

Abstract
AIM
To study the molecular mechanisms involved in the hepatoprotective effects of naringenin (NAR) on carbon tetrachloride (CCl4)-induced liver fibrosis.
METHODS
Thirty-two male Wistar rats (120-150 g) were randomly divided into four groups: (1) a control group (n = 8) that received 0.7% carboxy methyl-cellulose (NAR vehicle) 1 mL/daily p.o.; (2) a CCl4 group (n = 8) that received 400 mg of CCl4/kg body weight i.p. 3 times a week for 8 wk; (3) a CCl4 + NAR (n = 8) group that received 400 mg of CCl4/kg body weight i.p. 3 times a week for 8 wk and 100 mg of NAR/kg body weight daily for 8 wk p.o.; and (4) an NAR group (n = 8) that received 100 mg of NAR/kg body weight daily for 8 wk p.o. After the experimental period, animals were sacrificed under ketamine and xylazine anesthesia. Liver damage markers such as alanine aminotransferase (ALT), alkaline phosphatase (AP), γ-glutamyl transpeptidase (γ-GTP), reduced glutathione (GSH), glycogen content, lipid peroxidation (LPO) and collagen content were measured. The enzymatic activity of glutathione peroxidase (GPx) was assessed. Liver histopathology was performed utilizing Masson’s trichrome and hematoxylin-eosin stains. Zymography assays for MMP-9 and MMP-2 were carried out. Hepatic TGF-β, α-SMA, CTGF, Col-I, MMP-13, NF-κB, IL-1, IL-10, Smad7, Smad3, pSmad3 and pJNK proteins were detected via western blot.
RESULTS
NAR administration prevented increases in ALT, AP, γ-GTP, and GPx enzymatic activity; depletion of GSH and glycogen; and increases in LPO and collagen produced by chronic CCl4 intoxication (P < 0.05). Liver histopathology showed a decrease in collagen deposition when rats received NAR in addition to CCl4. Although zymography assays showed that CCl4 produced an increase in MMP-9 and MMP-2 gelatinase activity; interestingly, NAR administration was associated with normal MMP-9 and MMP-2 activity (P < 0.05). The anti-inflammatory, antinecrotic and antifibrotic effects of NAR may be attributed to its ability to prevent NF-κB activation and the subsequent production of IL-1 and IL-10 (P < 0.05). NAR completely prevented the increase in TGF-β, α-SMA, CTGF, Col-1, and MMP-13 proteins compared with the CCl4-treated group (P < 0.05). NAR prevented Smad3 phosphorylation in the linker region by JNK since this flavonoid blocked this kinase (P < 0.05).
CONCLUSION
NAR prevents CCl4 induced liver inflammation, necrosis and fibrosis, due to its antioxidant capacity as a free radical inhibitor and by inhibiting the NF-κB, TGF-β-Smad3 and JNK-Smad3 pathways.
Keywords: Fibrosis, Transforming growth factor-β, Naringenin, pSmad3, Smad3, JNK, Nuclear factor kappa, Carbon tetrachloride
Core tip: To study the effect of naringenin (NAR) on carbon tetrachloride (CCl4)-induced chronic liver fibrosis, male Wistar rats were administered 400 mg of CCl4/kg body weight and 100 mg of NAR/kg body weight for 8 wk. NAR prevented necrosis, cholestasis, oxidative stress, collagen accumulation and the increase in MMP activity caused by CCl4 administration. NAR completely prevented the increase in TGF-β, α-SMA, CTGF, Col-1, MMP-13, NF-κB, IL-1 and IL-10 protein levels caused by CCl4 administration and pSmad3 and pJNK activation. In conclusion, NAR prevents CCl4 induced liver fibrosis due to its antioxidant capacity and by inhibiting the TGF-β-Smad3, JNK-Smad3 and NF-κB pathways.
