

Abstract
Chemotherapy has been shown to enrich cancer stem cells in tumors. Recently, we demonstrated that administration of chemotherapy to human bladder cancer xenografts could trigger a wound-healing response that mobilizes quiescent tumor stem cells into active proliferation. This phenomenon leads to a loss of sensitivity to chemotherapy partly due to an increase in the number of tumor stem cells, which typically respond to chemotherapy-induced cell death less than more differentiated cells. Different bladder cancer xenografts, however, demonstrate differential sensitivities to chemotherapy, the basis of which is not understood. Using mathematical models, we show that characteristics of the tumor cell hierarchy can be crucial for determining the sensitivity of tumors to drug therapy, under the assumption that stem cell enrichment is the primary basis for drug resistance. Intriguingly, our model predicts a weaker response to therapy if there is negative feedback from differentiated tumor cells that inhibits the rate of tumor stem cell division. If this negative feedback is less pronounced, the treatment response is predicted to be enhanced. The reason is that negative feedback on the rate of tumor cell division promotes a permanent rise of the tumor stem cell population over time both in the absence of treatment, and even more so during drug therapy. Model application to data from chemotherapy-treated patient-derived xenografts indicates support for model predictions. These findings call for further research into feedback mechanisms that might remain active in cancers, and potentially highlight the presence of feedback as an indication to combine chemotherapy with approaches that limit the process of tumor stem cell enrichment.
Keywords: Mathematical models, mathematical oncology, drug resistance, tumor stem cells, feedback
Quick Guide to Equations and Assumptions
The central mathematical model tracks the following populations: Quiescent tumor stem cells, Q, actively proliferating tumor stem cells, S, transit amplifying tumor cells, T, differentiated tumor cells, D, and the concentration of wound-healing factors, W. This model therefore reflects the notion that a tumor is characterized by a basic cellular hierarchy that is similar to normal tissue. Based on previous work (1,2), the model is given by the following ordinary differential equations:
| (1) |
First, we describe the dynamics of tumor growth without treatment (Figure 1A). Actively cycling stem cells divide with a rate r1. With a probability p1, this is a self-renewal division, resulting in two stem cells, and with a probability (1−p1), this is a differentiating division, resulting in two transit amplifying cells. Although asymmetric stem cell division (resulting in one stem cell and one transit amplifying cell) is also well documented, explicitly introducing it in Eq. (1) leads to an equivalent mathematical formulation (see Supplementary Information in (1,2)).
Figure 1.
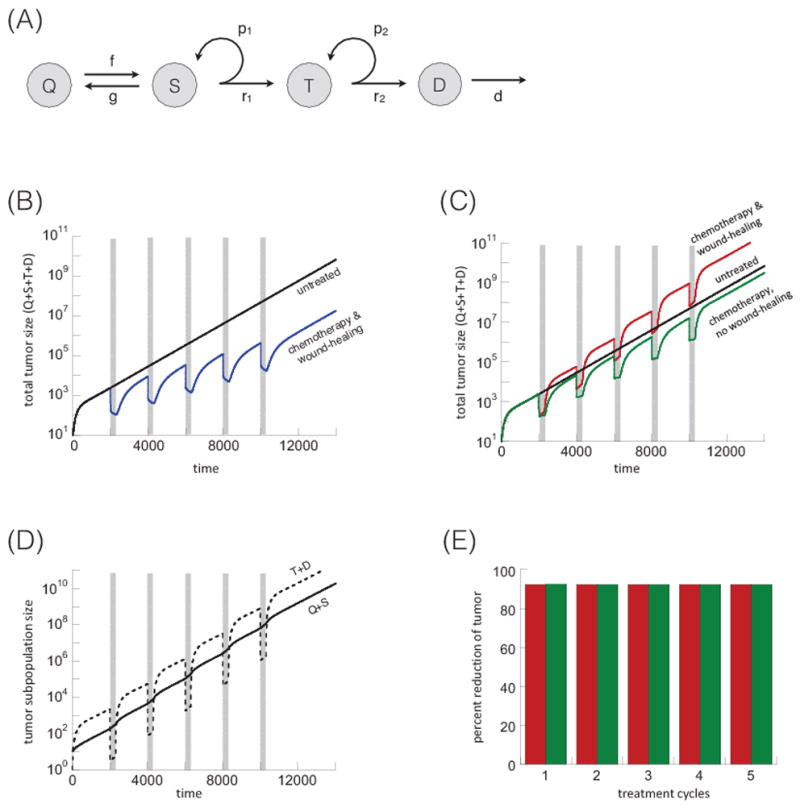
(A) Schematic representation of tumor dynamics without treatment (no negative feedback). Quiescent stem cells, Q, actively proliferating stem cells, S, transit amplifying cells, T, and differentiated cells, D. For explanation, see text. Treatment dynamics without negative feedback according to model (1). (B) Tumor treatment dynamics assuming that the treatment induced death rate of tumor stem cells is more pronounced that the treatment-induced stem cell expansion. Five treatment cycles are shown, indicated in grey. The black line is the untreated control, the blue line the chemotherapy-treated simulation. (C) Tumor treatment dynamics, assuming that the treatment-induced tumor stem cell expansion is more pronounced than the stem cell death rate during therapy. The black line shows the untreated control, the red line shows the chemotherapy-treated simulation, and the green line shows the same in the absence of a wound-healing response, corresponding to celecoxib administration. (D) Treatment dynamics showing the population of tumor stem cells (Q+S, solid line) and more differentiated cells (T+D, dashed line), based on the red line in panel C. (E) Percent of tumor reduction during each treatment cycle. This is shown for the simulations in panel C: in the presence (red) and absence (green) of the wound-healing response. Parameters were chosen as follows: r1=0.02; r2=0.05; p1=0.6; p2=0.45; δ=0.0025; f=0.005; g=0.001; α=1; ε=1; η=1; c1=1; c2=1. For (B) c3=0.02 and β=0.001. For (C–E): c3=0.002. For simulations with the wound-healing response, β=1. For simulations without a wound-healing response, β=0. Parameter values were chosen for the purpose of illustration, and time units are thus arbitrary. Timing and duration of treatment cycles was chosen to allow for the re-population of the more differentiated tumor cells from the tumor stem cells between treatments, which also applies to subsequent figures.
