL-10 status or LPS stimulation and classified 9,457 300-bp intervals into three categories by hierarchical clustering (Figure 1B, Supplementary Figure 1A). One class exhibited increased chromatin accessibility upon LPS stimulation (“inducible”; e.g. Nos2 promoter, Figure 1C), and another demonstrated reduced chromatin accessibility upon LPS stimulation (“reducible”; e.g. Tcf12 intron, Figure 1D). In the third category, chromatin was at baseline more accessible in Il10−/− BMMs and accessibility was exaggerated following stimulation (e.g. Tle4 promoter, Figure 1E). We denoted this third class as IL-10 Variable Accessible Regions (“IL10VARs”) since IL-10-deficiency drove the observed variable chromatin profile. The majority of sites were considered inducible (5,579; 59%), with fewer in the IL10VAR (2,883; 30%) and reducible (995; 11%) classes.
To annotate these three differential chromatin classes, we first tested their association with histone modifications found in active promoters and enhancers (H3K27ac, H3K4me1, H3K4me3) as well as occupancy of AP-1 and ETS family transcription factors (JunB, PU.1), other immunomodulatory transcription factors (Bcl-6 and NFκB), and RNA Polymerase II (RNA Pol II) (Figure 1F). These data were derived from recent studies of the chromatin-mediated response to LPS stimulation in WT BMMs[4–6]; Il10−/− BMMs were not considered in this work. Inducible and reducible sites showed concordant and dramatic changes in H3K27ac, JunB, and PU.1 binding upon stimulation, indicating that LPS-responsive regions gain or lose hallmarks of active enhancer elements. Interestingly, IL10VARs exhibited increased recruitment of JunB upon LPS stimulation, whereas H3K4me3, H3K27ac, and PU.1 signals remained enriched but unchanged, at least in WT BMMs.
To extend these results to the Il10−/− BMMs, we performed H3K27ac ChIP-qPCR at three exemplary IL10VAR loci and one control locus in both WT and Il10−/− cells (Supplementary Figure 1B–C). For two of these regions, H3K27ac was present in WT cells but enhanced in Il10−/− cells, as predicted by our FAIRE data. Further, we found that an inaccessible region devoid of H3K27ac in WT cells exhibited both accessible chromatin by FAIRE and robust H3K27ac in Il10−/− cells. Together these data indicate that changes in accessible chromatin associated with the lack of IL-10 can directly coincide with changes in histone modifications.
We next searched for over-enriched transcription factor motifs. All three classes were enriched with similar frequency for the PU.1 motif (Supplementary Figure 1D). The NFκB motif was selective to the inducible class, whereas the AP-1 motif was found in both inducible and IL10VARs (Supplementary Figure 1D). These results further support the transcription factor occupancy associations based on ChIP-seq data (Figure 1F) and identify PU.1 as a factor in all three classes. PU.1 is a known master regulator driving transcriptional responses in macrophages, and along with other transcription factors establishes tissue-specific characteristics of macrophage populations[15].
To determine whether each differential chromatin class targeted functionally related genes, we mapped chromatin regions to genes and searched for enriched pathways using GREAT[16], independent of underlying gene expression (Supplementary Figure 1E). The inducible class was enriched for pathways related to immune response and LPS-responsive genes, and others linked to chemokine and cytokine signaling, including Il12a, Il18, Ccl2–4, Ccr1–3, Ccr7, and Ccr9. Reducible regions were linked to genes implicated in leukocyte activation, and IL10VARs were linked to FoxP3 and Myc targets. Taken together, these data show that FAIRE-seq detects active regulatory elements in multiple classes linked to biologically meaningful pathways[17, 18].
Lastly, we determined whether chromatin alterations were concordant with gene expression changes in the same BMMs. First, independent of chromatin status, we found 2,715 genes upregulated and 1,496 downregulated by LPS in WT mice (adjusted p<1×10−5; Figure 2A), many of which are integral to macrophage biology (Figure 2B)[19]. Next, to link changes in chromatin accessibility to expression changes driven by LPS and IL-10 deficiency, we associated regions in the three chromatin classes to genes using GREAT[16], then computed the fraction of those genes more highly (positive log2 fold-change) or lowly (negative log2 fold-change) expressed due to LPS stimulation or IL-10-deficiency (Figure 2C). Unexpectedly, we found a nearly equal proportion of down-regulated and up-regulated genes linked to the inducible class upon LPS stimulation. Subdividing these up- and down-regulated genes, we observed that their promoters showed differential H3K27ac and RNA Pol II levels, with increasing H3K27ac enrichment coinciding with increasing gene expression (Supplementary Figure 1F). Furthermore, genes linked to inducible chromatin sites whose expression increased upon LPS stimulation consisted of canonical cytokine signaling pathway members, whereas those with decreased expression included known anti-inflammatory mediators, such as Mef2c, Mef2d, Cx3cr1, IL6r, and Myc. Mef2d directly mediates IL-10 expression levels[20], and Myc is a critical transcription factor for many cellular functions, including anti-inflammation in macrophages[18]. In contrast to these dichotomous inducible chromatin sites, genes linked to reducible sites and IL10VARs tended to show decreased expression (Figure 2C). Together, these data indicate that LPS stimulation tends to more frequently increase chromatin accessibility, but results in decreased expression of more genes. Our findings highlight the highly dynamic nature of macrophage chromatin accessibility regulated by cytokines and bacterial products, in agreement with recent studies[6]. It also suggests recruitment of both transcriptional activators and repressors to accessible chromatin, such as has been shown during macrophage differentiation[21]. Furthermore, these results suggest that IL-10-deficiency leads to baseline chromatin accessibility changes that may poise chromatin for recruiting transcription factors that drive the transcriptional differences and increased inflammation in these mice.
Fig. 2. Alterations in gene expression due to IL-10 status or bacterial stimulation affect many key cytokine genes.