With rate f, active stem cells enter quiescence, and quiescent stem cells become activate with rate g. Transit amplifying cells divide with a rate r2, and they are also assumed to have a certain self-renewal capacity. That is, upon division, self-renewal occurs with a probability p2, and differentiated tumor cells are generated with a probability (1−p2). Differentiated cells are assumed to die naturally with a rate δ. If the tumor stem cell self-renewal probability is p1<0.5, then the tumor fails to grow and goes extinct. Hence, for a growing tumor we assume p1>0.5. Further, we assume that the transit amplifying cells alone cannot maintain the tumor. Therefore, 0<p2<0.5.
Now, consider chemotherapy phases. Therapy is assumed to kill differentiated, transit amplifying, and active tumor stem cells with rates c1, c2, and c3, respectively. In agreement with the notion that tumor stem cells are less sensitive to chemotherapy, we assume that c3≪c1, c2. The chemotherapy-induced death of differentiated and transit-amplifying cells triggers the release of wound-healing signals with rates αc1 and αc2. The wound-healing factors are assumed to decay with a rate η, and to trigger the activation of quiescent tumor stem cells with a rate βw/(w+ε). Hence, the rate of quiescent tumor stem cell activation is a saturating function of the amount of wound-healing signals available. This is biologically plausible, although data to formally derive a formulation are currently unavailable. For a schematic representation of these assumptions, see Figure S1.
Note that in computer simulations, parameter values were chosen arbitrarily for the purpose of illustration. The aim was to explore conditions under which the model can and cannot qualitatively account for the experimentally observed patterns, and not to fit the model to specific data sets, which would be premature, given the lack of appropriate data and parameter measurements. Illustrated results are not particular to the parameter values chosen, but arise from an analytical understanding of the type of tumor stem cell models considered (3).
1. Introduction
Treatment resistance, which is caused by multiple intrinsic and extrinsic factors, is a major obstacle to cancer therapy (4–6). With targeted therapies, mutations can emerge that prevent the binding of the drug to its target. Cells can resist chemotherapy by different mechanisms, including by reduced drug influx into cells, increased activity of efflux pumps, the activation of detoxifying proteins, the activation of DNA repair mechanisms, or defects in signaling pathways that respond to the damage induced by cytotoxic chemotherapies. Stem cells in particular, are thought to show significantly decreased sensitivity to both chemotherapy and targeted drugs (7–9).
We recently investigated drug resistance of bladder cancer to chemotherapy using patient-derived xenografts in immunocompromised mice (10,11). Experiments were performed in which standard-of-care chemotherapy (i.e. a number of successive treatment cycles) was administered to the xenografts, and the dynamics of tumor growth response was monitored over time. With successive cycles, it was observed that the response to treatment became less and less pronounced, partly due to an expansion of stem cells within the tumor cell population. It was demonstrated that the chemotherapy-induced programmed cell death of more differentiated cells could cause the activation of quiescent tumor stem cells and their proliferation (11). This phenomenon was termed “tumor stem cell repopulation”, which is characterized by molecular signatures reminiscent of a wound-healing response, driven by the release of prostaglandin E2 (PGE2) and activation of its upstream enzyme cyclooxygenase-2 (COX2). In support of this, administration of the COX2 inhibitor celecoxib antagonized the PGE2-mediated wound response, and improved continuous response to chemotherapy. The emergence of specific drug resistant mutants during the time course of these experiments was not investigated. Our additional data further noted that different patient-derived xenografts showed different degrees of sensitivity to chemotherapy. The reasons for this differential sensitivity are not understood.
These experiments led us to hypothesize that the cellular hierarchy of the tumor (12,13) might be an important factor determining tumor sensitivity to chemotherapy. Tumors are known to retain certain features of normal tissue cell hierarchy (14). This is supported by the notion that they are maintained by a subpopulation of tumor stem cells (15), and that the bulk of the tumor contains more differentiated cancer cells that by themselves cannot maintain or initiate a new tumor. Recent work further suggests that positive and negative feedback loops regulating normal tissue might remain active to a certain extent in tumors (13). The aforementioned wound-healing response (10) is an example of a positive feedback loop. In addition, it has been postulated that negative feedback loops that regulate normal stem cell division might remain active to some degree in tumors, and that this can impact the resulting tumor growth pattern, as well as the fraction of tumor stem cells (2,3). In turn, the fraction of tumor stem cells might be an important determinant of the sensitivity of a tumor to therapy due to their intrinsic resistance to cytotoxic therapies.