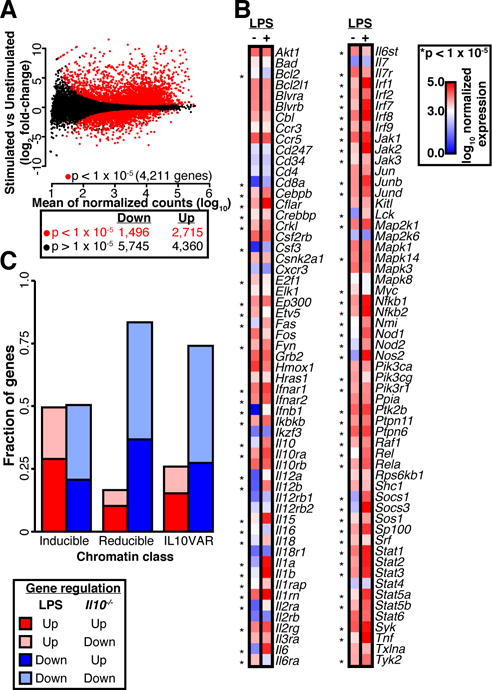
A. Differential expression analysis between LPS-stimulated and unstimulated BMMs for two biological replicates (red points, DEseq adjusted p < 1 × 10−5). B. Log10 expression of genes in the cytokine signaling pathway is plotted on a scale of 0 (blue) to 3.0 (white) to 5.0 (red), and genes with a significant alteration in expression (DEseq adjusted p < 1 × 10−5) are denoted with an asterisk. C. Fraction of genes linked to three regulatory element classes that were up- or down-regulated with respect to IL-10 status (Il10−/− vs WT) or LPS stimulation.
IL-10-deficient intestinal macrophages exhibit chromatin changes mimicking inflammation
In contrast to macrophages from other organs, intestinal macrophages under physiological conditions efficiently eradicate microbes that breach the intestinal epithelial barrier without mounting a potent inflammatory response. Mechanisms governing these functional adaptations of intestinal macrophages, however, remain incompletely elucidated, and the loss of this bacterial tolerance underlies chronic intestinal inflammatory diseases.
As with BMMs, we hierarchically clustered 7,957 300 bp regions with variable chromatin accessibility based on IL-10 status (WT vs Il10−/−) and presence of bacteria (germ-free vs conventionally-raised) in macrophages isolated from the lamina propria layer of the intestine (>95% purity, Supplementary Figure 2A). Similar to BMMs, we observed three distinct categories (inducible, reducible, and IL10VAR) (Supplementary Figure 2B, Figure 3A–B), however with markedly different proportions; the majority (4,724, 59%) were classified as IL10VARs, whereas in BMMs the majority (5,579, 57%) were inducible elements (Supplementary Figure 1A, Figure 1B). Importantly, intestinal macrophage IL10VARs were characterized by altered chromatin accessibility in Il10−/− mice even in the absence of intestinal bacteria and whose resulting chromatin landscape strongly mimicked that displayed by LPS-stimulated BMMs. We thus hypothesized that chromatin and transcriptional changes driven by IL-10-deficiency in intestinal macrophages heavily contribute to an inflammatory response typically seen in BMMs, but not intestinal macrophages (Figure 3B–D, Supplementary Figure 2C).
Fig. 3. Aberrant chromatin accessibility in Il10−/− intestinal macrophages from germ-free mice mimics that of LPS-stimulated bone marrow macrophages but is not cell-type-specific.
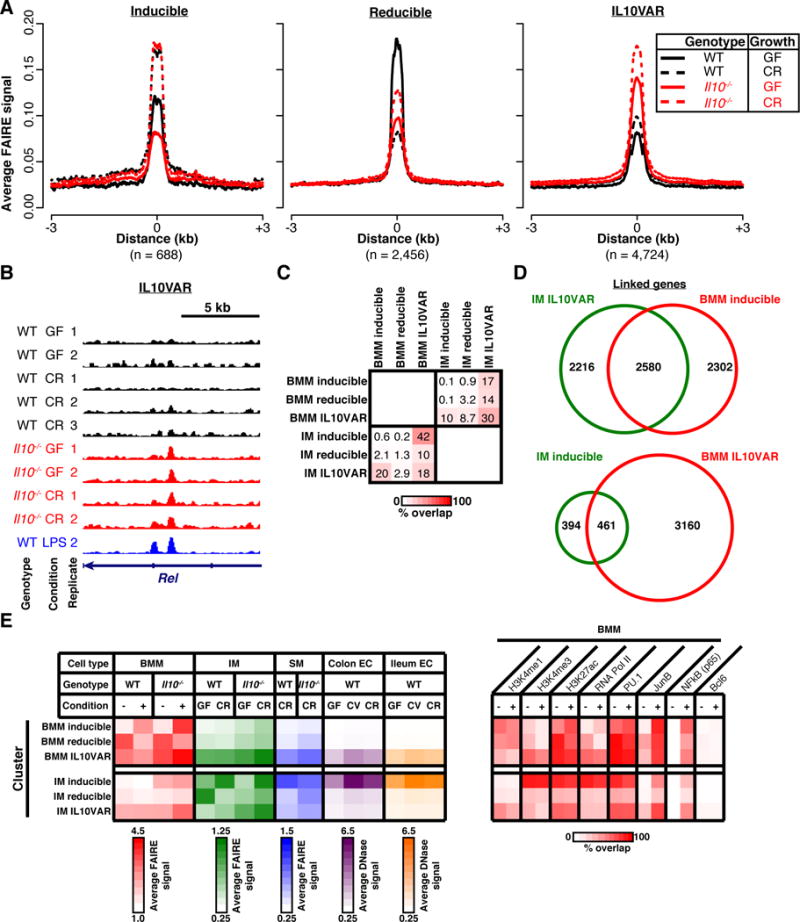
A. Average normalized FAIRE-seq signal across 2–3 biological replicates for WT (black) and Il10−/− (red) for both conventionally-raised (dotted) and germ-free (solid) intestinal macrophages is plotted around regions (±3 kb) classified as inducible (left), reducible (middle), or IL10VARs (right). B. Representative locus for IL10VARs. FAIRE-seq signal from WT LPS-stimulated BMMs (blue) included as a comparator for IL10VARs. C. Percentage of overlapping sites in three regulatory element classes from BMMs and intestinal macrophages. Overlap values presented reflect the percentage overlap relative to the given set of regions on the left. D. Fraction of common genes linked to intestinal macrophage IL10VAR and BMM inducible (top) or intestinal macrophage inducible and BMM IL10VAR (bottom). E. Annotation of regulatory element classes with averaged FAIRE-seq signal in BMM (red), intestinal macrophage (IM, green), and splenic macrophages (SM, blue), as well as published DNase-seq signal from colonic (purple) and ileal epithelial cells (orange). Summarized ChIP-seq data for transcription factors and histone modifications is presented on the right as percent overlap.
We investigated similarities between intestinal macrophage IL10VARs and LPS-responsive (inducible) regions in BMMs in several ways. First, we determined that 20% of intestinal macrophage IL10VARs directly overlapped BMM inducible regions (Figure 3C). Linking these regions to genes (GREAT), this concordance increased to 54% of genes held in common (Figure 3D). Given this overlap, many significantly enriched pathways associated with intestinal macrophage IL10VARs matched those enriched in the BMM inducible class, including cytokine and chemokine signaling, known perturbation by LPS, and known immune system functions (Supplementary Figure 2D). Furthermore, transcription factor motifs found within intestinal macrophage IL10VARs were highly consistent with those in BMM inducible regions, including AP-1, PU.1, CREB, RUNX, and IRF1 (Supplementary Figure 2E and Supplementary Figure 1D).