Here, we use mathematical models to investigate how aspects of the cellular hierarchy in tumors and feedback regulation can impact tumor sensitivity to chemotherapy, and relate our findings to previously published experimental data.
2. Materials and Methods
The mathematical and computational models are described in the Results section, and in the Quick Guide to Equations and Assumptions
We also present experimental data that show tumor growth and response to gemcitabine-cisplatin (GC) chemotherapy of bladder cancer patient-derived xenografts. These data were generated in the context of our previous study (10), where a detailed description of experimental design can be found. Patient-derived xenografts were established from human bladder cancer tissues as previously described (10,16,17). When xenograft tumors reached a palpable size, mice were randomized into different experimental groups: vehicle control (12.5% DMSO in saline) group; gemcitabine-cisplatin (GC) chemotherapy treatment group; Celecoxib treatment group; and Celecoxib+GC combinatory treatment group. Cisplatin (6 mg/kg) was applied after the first gemcitabine (40 mg/kg) treatment on day 2, followed by three consecutive treatments of gemcitabine on day 5, 8, and 11 (i.e. GC/G/G/G). In Celecoxib combination treatment experiments, mice were pretreated with Celecoxib (5 mg/kg; daily) via I.P. injection 2 days prior to the initiation of chemotherapy, followed by daily I.P. administration of Celecoxib during chemotherapy or vehicle treatment cycle.
3. Results
3.1. Mathematical model of the wound-healing response during chemotherapy
We present a mathematical model of stem-cell-driven tumor growth that takes into account both quiescent and activated tumor stem cells, and that describes the wound-healing response during chemotherapy. The mathematical model is described in detail in the Quick Guide to Equations and Assumptions, and schematically in Figure 1A. In this model, the tumor cell population converges to exponential growth in the absence of treatment (Figures 1B and 1C, black line). The following explores the tumor dynamics during chemotherapy cycles. Figure 1B plots total tumor size (Q+S+T+D) over the time course of five treatment cycles. The black line shows tumor growth without chemotherapy treatment, i.e. the control scenario. The blue line assumes chemotherapy treatment in the presence of a wound-healing response. For the treatment scenario, we first observe a fast reduction of the overall tumor cell population during treatment, resulting from the death of differentiated and transit amplifying tumor cells. This fast reduction phase is followed by a significant slow-down of the therapeutic response, because tumor stem cells die with a lower rate during therapy, and because the wound-healing response drives quiescent stem cells into proliferation counteracting the effect of cell death. After each treatment cycle, tumor load remains significantly lower compared to the untreated simulation. The reason is that although chemotherapy results in a wound-healing response and tumor stem cell repopulation, this is more than canceled out by therapy-induced tumor stem cell death. If we assume that treatment-induced stem cell death is a less dominant force compared to tumor stem cell repopulation, then a different kind of dynamics occur (Figure 1C, red line). Now, tumor size can reach higher levels following treatment, compared to the untreated simulation, because the wound-healing response induces a faster expansion of the cancer stem cell pool than occurs during natural growth. This is reflected by an accelerated phase of tumor expansion following the initial decline during treatment (Figure 1C red line). This effect is prevented in simulations where the wound-healing response is disabled (β=0, Figure 1C green line), which models celecoxib administration in (10).
Figure 1D plots the subpopulations of stem cells (Q+S) and non stem cells (T+D) for the simulation depicted by the red line in Figure 1C (other simulations exhibit similar patterns). During treatment, the tumor stem cell population becomes dominant. However when treatment stops, the proportion of tumor stem cells re-equilibrates to pre-treatment levels (for a mathematical motivation of these dynamics see the Supplementary Materials). Thus, in the long run, chemotherapy does not alter the relative fraction of the tumor stem cell population. As a consequence, the percent of the tumor that is removed during each treatment cycle remains constant (Figure 1E). In other words, with each treatment cycle, the degree to which the tumor responds to chemotherapy remains unchanged.
3.2. Adding negative feedback regulation to the model
Tumors are thought to retain certain features of normal tissue cell hierarchies, including the presence of regulatory feedback loops between tumor cells at various differentiation stages (2). The wound-healing response is an example of a positive feedback (10). Also, differentiated cells exert negative feedback on the stem cell division rate in healthy tissue. Such regulatory mechanisms are likely retained in some cancers, and this might be required to account for certain tumor growth patterns found in the literature (2,18).
To incorporate negative feedback we assume that differentiated tumor cells release factors that inhibit the division rate of tumor stem cells and transit amplifying cells (1,19). The model is again given by equation (1), but in addition, we assume that the division rate of tumor stem cells and transit amplifying cells is a decreasing function of the number of differentiated cells. More precisely, , where r1(0) and r2(0) are the division rates without negative feedback and the parameters k and h control the strength of the inhibitory signals (for a schematic representation see Figure 2A). The modeling of the division rates using Hill equations builds on previous work on feedback regulation by others and us (1–3,19–23).
Figure 2.

(A) Schematic representation of tumor dynamics (with negative feedback). The basic dynamics are the same as in Figure 1A. In addition, we model negative feedback from differentiated cells on the cell division rates of tumor stem cells and transit amplifying cells. (B) Negative feedback and its effect on tumor growth. The black line depicts tumor growth without of negative feedback, simulated by model (1). The green and red lines show tumor growth dynamics in the presence of weak and strong feedback, respectively. (C) When there is negative feedback, the stem cell fraction increases with tumor size (blue lines; Q+S dashed line, T+D solid line). When there is no feedback the stem cell fraction remains constant (black lines). Parameters were chosen as follows. In panel (A): r1=0.02; r2=0.05; p1=0.7; p2=0.45; δ=0.00025; f=0.005; g=0.001; α=1; ε=1; ε=1; h=0.01; β=c1=c2=c3=0; for weak negative feedback, k=0.2; for strong negative feedback, k=1. For absence of feedback, h=0. For panel (C): k=0.3 and p=0.6 (all other parameters are the same as in (A); h=0 for no feedback).
Note that while it is assumed that the level of negative feedback factors produced by the differentiated cells is simply proportional to the number of differentiated cells (they are in a quasi-steady state), the concentration of wound-healing factors is explicitly taken into account in the model (variable W). Negative feedback factors are secreted while cells are alive, and their main function in healthy tissue is to promote homeostasis (1,2). For this reason, their concentration should be roughly proportional to the number of differentiated cells present in the system, justifying the quasi-steady state assumption (1,19). In contrast, the wound-healing signals are released upon chemotherapy-induced cell death. Therefore, to be effective, the wound-healing factors must persist for a time period after the cell has died, which is why they are explicitly described.
Tumor growth with this type of negative feedback was previously examined with a simpler model that only distinguished between tumor stem cells and differentiated cells (2,3). It was found that the level of feedback can fundamentally impact the tumor growth law. Without negative feedback, the tumor grows exponentially, and the fraction of tumor stem cells converges to a constant over time. This growth pattern was termed “uninhibited tumor growth”. When inhibition is present, however, the tumor grows at a slower sub-exponential rate, resulting in a pattern called “inhibited tumor growth” (2). With inhibited growth, it was also found that stem and differentiated cells grow at different rates: tumor stem cells grow asymptotically as t1+1/k, and differentiated cells as t1/k (3). Thus, with negative feedback, tumor growth over time follows a power law, and the growth rate is determined by the strength of the feedback signal, k. Hence, with negative feedback the tumor stem cell population grows at a faster rate than the differentiated cell population, and this leads to a steady increase in the fraction of the tumor stem cell over time. The intuitive reason for the faster growth of the tumor stem cell population is as follows. As the tumor becomes larger, so does the population of differentiated cells. This leads to stronger inhibition of the rate of stem cell division, which in turn reduces the rate at which both stem cells and differentiated cells are generated. While the rate at which differentiated cells are generated slows down as the tumor grows, the death rate of differentiated cells is assumed to remain unchanged in the model. At the same time, it is assumed that tumor stem cells do not experience a significant rate of death. In this setting, the overall growth rate of the differentiated cell population slows down more over time than that of tumor stem cells.
These kinds of dynamics are also seen in our current feedback model. Negative feedback results in sub-exponential, “inhibited”, growth, with stronger feedback leading to slower growth (Figure 2B). When negative feedback is present, the fraction of tumor stem cell increases with tumor size (Figure 2C, blue lines). In contrast, when there is no negative feedback the fraction of stem cells remains constant (Figure 2C, black lines). With feedback both stem and transit amplifying cells grow asymptotically as t1+1/k, while differentiated tumor cells grow as t1/k. Hence, in the long-term, both stem cells and transient amplifying cells grow faster than the differentiated cell population, and the fractions of stem cells and transit amplifying cells converge toward positive numbers (not shown).
Figure 3A shows the effect of chemotherapy in the model with negative feedback. The blue line simulates treatment with the wound-healing response, the black line the untreated tumor. During treatment, the death of differentiated and transit-amplifying cells first results in a fast phase of tumor decline. When most of the transit amplifying and differentiated cells are gone, the continued reduction in tumor size is then driven by stem cell death, which occurs at a much slower rate, producing a slow-down in tumor decline. This slow-down is also caused by the wound-healing response, and by the release from negative feedback on cell division, resulting from the disappearance of the differentiated cells that emit the feedback factors. Both processes contribute to tumor stem cell repopulation. This simulation assumes that treatment-induced stem cell death is dominant over stem cell repopulation. Hence, when treatment stops, the treated tumor size remains lower than the size of the untreated tumor (Figure 3A). This is not the case if we assume that the process of stem cell repopulation is dominant over treatment-induced stem cell death. In this case, tumor stem cell repopulation causes a pronounced expansion in the number of stem cells during treatment (Figure 3C), which can result in the treated tumor size becoming larger than the untreated control (Figure 3B, red line)
Figure 3.

Treatment dynamics in the presence of negative feedback on cell division in model (2). (A) Tumor treatment dynamics assuming that the treatment induced death rate of tumor stem cells is more pronounced that the treatment-induced stem cell expansion. Five treatment cycles are shown, indicated in grey. The black line is the untreated control, the blue line the chemotherapy-treated simulation. (B) Tumor treatment dynamics, assuming that the treatment-induced tumor stem cell expansion is more pronounced than the stem cell death rate during therapy. The black line shows the untreated control, the red line shows the chemotherapy-treated simulation, and the green line shows the same in the absence of a wound-healing response, corresponding to celecoxib administration. (C) Treatment dynamics showing the population of tumor stem cells (Q+S, solid line) and more differentiated cells (T+D, dashed line), based on the red line in panel B. (D) Same as panel (B), but with weaker feedback inhibition. Only the untreated simulation and the chemotherapy simulation in the presence of a wound-healing response are shown. (E) Percent of tumor reduction during each treatment cycles. The color codes correspond to the simulations shown in the corresponding colors in panels (B) and (D). Parameters were chosen as follows: r1=0.02; r2=0.05; p1=0.7; p2=0.45; δ=0.0025; f=0.005; g=0.001; α=1; ε=1; η=1; h=0.01; c1=1; c2=1, For (A) c3=0.02, β=0.001, k=1. For (B–D): c3=0.002. For simulations with the wound-healing response, β=1. For simulations without a wound-healing response, β=0. For stronger feedback, k=1; for weaker feedback, k=0.2.