We next examined gene expression patterns of intestinal macrophages both confined to the cytokine pathway (Supplementary Figure 3A) as well as globally (Supplementary Figure 3B–C). We noticed that although the baseline expression pattern was altered in germ-free intestinal macrophages from Il10−/− mice, unlike at the chromatin level, it did not mimic LPS-stimulated WT BMMs. Instead, this expression phenotype was only apparent in intestinal macrophages from conventionally-raised Il10−/− mice, where the aberrant inflammatory response is manifested[8, 11, 14, 22]. For example, nearly all interleukin genes exhibited a relative expression decrease at baseline in germ-free Il10−/− intestinal macrophages, but were massively upregulated in conventionally-raised Il10−/− intestinal macrophages (Supplementary Figure 3A). When comparing gene expression globally, IL-10-associated expression changes in intestinal macrophages correlated more strongly with expression changes in BMMs driven by stimulation (r = 0.15) than with those driven by IL-10-deficiency in BMMs (r = 0.1) (Supplementary Figure 3B). However, only the conventionally-raised intestinal macrophages from Il10−/− mice displayed a high similarity to LPS-stimulated BMMs with respect to these gene expression changes (Supplementary Figure 3C).
Together, our findings suggest that IL-10-deficiency alone is sufficient to poise intestinal macrophage chromatin for an inflammatory response to intestinal bacteria, even before microbial colonization and subsequent manifestation of inflammation. This raises the possibility that in the absence of IL-10, intestinal macrophages lose their hypo-responsiveness to the intestinal bacteria and acquire a chromatin accessibility pattern consistent with LPS-stimulated BMMs, poising cells to respond inappropriately through unabated intestinal inflammation. This model is supported by well-described phenotypic differences between intestinal and non-intestinal macrophages[3, 14, 19, 23, 24]. However, our data suggests chromatin is a key mediator of this microbial response.
Regions of dynamic macrophage chromatin are associated with enhancers but are not cell-type-specific
Camp et al.[25] recently described changes in chromatin accessibility and gene expression in intestinal (colonic and ileum) epithelial cells of conventionally-raised and germ-free mice, and germ-free mice intentionally conventionalized with the intestinal bacteria. They reported few significant accessibility changes with bacterial colonization. We questioned whether differential chromatin regions in macrophages exhibited altered chromatin accessibility in epithelial cells. In addition, we wondered whether IL-10-deficiency-driven alterations in chromatin were observed in macrophages from other organs. For the latter, we therefore generated and assessed chromatin accessibility data in splenic macrophages from the same conventionally-raised mice described above as an in vivo control of macrophage cellular environment. We integrated these chromatin data with the occupancy of histone modifications (H3K27ac, H3K4me1, and H3K4me3) and transcription factors (PU.1, JunB, NFκB, Bcl-6 and RNA polymerase II (Pol2)) in BMMs in the presence or absence of LPS to more comprehensively annotate the macrophage regulatory landscape (Figure 3E). We found that IL-10-deficiency also altered the baseline chromatin accessibility of splenic macrophages, suggesting similar regulatory defects in the absence of basal IL-10 production. Interestingly, inducible sites in intestinal macrophages (almost half of which overlap BMM IL10VARs, Figure 3C) also exhibited changes in epithelial cells isolated from WT germ-free mice compared to conventionally-raised and conventionalized mice, suggesting these bacteria-responsive dynamic regions may not be cell-type-specific. Based on consistent PU.1 occupancy in the presence or absence of LPS stimulation in all intestinal macrophage and BMM differential chromatin regions, our data suggest that PU.1 may recruit pro-inflammatory (AP-1, NFκB) factors necessary to facilitate the bacterial response, a model that has been previously proposed[5, 6].
Proof-of-concept that chromatin alterations may also occur in human Crohn’s Disease
As shown above (Figure 3), IL-10-deficiency sufficiently poised intestinal macrophage chromatin for an inappropriate inflammatory response even in the absence of intestinal bacteria. We wondered whether specific sites of chromatin alterations identified here were representative of changes associated with loss of macrophage tolerance in humans as well. We focused on IL10VARs in intestinal macrophages to assess specifically those sites linked with genetic variation. To test the conservation of chromatin changes in human Crohn’s Disease, we generated FAIRE-seq data from colonic mucosal biopsies from two Crohn’s Disease and two control patients. These patients were genotyped and did not harbor variants near IL10 or IL10RB previously linked with Crohn’s Disease[26] and had no differences in IL-10 expression as measured by RNA-seq on the same colonic mucosal tissue samples (Figure 4A). We next converted the coordinates of the 4,724 intestinal macrophage IL10VARs over to the reference human genome. As genomic regulatory regions can change location during evolution[27], we evaluated 300-bp windows within a 10 kb region centered around the midpoint of each mapped regulatory element for changes in FAIRE signal between Crohn’s Disease and control colon samples. We found 327 windows with differential FAIRE signal, a significant enrichment relative to random chance (permutation, p<0.001; Figure 4B). Of these, 304 (93%) exhibited increased accessibility in Crohn’s Disease patients. This significant linkage between syntenic human-mouse regions was not unique to IL10VARs, however, and was also seen for inducible and reducible classes as well (Figure 4B). As a class, syntenically-mapped IL10VARs were again significantly enriched near key cytokine pathway and pro-inflammatory genes, including IL7R (Figure 4C–D), NFKB1, MYD88, as well as a component of Mediator, MED19. These sites and others suggest that human Crohn’s Disease patients may also have chromatin alterations associated with an inappropriate immune response.
Fig. 4. IL10VARs display enrichment for differential chromatin in syntenic regions in human tissue and allele specific protein binding.