When negative feedback is present, the fraction of tumor stem cells does not re-equilibrate to pre-treatment levels. Instead it remains elevated in the long term, and this occurs after each treatment cycle (Figure 3C). This is not a feature of the particular parameter set used in this simulation, but arises from the properties of inhibited growth outlined above and worked out in (3). As a consequence the percent of the tumor that is removed with each new treatment cycle decreases (Figure 3E). Hence, with each treatment cycle, the response to chemotherapy is diminished.
Next, assume that the wound-healing response is absent during chemotherapy (Figure 3B, green line). In this case, the increase in tumor load relative to the untreated simulation is less pronounced. Also, the loss of the tumor response with each treatment cycle is smaller, i.e. the percent of tumor killing declines less over successive treatment cycles (Figure 3E, green bars). It is noteworthy that without negative feedback, the wound-healing response did not change the percentage of the tumor killed by each treatment cycle (Figure 1E; red vs. green). But, when negative feedback is present, the wound-healing response contributes markedly to a loss of response over successive rounds of treatment [Figure 3E; compare red (with wound-healing response) with green bars (no wound-healing response)]. The reason is that with negative feedback, the increase in the fraction of stem cells that occurs with each treatment cycle and that is promoted by the wound-healing response is not fully reversed once the cycle ends; as a consequence, after each cycle the fraction of tumor stem cells remains permanently elevated (Figure 3C; solid black line). This trend is not observed in the model without negative feedback, where the fraction of tumor stem cells is predicted to re-equilibrate following a disturbance brought about by the wound-healing response (compare Figures 1D and 3C; solid black line). This is again not dependent on the particular parameters chosen in the simulations (see Supplementary Materials). Thus, in the model, negative feedback and the wound-healing response act synergistically to reduce the effect of chemotherapy with each treatment cycle.
Finally, consider the treatment response when the strength of negative feedback on the rate of stem cell division is lower (Figure 3D, characterized by k=0.2 rather than k=1). This simulation includes the wound-healing response, and is depicted by the beige curve. We observe similar dynamics, although the overall tumor growth rate is faster, both with and without chemotherapy, due to reduced feedback. It is, however, interesting to look at the percent of tumor reduction for each treatment cycle, shown by beige bars in Figure 3E. Note that compared to the simulations with strong feedback inhibition (red and green bars), the simulation with weaker negative feedback (beige bar) results in a better response to chemotherapy even in the first treatment cycle. Similarly, the decline in the treatment response with each chemotherapy cycle is much less pronounced for weaker feedback inhibition (Figure 3E). In sum the presence of negative feedback correlates with slower tumor growth and reduced sensitivity to chemotherapy.
3.3. Spatial tumor growth models
The models considered so far do not take into account space (24,25). Therefore, we now consider a spatially stochastic agent-based model, based on reference (26). We assume that cells can occupy any site of a 3-dimensional rectangular lattice, and that each lattice site can host at most one cell at a time (Figure 4A). For a cell to divide, there must be a free lattice point adjacent to it to place one of the two daughter cells produced during cell division. We use a stochastic simulation algorithm, where the probabilities of cell division, self-renewal, differentiation and death correspond to our previous non-spatial models.
Figure 4.

Spatial dynamics. (A) Three dimensional representation of a tumor. (B) Cross section of a tumor 3D tumor. A large number of stem cells (blue and red) are “trapped” in the tumor mass where they are unable to divide. (C) A tumor during treatment. The killing of transit and differentiated cells frees up space, which allows formerly trapped stem cells to divide. (D) Tumor dynamics during three treatment cycles, indicated in grey. Red: intact wound-healing response. Green: No wound-healing response. Black: No treatment. (See Figure S2 for simulations where the treated tumor remains consistently smaller than the untreated tumor.) (E) Percent of tumor reduction during the three treatment cycles. (F) Fraction of stem cells in the tumor population (Q+S)/(Q+S+T+D) for the treated tumor with wound-healing response. Parameters were chosen as follows: r1=r2=10; p1=0.55; p2=0.45; δ=0.00025; f=0.1; g=0.01; α=1; ε=1; η=0.02; h=2; β=0.5; c3=0.001. Panels A–C (weak feedback): c1=c2=20, k=0.2. Panels D–F (strong feedback): c1=c2=0.1, k=1.