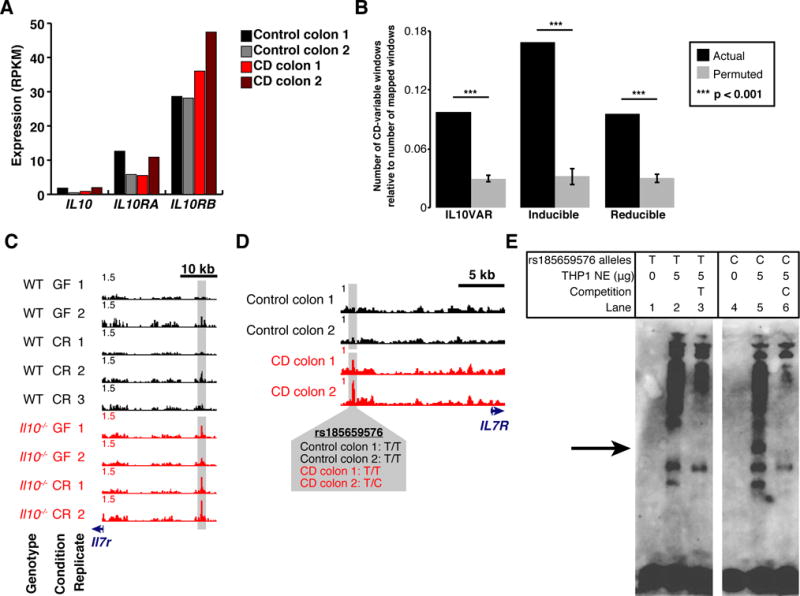
A. Expression levels (RPKM) of IL10, IL10RA, and IL10RB for each human patient biopsied as measured by RNA-seq. B. Regions falling into one of three regulatory element classes from intestinal macrophages were mapped to the human genome and selected based on CD-selective chromatin accessibility in mucosal biopsies from CD and non-CD patients. The number of final CD-variable windows relative to the number of windows that successfully mapped to the human genome is compared to a permuted control for each regulatory element class. P-values were derived empirically based on 1,000 permutations. Error bars represent standard deviation around the mean. C–D. Normalized FAIRE-seq signal at Il7r (mouse, C) and IL7R (human, D) depicting conserved enrichment in mouse intestinal macrophages and human mucosal biopsies. Crohn’s Disease (CD) and control patient genotypes for overlapping variant rs185659576 are also presented. E. Electrophoretic Mobility Shift Assay (EMSA) with human THP-1 macrophage nuclear extract utilizing complementary oligonucleotides containing either the T (reference) or C (alternate) alleles of rs185659576. Protein complex binding specifically to the C allele is demarcated with an arrow.
One interesting site demonstrated Crohn’s Disease-selectivity, overlapped a site of genetic heterogeneity, and was upstream of the interleukin-7 receptor gene (IL7R) (Figure 4C–D). One Crohn’s Disease patient was heterozygous at rs185659576, a T/C variant not previously reported to have disease relevance, whereas the controls and other Crohn’s Disease patient were homozygous for the T allele. We tested for allelic effects on protein binding by using biotin-labeled probes surrounding the C or T alleles, incubated with human macrophage (THP-1) nuclear lysate and subjected to electrophoretic mobility shift assays (EMSA) (Figure 4E). Band shifts indicative of multiple complexes were observed. Moreover, a protein complex (Figure 4E, arrow) was observed when the C allele was present but not the T allele, suggesting differential protein binding dependent on the rs185659576 allele. Competition of labeled C-allele probe with excess unlabeled C-allele probe more efficiently competed away allele-specific bands than excess unlabeled T-allele probe, demonstrating allele-specificity of the protein-DNA complexes. Together, these data suggest that an unknown transcriptional regulator binds preferentially to the rs185659576-C allele in macrophages. Collectively, these results provide a proof-of-concept for using chromatin accessibility important in intestinal macrophage biology and colitis pathogenesis to identify and prioritize regions for further experimental investigation that may be directly driving susceptibility to human Crohn’s Disease.
Ectopic IL-10 supplementation minimally effects aberrant chromatin associated with IL-10-deficiency
The spontaneous onset of intestinal inflammation in IL-10-deficient mice may be reversed by treatment with recombinant IL-10 soon after weaning[28]. Exogenous IL-10 also attenuates the pro-inflammatory transcriptional output in bone-marrow-derived macrophages. However, use of recombinant IL-10 for the treatment of Crohn’s Disease has been largely unsuccessful[29] with little understanding for the basis of this failure. IL10VARs exhibited increased chromatin accessibility at baseline and were associated with expression changes in key inflammation-related genes in response to microbial stimulation (Figure 1, Figure 3, Supplementary Figures 2–3). To investigate whether IL-10 supplementation in vitro can revert the Il10−/− BMM chromatin profile to a normal state, we performed FAIRE-seq on Il10−/− BMMs stimulated with IL-10 and/or LPS. Surprisingly, chromatin accessibility at <5% of the 2,883 IL10VARs in BMMs was affected by the addition of IL-10 (Figure 5), and was unchanged at nearly all genes with known expression changes (data not shown). Chromatin accessibility was also unaffected at reducible regions (Supplementary Figure 4A), but was more pronounced at inducible regions (Supplementary Figure 4B), with a small subset near genes with IL-10-mediated expression changes. These data suggest IL-10 may transiently modulate expression and chromatin structure of certain target genes (e.g. Irf1, Figure 5B), however at most IL-10-dependent loci, exogenous IL-10 does not significantly remodel chromatin. We propose that the absence of IL-10 during development drives the formation of a chromatin landscape that leads intestinal macrophages to respond aberrantly to bacterial stimulation that cannot be corrected by IL-10 after differentiation. This model is supported by the development of early and severe Crohn’s Disease in patients with genetic dysregulation of IL-10 signaling[30], the lack of therapeutic response to recombinant IL-10[29], and increased success with earlier IL-10 therapeutic intervention[28].
Fig. 5. Ectopic IL-10 does not revert aberrant baseline chromatin structure of Il10−/− macrophages.
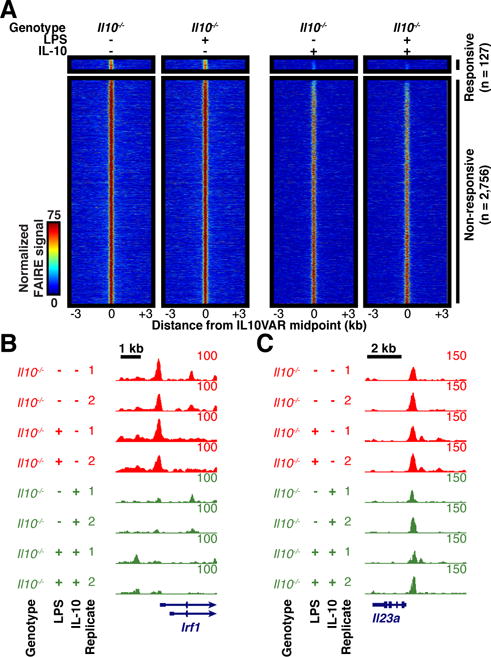
A. Normalized FAIRE-seq signal for BMMs with and without LPS stimulation and ectopic IL-10 supplementation within 3 kb of IL10VARs divided into responsive or non-responsive based on effect from ectopic IL-10. B–C. FAIRE-seq signal without (red) and with (green) ectopic IL-10 at responsive (B) and non-responsive (C) regions.
DISCUSSION
Chromatin accessibility influences cellular identity, providing access for transcription factors and setting the stage for responses to changing cellular conditions and external stimuli. For each cell, the chromatin landscape evolves throughout differentiation[31–34], finely tuning regulatory programs. Closely related cells, such as macrophages from different tissues, will share a large fraction of their accessible regions associated with common functions, but their differences highlight unique regulatory programs driven by their environments and their normal responses. This was demonstrated in a recent study showing macrophage gene expression and chromatin differences depending on the microenvironment[35], despite a common progenitor[36]. Further, Lavin et al. showed that the differentiated macrophage chromatin landscape could be re-programmed after changing the microenvironment. This is likely due, at least in part, to the tissue-restricted expression of critical transcription factors that govern tissue macrophage function, including members of the GATA, RUNX, and MEF2 families[35]. Microenvironment and tissue-restricted transcription factors therefore likely play prominent roles in properly establishing chromatin during differentiation through subtle alterations of the common canvas in progenitor cells, leading to significantly different behaviors, a concept proposed in a study of chromatin during Drosophila development[32].
Intestinal macrophages respond to bacteria very differently compared to macrophages from other tissues. We provide strong evidence here that chromatin plays a critical role in maintaining this cellular identity and function. The establishment of a proper chromatin landscape may depend on IL-10 early in development, even though IL-10 is not known to be a classic chromatin remodeler, pioneer factor, or even a factor that directly interacts with chromatin. We believe this may have relevance for the development of therapies for chronic intestinal inflammatory diseases. Drugs that simply target aberrantly expressed genes may fail if restoration of gene activity does not correct abnormal function due to an improper chromatin environment in which to operate, as is the case with current management strategies for patients with Crohn’s Disease[37]. In humans, mutations in IL-10 receptor genes (IL10RA, IL10RB) are associated with early, severe Crohn’s Disease[30], indicating that IL-10 activity is relevant in human disease, but are rare. Interestingly, clinical trials have explored recombinant IL-10 therapy in Crohn’s Disease with little success[29]. Various reasons have been cited for this lack of therapeutic efficacy, including dosage, timing of IL-10 therapy and resulting IL-10 concentration in the intestine, and the large degree of disease phenotype heterogeneity among patients[38]. Our results suggest an alternative, complementary explanation. If macrophage cellular identity has been stably altered at the chromatin level, simply restoring IL-10 levels may not be sufficient, especially in the long term. More generally, if factors essential for proper chromatin programming during development or differentiation are missing, their lack of chromatin-modifying activity may preclude their efficacy as a sole therapy later in time. Factors that drive differentiation are known to promote and suppress transcription, thus This model is supported by a longitudinal study of disease progression in mice, where recombinant IL-10 therapy administered to weanlings prevented the onset of intestinal inflammation, whereas adult mice only modestly exhibited reduced progression[28]. Future studies can examine macrophage chromatin dynamics during development. However, proper temporal expression of critical tissue-specific transcription factors and other genes is also likely to play a role in establishing appropriate cellular identities during differentiation, and may very well occur upstream of structural changes to chromatin.
We also believe that this has important implications for how underlying genetics should be investigated for contributions to disease, and importantly here chronic intestinal inflammatory diseases. Not only should variant effects be investigated in the end disease cell, but also how they may contribute the establishment of the epigenetic program. In complex human disease, contributions from multiple genetic variants may result in subtle chromatin changes that when combined with other environmental factors may be principal drivers behind increased disease susceptibility. Genetic predisposition to intestinal inflammation, regardless of whether IL-10 signaling is involved, may therefore result from a poised chromatin state that drives an aberrant transcriptional response to stimuli in a tissue specific context. Neither human Crohn’s Disease patients we analyzed had genotypes that indicated IL-10 signaling had been affected, yet they exhibited chromatin changes similar to those found in our IL-10-deficient mouse colitis models. These findings support the well-described differences in transcriptional responses to microbial stimulation between intestinal macrophages and other non-intestinal macrophage populations[2, 39]. Loss of this perpetual hypo-responsiveness to the intestinal bacteria is also observed in intestinal macrophages from patients with Crohn’s Disease compared to intestinal macrophages from control patients[3].
We found that IL-10-deficiency can profoundly influence establishment of the proper macrophage chromatin environment, effectively altering cellular identity resulting in aberrant behavior. We found that in the presence of bacteria, IL-10-deficient intestinal macrophages adopted chromatin and gene expression patterns characteristic of an inflammatory response. In IL-10-deficient mice raised germ-free, which do not exhibit intestinal inflammation, chromatin accessibility patterns indicative of inflammation were also present but lacked the corresponding gene expression changes.
Additionally, our work shows an association between sites in mice reflective of a baseline alteration in chromatin driven by IL-10 deficiency and similar Crohn’s Disease-specific changes in syntenic regions in human colon tissue, despite the fact that the underlying genetic changes are not the same. Based on these results, we postulate that multiple genetic mechanisms may drive a common core chromatin signature reflective of chronic inflammation. It is critical, therefore, to understand how all types of genetic and environmental variation is changing the chromatin landscape leading to increased disease susceptibility.
Our findings also have direct implications for the development of novel therapeutic strategies for Crohn’s Disease and possibly other chronic inflammatory diseases that result from a strong interplay between genetics and the environment. Future studies that uncover the mechanisms of regulatory aberrations in chronic inflammatory disorders should direct the exploration of new therapeutic strategies towards stimulation or inhibition of the appropriate intestine-specific macrophage response, especially in macrophages isolated from the healthy and inflamed human intestine.
MATERIALS AND METHODS
Reagents, mice, and macrophages
IL-10 was purchased from BD PharMingen (San Diego, CA), LPS was obtained from Sigma-Aldrich (St. Louis, MO). WT and Il10−/− mice on a C57BL/6 background and controls were purchased from The Jackson Laboratory (Bar Harbor, ME). WT and Il10−/− mice on the C57BL/6 background were matched for age in all experiments. GF mice (WT and Il10−/−) were derived by embryo transplant and maintained at the National Gnotobiotic Resource Center at the University of North Carolina. Mice were housed in accordance with guidelines from the American Association for Laboratory Animal Care and Research Protocols, and the Institutional Animal Care and Use Committee of the University of North Carolina approved experiments (IACUC #12-199).