The conclusions remain robust in the spatial model. If stem cell repopulation during therapy is dominant over stem cell death, then after multiple treatment cycles the tumor load can be higher compared to the untreated simulation (Figure 4D). Conversely, if stem cell death is dominant over stem cell repopulation, post-therapy tumor sizes remain smaller than those that occur without treatment (Figure S2B; Supplementary Materials). As before, when negative feedback is present, the fraction of stem cells remains elevated after each round of chemotherapy (Figure 4F). As a consequence the percent reduction of tumor decreases with each new treatment cycle (Figure 4E). This effect is more pronounced when the wound-healing response is also present. Tumor dynamics for weak and non-existent negative feedback are discussed in the Supplementary Materials (Figure S2).
The spatial model identifies an additional mechanism that can contribute to the rise in the stem cell fraction during chemotherapy. In a solid tumor a number of stem cells are trapped in the tissue mass where they are unable to divide and cannot contribute to growth (Figure 4B). When treatment is administered, the killing of transit amplifying and differentiated cells frees up space, which allows these formerly trapped stem cells to divide (Figure 4C). This type of dynamics has been described before (27,28). Furthermore, by targeting transit amplifying and differentiated cells preferentially, treatment actively selects for self-renewing stem cells. Hence, during treatment the number and spatial distribution of active (diving) stem cells increases. When treatment stops, the greater number and wider spatial distribution of active stem cells becomes an additional driver of fast tumor regrowth. Thus, in a spatial setting a treatment that largely spares cancer stem cells can have a pruning effect on the tumor that ultimately promotes greater tumor repopulation.
3.4. Application of models to experimental data from bladder cancer patient-derived xenografts
To explore whether there is support for our theory in data, we consider experiments in which bladder cancer patient-derived xenografts were treated with chemotherapy (10). Different xenograft lines displayed different degrees of sensitivity to treatment, and were classified as “resistant”, “intermediate”, and “sensitive” according to qualitative assessment of treatment responses (Figure 5). For the sensitive tumor (Figure 5A), two treatment cycles are shown, where tumor size was reduced substantially by 83% and 73% respectively. For the intermediate xenograft (Figure 5B), the percent of the tumor killed by the drug was 59% and 53% in the first two treatment cycles, but dropped to 33% in the third treatment cycle (tumor sizes were not followed for long enough after the fourth treatment cycle to determine the extent of the response). For the resistant xenograft line (Figure 5C) the responses in the first two treatment cycles were even lower (12% and 23% respectively), and declined even further during treatment cycles 3 and 4 (3.7% and 3.1%).
Figure 5.

Experimental data documenting treatment dynamics of patient-derived bladder cancer mouse xenografts (A–C), and the dynamics of tumor growth without treatment (D). The tumors were treated with the chemotherapy regime gemcitabine and cisplatin (GC), treatment cycles are indicated in grey. Xenografts from three patient-derived cell lines are shown and designated as “sensitive”, “intermediate”, and resistant, based on qualitative observations. The “resistant” cell line data are re-plotted from Kurtova et al. (10), the data for the “intermediate” and “sensitive” xenografts were collected in the context of the same study but have not been previously published. Details of the experimental designs and the number of repeats are given in Kurtova et al. (10).
Our models predict that increased drug resistance can be explained by the presence of feedback inhibition on the rate of tumor stem cell division. This in turn should be reflected in the corresponding tumor growth patterns without treatment (Figure 5D). According to the model, when negative feedback is present, tumor growth should follow a power law, and this assumption (a power law for tumor growth) fits all xenograft data better than an exponential law (indicated by higher coefficients of determination for power than exponential models, containing the same number of parameters). Using regression procedures, the estimated powers were found to be 3.821, 2.797, and 2.768 for the sensitive, intermediate, and resistant xenograft lines. According to t-tests, the sensitive xenograft has a significantly larger power than the intermediate (p=0.00015) or the resistant (p=0.017) xenograft. If the bulk of the tumor is made up of differentiated cells, then, according to theory, the power law for tumor growth should in the long term be approximately given by 1/k (3), and it is thus directly related to the strength of negative feedback on cell division. It is unclear, however, whether the tumor has grown long enough to reach this asymptotic behavior, and hence it is uncertain whether the estimated powers in the data can be used to establish the value of k. Nevertheless, it is likely that a smaller power indicates the presence of stronger negative feedback. This suggests that the intermediate and resistant cell lines might be characterized by significantly stronger negative feedback on cell division than the sensitive cell line, which would be in accordance with our theory. The resistant and intermediate xenografts display not only an overall heightened resistance, but also a reduction in the percent of the tumor killed with each treatment cycle, as predicted by the model. In contrast, the sensitive xenograft, which might be characterized by weaker feedback on cell division, displays the highest sensitivity to chemotherapy. Whether the percentage of the tumor killed remains robust with repeated treatment cycles is currently unclear because only two treatment cycles were performed for this xenograft.
We note that the resistant xenograft shows a lower treatment response than the intermediate one although the estimated powers of tumor growth are not statistically different. This might appear contradictory. Treatment, however, was initiated at a higher tumor size in the resistant compared to the intermediate xenograft. When negative feedback is present, the stem cell fraction is predicted to be higher for larger tumor sizes (3), which might explain the observed reduced treatment response in the resistant xenograft.
While the discussed experimental data contain signatures that support our theory, this is by no means conclusive because these experiments were performed in the context of a previous study and not with the aim to test the theory presented here. Extensive, more detailed experiments are required that are specifically designed for theory testing and for estimation of crucial parameters, which is subject of future work. The value of our current results is the generation of novel insights about the dynamics of stem cell-based chemotherapy resistance that is supported by limited, currently available data. This provides the basis upon which to build future work, which would not be available without the modeling results presented here.