Macrophage isolation and stimulation
Bone marrow-derived macrophages (BMMs) were harvested, cultured and stimulated as described [11]. Macrophages were stimulated with 100 ng/mL LPS where denoted. For Il10−/− macrophages supplemented with IL-10, cells were pre-treated with IL-10 (10 ng/mL) for 24 hours. Lamina propria mononuclear cells were isolated from mouse colon by an enzymatic method and density gradient centrifugation, as described previously [40]. Splenocytes were isolated as previously described [41]. Lamina propria mononuclear cells and splenocytes were further separated into CD11b+ cells using anti-CD11b microbeads (Miltenyi Biotec, Auburn, CA). Purity was >90% by flow cytometric analysis (Supplementary Figure 2A).
Creation of a mouse genome blacklist filter
Certain types of genomic regions are known to induce artifactual signal caused by experimental or technical biases, and thus it has been suggested that these regions be masked or ignored[42]. Mouse blacklists were therefore created in a fashion consistent with ENCODE[43] for the human genome, including problematic satellite repetitive elements (CENSTAT, GSAT, MurSAT, and SYNREP), regions with sequence homology to mitochondrial DNA (NumtS), rRNA, and regions on chrX with strong sequence homology to chrY. The effects of these types of genomic elements on sequencing data has been previously discussed[42].
Formaldehyde-assisted isolation of regulatory elements (FAIRE) and hierarchical clustering of differentially open chromatin
FAIRE was performed as previously described[13] in biological duplicate or triplicate as indicated. Sequencing was performed using 50-bp single-end reads (Illumina HiSeq 2000). Reads were filtered such that their identical sequence could appear only up to five times, and only reads with quality score greater than 20 for 90% of bases were retained. Reads were then were filtered using TagDust [44] and aligned to the reference mouse genome (mm9) with GSNAP [45] using a k-mer size of 15, indel penalty of 5, and allowing 1 mismatch. Reads aligning to more than four locations or overlapped blacklisted regions of the mouse genome were removed. Reads were counted in 300-bp windows sliding by 100 bp across the genome and normalized for sequencing depth. Windows were then intersected with the union set of the top 50,000 peaks identified for each sample using Fseq [46]. For clustering analyses, only windows with variable signal (row standard deviation > 0.75) were retained. Overlapping windows were handled by retaining the one window for a given region with the maximal standard deviation. Remaining windows were then hierarchically clustered and plotted. Feature intersections were computed using BEDTools [47]. For comparison with exogenous IL-10, quantile normalization of wiggle files was carried out using DANPOS [48]. Regions were split into “responsive” and “non-responsive” based on whether a peak called in the +IL-10 conditions fell into the union set of the top 50,000 peaks called for all control samples.
Chromatin immunoprecipitation and quantitative PCR
Chromatin immunoprecipitation (ChIP) was performed on both WT and Il10−/− with the ChIP-IT Express kit (ActiveMotif, Carlsbad, CA) according to the manufacturer’s recommendations. Briefly, 10×106 BMMs were washed with complete growth media, and fixed with 1% formaldehyde for 10 minutes at room temperature. IPs were carried out using a polyclonal H3K27ac antibody (ab4729; Abcam, Cambridge, MA). Real-time PCR was then performed with the SensiFAST SYBR Hi-ROX Kit (Bioline, Boston, MA) as recommended, using primers for the four regions (below); input DNA was diluted 10-fold prior to PCR. ChIP enrichment values were compared to input controls and converted to percentage of input DNA immunoprecipitated using 2−ΔCt. These percentage input values were then averaged for three independent biological replicates and standard error was computed.
Primer sequences used were as follows:
|
| ||
| Primer set name | Forward | Reverse |
|
| ||
| Suz12 | 5′TGGTGGGGGAGTTAAATATCTG | 5′ATGACAGGTGCTTTTGAGGTTT |
|
| ||
| Irf1 | 5′GAGTTCCTACTGCCCCGTATTT | 5′TATGTCCCTGTACCGCCTTACT |
|
| ||
| Steap3 | 5′CTCAGCAGTTTGTGCTGATAGG | 5′GTGCCCTTATCTACCATTTCCA |
|
| ||
| Cacng8 (neg. control) | 5′AGGCCATGTTTGGGATACTG | 5′AAAGCAGAAGCGGAACTCAC |
|
| ||
Transcript abundance measurements and differential expression using RNA-seq
RNA was isolated using Qiagen RNeasy Mini Kit (Valencia, CA). This kit uses a column-based DNase treatment to eliminate DNA contamination. RNA was sequenced using paired-end 50bp reads (Illumina Hiseq 2000). Reads were filtered using TagDust (Lassmann et al. 2009) and only reads with quality score greater than 20 for 90% of bases were retained. Reads were then aligned to the reference mouse genome (mm9) with GSNAP [45] using parameters described above. Reads aligning to blacklisted regions of the mouse genome were removed. Transcript abundance was estimated by computing RPKM [49] using RefSeq gene models aggregated by gene symbol. For differential expression analyses, raw counts over RefSeq exons were used, then compared across samples using DESeq [50] at an adjusted p-value threshold of p < 1×10−5.
Analysis of ChIP-seq and DNase-seq data
Sequencing reads were obtained from the Short Read Archive and re-processed by filtering using TagDust [44] and aligning to the reference mouse genome (mm9) with Bowtie [51] using default parameters. Reads aligning to blacklisted regions of the mouse genome were removed. Remaining reads were extended in silico to an assumed fragment length of 250 bp. DNase data were obtained from Camp et al. [25].
Motif analysis
Significantly enriched known transcription factor (TF) motifs were identified using HOMER [15]. The 300-bp flanking region was used as local background. The motifs detected de novo are presented in Supplementary Figure 1 and Supplementary Figure 2. Highly similar entries were merged.
Ontologies associated with differentially open chromatin
Regions from Clusters 1–3 were associated with Gene Ontologies using GREAT [16]. The top term for a given ontology with q < 1 × 10−5 was presented.
Patient Population
CD patients from the adult IBD Center at University of North Carolina were included in this study. A total of 4 samples (2 CD and 2 non-CD controls) were studied using FAIRE-seq. This study received IRB approval at UNC Chapel Hill (Protocol # 10-0355).
Genotyping and analysis of human FAIRE-seq data
Genotypes for two non-IBD and two CD individuals were assayed on the Illumina Immunochip. QC and removal of poor quality SNPs was performed using a standard cluster file from Illumina. Genotype imputation was performed with MaCH-admix [52], and sex-specific, custom genomes were created for each individual using genotype calls for all genotyped and imputed variants. FAIRE-seq reads with quality scores greater than 20 for 90% of bases were retained. High quality reads were aligned to their respective personalized genomes using the SNP-tolerant GSNAP software [45].