An uncertain aspect of the model concerns the long-term effect of therapy. If the rate of stem cell death during treatment was larger than the rate of treatment-induced stem cell expansion/repopulation, the treated tumor always remained smaller than the untreated control simulation. In contrast, if the rate of treatment-induced stem cell expansion/repopulation was more pronounced than the rate of stem cell death, treatment cycles could lead to post-therapy tumor sizes that are bigger than those occurring without treatment. This makes intuitive sense: If treatment enriches the stem cell population, and if stem cells maintain the tumor bulk, then an enriched stem cell pool after therapy gives rise to an overall larger tumor. This, however, has not been observed in the bladder cancer xenograft data. Additional experiments and parameter estimates are required to investigate whether the bladder cancer system is characterized by parameters that do not allow for this outcome, or whether it is a possibility. Interestingly, experiments with ovarian cancer mouse xenografts (29) have shown that chemotherapy (cisplatin and paclitaxel) led to the enrichment of cells with tumor stem cell markers, and that this resulted in significantly higher tumor burdens during subsequent growth, compared to untreated xenografts, thus supporting the biological relevance of this particular model prediction.
4. Discussion and Conclusion
The mathematical models identified the presence of negative feedback loops in tumors as crucial determinants of tumor sensitivity to chemotherapy in the context of stem-cell-based resistance. When negative feedback is absent, the enrichment of tumor stem cells during therapy was predicted to be only temporary, reverting back to pre-treatment levels upon treatment cessation. As a consequence, treatment is unaltered during successive therapy cycles. In contrast, when negative feedback is present, the increase in the fraction of tumor stem cells that occurs during a therapy round is never fully reversed. This in turn leads to a loss of chemotherapeutic response over repeated treatment cycles.
In normal tissue, feedback loops play a fundamental role in promoting homeostasis and the rapid regeneration after an injury (1,30,31). In specific cancers some of these feedback loops are lost (32,33), while others are fully or partially maintained (e.g. the wound-healing response (10)). No studies have been conducted that systematically characterize the negative feedback loops still present in specific cancers, although it has been suggested that negative feedback has to be invoked to explain certain experimentally observed tumor growth patterns (18). The results reported here further emphasize the need to investigate which aspects of healthy tissue cell hierarchies remain active in tumors (23), and how this relates to treatment outcome. Not all feedback mechanisms, however, enable permanent shifts in tumor stem cell fractions after therapy, and further mathematical work should take a more comprehensive approach (34) to define possible feedback patterns that can and cannot induce this effect.
While our results indicate that chemotherapy resistance phenotype could be associated with the presence of feedback, this is not meant to imply that the feedback mechanisms should be eliminated to increase sensitivity to drugs. Indeed, this would be counter-productive. This finding simply indicates that tumor stem-cell-based resistance might be a more significant problem in relatively slowler growing tumors, characterized by the existence of negative feedback, rather than in faster growing tumors that lack negative feedback inhibition. If confirmed, a test for the presence of this kind of negative feedback could indicate whether or not therapeutic strategies should be considered that limit tumor stem cell expansion during chemotherapy (such as celecoxib in bladder cancer). It further highlights the fact that slower growing tumors not necessarily correlate with better prognosis and that specific treatment regimens may need to be identified to target them differently.
Finally, as with all studies based on mathematical modeling, it must be remembered that conclusions depend on assumptions, and that assumptions are always characterized by a degree of uncertainty. We have, however, shown that conclusions remain robust both in the context of ordinary differential equation models (which do not take into account space), and in spatially explicit stochastic 3D models of tumor growth. Therefore, the results are sufficiently convincing to justify further experimental work to investigate these dynamics and feedback mechanisms in more detail.
Major Findings.
We show that characteristics of the tumor cell hierarchy can be crucial for determining the sensitivity of tumors to drug therapy, assuming that stem cell enrichment is the primary basis for drug resistance.
Our model predicts a weaker response to therapy if there is negative feedback from differentiated tumor cells that inhibits the rate of tumor stem cell division. If this negative feedback is less pronounced, the treatment response is predicted to be enhanced.
Preliminary model application to existing data from chemotherapy-treated patient-derived xenografts indicates support for model predictions, and defines what needs to be measured in future work to test these predictions further.
If confirmed, a test for the presence of this kind of negative feedback could indicate whether or not therapeutic strategies should be considered that limit tumor stem cell expansion during chemotherapy.
Acknowledgments
Financial support: This work was funded in part by NIH grant U01 CA187956. This grant was awarded to Dominik Wodarz, and Ignacio Rodriguez-Brenes receives funds from it.
Footnotes
Conflict of Interest Statement: None of the authors has a conflict of interest.
References
- 1.Lander AD, Gokoffski KK, Wan FY, Nie Q, Calof AL. Cell lineages and the logic of proliferative control. PLoS Biol. 2009;7:e15. doi: 10.1371/journal.pbio.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez-Brenes IA, Komarova NL, Wodarz D. Evolutionary dynamics of feedback escape and the development of stem-cell-driven cancers. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18983–8. doi: 10.1073/pnas.1107621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez-Brenes IA, Wodarz D, Komarova NL. Characterizing inhibited tumor growth in stem-cell-driven non-spatial cancers. Mathematical biosciences. 2015;270:135–41. doi: 10.1016/j.mbs.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Gottesman MM. Mechanisms of cancer drug resistance. Annual review of medicine. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 5.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 6.Druker BJ. Overcoming resistance to imatinib by combining targeted agents. Mol Cancer Ther. 2003;2:225–6. [PubMed] [Google Scholar]
- 7.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 8.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 9.Abdullah LN, Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clinical and translational medicine. 2013;2:3. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015;517:209–13. doi: 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan KS. Molecular Pathways: Targeting Cancer Stem Cells Awakened by Chemotherapy to Abrogate Tumor Repopulation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22:802–6. doi: 10.1158/1078-0432.CCR-15-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egeblad M, Nakasone ES, Werb Z. Tumors as Organs: Complex Tissues that Interface with the Entire Organism. Dev Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Werner B, Scott JG, Sottoriva A, Anderson AR, Traulsen A, Altrock PM. The Cancer Stem Cell Fraction in Hierarchically Organized Tumors Can Be Estimated Using Mathematical Modeling and Patient-Specific Treatment Trajectories. Cancer research. 2016;76:1705–13. doi: 10.1158/0008-5472.CAN-15-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ho PL, Kurtova A, Chan KS. Normal and neoplastic urothelial stem cells: getting to the root of the problem. Nature reviews Urology. 2012;9:583–94. doi: 10.1038/nrurol.2012.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell stem cell. 2012;10:717–28. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14016–21. doi: 10.1073/pnas.0906549106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Volkmer JP, Sahoo D, Chin RK, Ho PL, Tang C, Kurtova AV, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2078–83. doi: 10.1073/pnas.1120605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez-Brenes IA, Komarova NL, Wodarz D. Tumor growth dynamics: insights into evolutionary processes. Trends in ecology & evolution. 2013;28:597–604. doi: 10.1016/j.tree.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Lo WC, Chou CS, Gokoffski KK, Wan FY, Lander AD, Calof AL, et al. Feedback regulation in multistage cell lineages. Math Biosci Eng. 2009;6:59–82. doi: 10.3934/mbe.2009.6.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marciniak-Czochra A, Stiehl T, Ho AD, Jager W, Wagner W. Modeling of asymmetric cell division in hematopoietic stem cells--regulation of self-renewal is essential for efficient repopulation. Stem cells and development. 2009;18:377–85. doi: 10.1089/scd.2008.0143. [DOI] [PubMed] [Google Scholar]
- 21.Bocharov G, Quiel J, Luzyanina T, Alon H, Chiglintsev E, Chereshnev V, et al. Feedback regulation of proliferation vs. differentiation rates explains the dependence of CD4 T-cell expansion on precursor number. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:3318–23. doi: 10.1073/pnas.1019706108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Lander AD, Nie Q. A reaction-diffusion mechanism influences cell lineage progression as a basis for formation, regeneration, and stability of intestinal crypts. BMC systems biology. 2012;6:93. doi: 10.1186/1752-0509-6-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Konstorum A, Hillen T, Lowengrub J. Feedback Regulation in a Cancer Stem Cell Model can Cause an Allee Effect. Bulletin of mathematical biology. 2016;78:754–85. doi: 10.1007/s11538-016-0161-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anderson AR, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. 2006;127:905–15. doi: 10.1016/j.cell.2006.09.042. [DOI] [PubMed] [Google Scholar]
- 25.Korolev KS, Xavier JB, Gore J. Turning ecology and evolution against cancer. Nature reviews Cancer. 2014;14:371–80. doi: 10.1038/nrc3712. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez-Brenes IA, Wodarz D, Komarova NL. Stem cell control, oscillations, and tissue regeneration in spatial and non-spatial models. Frontiers in oncology. 2013;3:82. doi: 10.3389/fonc.2013.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enderling H, Anderson AR, Chaplain MA, Beheshti A, Hlatky L, Hahnfeldt P. Paradoxical dependencies of tumor dormancy and progression on basic cell kinetics. Cancer research. 2009;69:8814–21. doi: 10.1158/0008-5472.CAN-09-2115. [DOI] [PubMed] [Google Scholar]
- 28.Hillen T, Enderling H, Hahnfeldt P. The tumor growth paradox and immune system-mediated selection for cancer stem cells. Bulletin of mathematical biology. 2013;75:161–84. doi: 10.1007/s11538-012-9798-x. [DOI] [PubMed] [Google Scholar]
- 29.Abubaker K, Latifi A, Luwor R, Nazaretian S, Zhu H, Quinn MA, et al. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Molecular cancer. 2013;12:24. doi: 10.1186/1476-4598-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 31.Daluiski A, Engstrand T, Bahamonde ME, Gamer LW, Agius E, Stevenson SL, et al. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat Genet. 2001;27:84–8. doi: 10.1038/83810. [DOI] [PubMed] [Google Scholar]
- 32.Lee J, Son MJ, Woolard K, Donin NM, Li A, Cheng CH, et al. Epigenetic-mediated dysfunction of the bone morphogenetic protein pathway inhibits differentiation of glioblastoma-initiating cells. Cancer cell. 2008;13:69–80. doi: 10.1016/j.ccr.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 34.Sun Z, Komarova NL. Stochastic control of proliferation and differentiation in stem cell dynamics. Journal of mathematical biology. 2015;71:883–901. doi: 10.1007/s00285-014-0835-2. [DOI] [PubMed] [Google Scholar]