Cross-species analysis
Windows falling into one of the three classes from mouse analyses were converted to coordinates in the human genome using LiftOver with a conservation threshold of 0.1[27]. These new regions were then extended 5 kb on either side of the region midpoint. New 300 bp windows sliding by 100 bp were then constructed across these 10 kb regions, and normalized FAIRE-seq enrichment for the two control and two CD individuals was computed. Regions with variable open chromatin (row standard deviation > 0.25) were then detected. Overlapping windows were handled by retaining the one window for a given region with the maximal standard deviation. Permutations were computed by shuffling a given class of regions across the uniquely mappable mouse genome, and then repeating this cross-species process 1,000 times and counting how many variable windows were detected relative to the number that converted successfully. P-values for permutations were derived empirically based on how many times the shuffled set of mouse windows resulted in more patient-variable regions than the actual regions from our three clusters.
Electrophoretic mobility shift assay (EMSA)
Complementary oligonucleotides (5′-CTTATATATGTAA[C/T]ATATG-3′) were designed around the variant rs185659576 (Integrated DNA Technologies, Coralville, IA). Total nuclear protein of human monocyte THP-1 cells was extracted using a NE-PER Extraction Kit (Thermo Scientific, Waltham, MA), and the protein concentration was measured with the BCA Protein Assay (Thermo Scientific). The LightShift Chemiluminescent EMSA Kit (Thermo Scientific) was used following the manufacturer’s protocol, and all incubations were carried out at room temperature. The biotin-labeled probe-protein binding reaction was incubated for 25 min and contained: 1× binding buffer, 1 μg poly(dI-dC), 5 μg nuclear extract, and 200 fmol 5′-biotin-labeled double-stranded oligonucleotide probe in a final 20 μL reaction volume. The unlabeled probe-protein competition reaction contained 100-fold excess of unlabeled probe and was incubated with the nuclear extract for 15 minutes prior to adding the biotin-labeled probe and incubating for another 25 minutes. The DNA-protein complexes were resolved using 6% DNA retardation electrophoresis gels (Life Technologies, Carlsbad, CA) in 0.5× TBE (Lonza, Basel, Switzerland). The gels were transferred to Biodyne Nylon Transfer Membranes (Thermo Scientific). After UV crosslinking, chemiluminescence was used to visualize the protein-probe complexes.
Supplementary Material
Acknowledgments
We thank J. Rawls and J. Camp for advice and assistance with their published data from epithelial cells, as well as members of the Furey and Sheikh labs, D. McKay, and S. Pott for insightful comments. This work was supported by American Gastroenterological Association (AGA) Research Scholar Award (SZS), Broad Medical Research Program (SZS), Crohn’s and Colitis Foundation of America’s Career Development Award (SZS) and Microbiome Consortium (RBS), R01-ES024983 from NIEHS (MW, SZS and TSF), R01-DK072193 and R01-DK093757 from NIDDK (KLM), P01-DK094779 from NIDDK (RBS and SZS), P40-OD101995 from NIH (RBS), P30-DK034987 from NIDDK (RBS and SZS), and SHARE from the Helmsley Trust (RBS). The GEO accession number is GSE70517. The record is currently private, however reviewers can access the series at the following link: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=ohwxwuogtjafhmd&acc=GSE70517
Abbreviations
- CD
Crohn’s disease
- IBD
inflammatory bowel diseases
- LP
lamina propria
- FAIRE
Formaldehyde Assisted Isolation of Regulatory Elements
- EMSA
Electrophoretic Mobility Shift Assay
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare no commercial or financial conflict of interest.
References
- 1.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 2.Sheikh SZ, Plevy SE. The role of the macrophage in sentinel responses in intestinal immunity. Curr Opin Gastroenterol. 2010;26:578–582. doi: 10.1097/MOG.0b013e32833d4b71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kamada N, Hisamatsu T, Okamoto S, Chinen H, Kobayashi T, Sato T, Sakuraba A, Kitazume MT, Sugita A, Koganei K, Akagawa KS, Hibi T. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118:2269–2280. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, Dent AL, Tangirala RK, Evans RM. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev. 2010;24:2760–2765. doi: 10.1101/gad.1998010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghisletti S, Barozzi I, Mietton F, Polletti S, De Santa F, Venturini E, Gregory L, Lonie L, Chew A, Wei CL, Ragoussis J, Natoli G. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 6.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 7.Krause P, Morris V, Greenbaum JA, Park Y, Bjoerheden U, Mikulski Z, Muffley T, Shui JW, Kim G, Cheroutre H, Liu YC, Peters B, Kronenberg M, Murai M. IL-10-producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL-23 synthesis. Nat Commun. 2015;6:7055. doi: 10.1038/ncomms8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shouval DS, Biswas A, Goettel JA, McCann K, Conaway E, Redhu NS, Mascanfroni ID, Al Adham Z, Lavoie S, Ibourk M, Nguyen DD, Samsom JN, Escher JC, Somech R, Weiss B, Beier R, Conklin LS, Ebens CL, Santos FG, Ferreira AR, Sherlock M, Bhan AK, Muller W, Mora JR, Quintana FJ, Klein C, Muise AM, Horwitz BH, Snapper SB. Interleukin-10 receptor signaling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity. 2014;40:706–719. doi: 10.1016/j.immuni.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun. 1998;66:5224–5231. doi: 10.1128/iai.66.11.5224-5231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheikh SZ, Hegazi RA, Kobayashi T, Onyiah JC, Russo SM, Matsuoka K, Sepulveda AR, Li F, Otterbein LE, Plevy SE. An anti-inflammatory role for carbon monoxide and heme oxygenase-1 in chronic Th2-mediated murine colitis. J Immunol. 2011;186:5506–5513. doi: 10.4049/jimmunol.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheikh SZ, Matsuoka K, Kobayashi T, Li F, Rubinas T, Plevy SE. Cutting edge: IFN-gamma is a negative regulator of IL-23 in murine macrophages and experimental colitis. J Immunol. 2010;184:4069–4073. doi: 10.4049/jimmunol.0903600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, Mackey LC, Hansen JJ, Moeser AJ, Rawls JF, Borst LB, Otterbein LE, Plevy SE. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology. 2013;144:789–798. doi: 10.1053/j.gastro.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simon JM, Giresi PG, Davis IJ, Lieb JD. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nature protocols. 2012;7:256–267. doi: 10.1038/nprot.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi T, Matsuoka K, Sheikh SZ, Russo SM, Mishima Y, Collins C, deZoeten EF, Karp CL, Ting JP, Sartor RB, Plevy SE. IL-10 regulates Il12b expression via histone deacetylation: implications for intestinal macrophage homeostasis. J Immunol. 2012;189:1792–1799. doi: 10.4049/jimmunol.1200042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manrique SZ, Correa MA, Hoelzinger DB, Dominguez AL, Mirza N, Lin HH, Stein-Streilein J, Gordon S, Lustgarten J. Foxp3-positive macrophages display immunosuppressive properties and promote tumor growth. J Exp Med. 2011;208:1485–1499. doi: 10.1084/jem.20100730. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI, Mantovani A, Locati M. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012;119:411–421. doi: 10.1182/blood-2011-02-339911. [DOI] [PubMed] [Google Scholar]
- 19.Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253–2263. doi: 10.4049/jimmunol.169.5.2253. [DOI] [PubMed] [Google Scholar]
- 20.Yang S, Gao L, Lu F, Wang B, Gao F, Zhu G, Cai Z, Lai J, Yang Q. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J Neuroinflammation. 2015;12:33. doi: 10.1186/s12974-015-0258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laslo P, Spooner CJ, Warmflash A, Lancki DW, Lee HJ, Sciammas R, Gantner BN, Dinner AR, Singh H. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 22.Uno JK, Rao KN, Matsuoka K, Sheikh SZ, Kobayashi T, Li F, Steinbach EC, Sepulveda AR, Vanhaesebroeck B, Sartor RB, Plevy SE. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110delta. Gastroenterology. 2010;139:1642–1653. 1653 e 1641–1646. doi: 10.1053/j.gastro.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi T, Matsuoka K, Sheikh SZ, Elloumi HZ, Kamada N, Hisamatsu T, Hansen JJ, Doty KR, Pope SD, Smale ST, Hibi T, Rothman PB, Kashiwada M, Plevy SE. NFIL3 is a regulator of IL-12 p40 in macrophages and mucosal immunity. J Immunol. 2011;186:4649–4655. doi: 10.4049/jimmunol.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takada Y, Hisamatsu T, Kamada N, Kitazume MT, Honda H, Oshima Y, Saito R, Takayama T, Kobayashi T, Chinen H, Mikami Y, Kanai T, Okamoto S, Hibi T. Monocyte chemoattractant protein-1 contributes to gut homeostasis and intestinal inflammation by composition of IL-10-producing regulatory macrophage subset. J Immunol. 2010;184:2671–2676. doi: 10.4049/jimmunol.0804012. [DOI] [PubMed] [Google Scholar]
- 25.Camp JG, Frank CL, Lickwar CR, Guturu H, Rube T, Wenger AM, Chen J, Bejerano G, Crawford GE, Rawls JF. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 2014;24:1504–1516. doi: 10.1101/gr.165845.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D’Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, International IBDGC, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibata Y, Sheffield NC, Fedrigo O, Babbitt CC, Wortham M, Tewari AK, London D, Song L, Lee BK, Iyer VR, Parker SC, Margulies EH, Wray GA, Furey TS, Crawford GE. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012;8:e1002789. doi: 10.1371/journal.pgen.1002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berg DJ, Davidson N, Kuhn R, Muller W, Menon S, Holland G, Thompson-Snipes L, Leach MW, Rennick D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–1020. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schreiber S, Fedorak RN, Nielsen OH, Wild G, Williams CN, Nikolaus S, Jacyna M, Lashner BA, Gangl A, Rutgeerts P, Isaacs K, van Deventer SJ, Koningsberger JC, Cohard M, LeBeaut A, Hanauer SB. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology. 2000;119:1461–1472. doi: 10.1053/gast.2000.20196. [DOI] [PubMed] [Google Scholar]
- 30.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, Hatscher N, Pfeifer D, Sykora KW, Sauer M, Kreipe H, Lacher M, Nustede R, Woellner C, Baumann U, Salzer U, Koletzko S, Shah N, Segal AW, Sauerbrey A, Buderus S, Snapper SB, Grimbacher B, Klein C. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKay DJ, Lieb JD. A common set of DNA regulatory elements shapes Drosophila appendages. Dev Cell. 2013;27:306–318. doi: 10.1016/j.devcel.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, Moon RT, Stamatoyannopoulos J, Murry CE. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas S, Li XY, Sabo PJ, Sandstrom R, Thurman RE, Canfield TK, Giste E, Fisher W, Hammonds A, Celniker SE, Biggin MD, Stamatoyannopoulos JA. Dynamic reprogramming of chromatin accessibility during Drosophila embryo development. Genome Biol. 2011;12:R43. doi: 10.1186/gb-2011-12-5-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, Amit I. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanai H, Hanauer SB. Assessing response and loss of response to biological therapies in IBD. Am J Gastroenterol. 2011;106:685–698. doi: 10.1038/ajg.2011.103. [DOI] [PubMed] [Google Scholar]
- 38.Herfarth H, Scholmerich J. IL-10 therapy in Crohn’s disease: at the crossroads. Treatment of Crohn’s disease with the anti-inflammatory cytokine interleukin 10. Gut. 2002;50:146–147. doi: 10.1136/gut.50.2.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steinbach EC, Plevy SE. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm Bowel Dis. 2014;20:166–175. doi: 10.1097/MIB.0b013e3182a69dca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobayashi T, Steinbach EC, Russo SM, Matsuoka K, Nochi T, Maharshak N, Borst LB, Hostager B, Garcia-Martinez JV, Rothman PB, Kashiwada M, Sheikh SZ, Murray PJ, Plevy SE. NFIL3-deficient mice develop microbiota-dependent, IL-12/23-driven spontaneous colitis. J Immunol. 2014;192:1918–1927. doi: 10.4049/jimmunol.1301819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinbach EC, Gipson GR, Sheikh SZ. Induction of Murine Intestinal Inflammation by Adoptive Transfer of Effector CD4+CD45RBhigh T Cells into Immunodeficient Mice. J Vis Exp. 2015 doi: 10.3791/52533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carroll TS, Liang Z, Salama R, Stark R, de Santiago I. Impact of artifact removal on ChIP quality metrics in ChIP-seq and ChIP-exo data. Front Genet. 2014;5:75. doi: 10.3389/fgene.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lassmann T, Hayashizaki Y, Daub CO. TagDust-a program to eliminate artifacts from next generation sequencing data. Bioinformatics. 2009;25:2839–2840. doi: 10.1093/bioinformatics/btp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyle AP, Guinney J, Crawford GE, Furey TS. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, Li W. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 2013;23:341–351. doi: 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 50.Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell PJ, Stephens PJ, Pleasance ED, O’Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, Teague JW, Menzies A, Goodhead I, Turner DJ, Clee CM, Quail MA, Cox A, Brown C, Durbin R, Hurles ME, Edwards PA, Bignell GR, Stratton MR, Futreal PA. Bowtie: An ultrafast memory-efficient short read aligner Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu EY, Li M, Wang W, Li Y. MaCH-admix: genotype imputation for admixed populations. Genet Epidemiol. 2013;37:25–37. doi: 10.1002/gepi.21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.