INTRODUCTION
It is known that various agents, including infections, ethanol ingestion, drug intoxication or malnutrition, result in liver damage. When the damage is prolonged, liver fibrosis occurs. Fibrosis is the result of excessive accumulation of extracellular matrix (ECM) proteins after chronic liver damage. Advanced liver fibrosis leads to cirrhosis, which is characterized by scar tissue, loss of parenchymal architecture and organ failure[1,2].
The most important profibrogenic pathways involve transforming growth factor (TGF)-β and plateled-derived growth factor (PDGF), which induce the transdifferentiation of hepatic stellate cells (HSCs) from a non-proliferating cell type into a proliferating activated phenotype. This activation results in α-smooth-muscle actin (α-SMA) and collagen expression, which in turn lead to fibrosis[3,4].
Traditionally, TGF-β has been considered to exert profibrogenic actions by binding to its receptor, which in turn phosphorylates transcription factors called R-Smads (Smad2 and Smad3) at its C-terminal. This is the canonical pathway, generally described as the main fibrogenic pathway, especially when Smad3 is phosphorylated at its C-terminus (pSmad3C)[5-8]. There is also a non-canonical pathway, in which PDGF activates JNK, which in turn phosphorylates Smad3 in the linker domain to generate pSmad3L, which rapidly translocates to the nucleus where it stimulates HSC proliferation[9,10]. These pathways are critical for the induction of ECM deposition and the development of fibrosis and cirrhosis. Therefore, the canonical and non-canonical pathways constitute valuable targets in the development new and effective drugs to prevent liver fibrosis.
On the other hand, naringenin (NAR), 4’,5,7-trihydroxy flavanone (Figure 1), is a flavonoid that is widely distributed in cherries, cocoa, grapes, tangelos, blood oranges, lemons, grapefruit, tangerines and tomatoes[11-13]; it has been demonstrated to prevent acute liver damage induced by alcohol, carbon tetrachloride (CCl4), lipopolysaccharide or heavy metals[14-17]. In addition, there is some experimental evidence on the anticancer properties of NAR[18,19].
Figure 1.
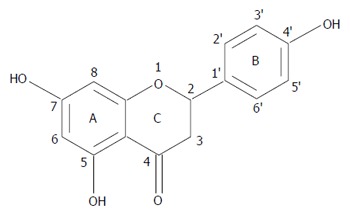
Chemical structure of naringenin.
Some reports have shown evidence of the antifibrotic properties of NAR. Lee et al[19] showed some histological evidence that NAR prevents collagen deposition in dimethyl nitrosamine-induced liver damage in rats, but no mechanism was evaluated. Du et al[20] reported an antifibrotic effect of NAR in lung tissue that was associated with the downregulation of TGF-β activity. In a study with cultured rat HSCs, NAR prevented ECM deposition induced by TGF-β through the downregulation of Smad3 protein levels[8]. However, to the best of our knowledge, there are no studies on the antifibrotic effect of NAR on liver cirrhosis. Therefore, the above information prompted us to investigate whether NAR was able to prevent CCl4- induced liver cirrhosis in the rat and to elucidate if the mechanism of action of the flavonoid was associated with an inhibition of the canonical and/or non-canonical TGF-β pathways and/or the inflammatory pathway (NF-κB) and/or through antioxidant mechanisms.
In fact, we demonstrate for the first time that NAR is able to prevent CCl4 -cirrhosis by inhibiting the canonical and non-canonical pathways, including the prevention of pSmad3L phosphorylation by JNK, by exerting anti-inflammatory properties that block the NF-κB inflammatory cascade, and via an antioxidant mechanism. These effects lead to the inhibition of HSC transdifferentiation, the downregulation of ECM synthesis, and the prevention of necrosis, cholestasis and distortion of the hepatic architecture. As a result, experimental cirrhosis was prevented.
MATERIALS AND METHODS
Ethics statement and animal treatment
Wistar male rats were used and maintained on a standard diet of rat chow with free access to drinking water. Four or five animals were housed in each polycarbonate cage under controlled conditions (22 ± 2 °C, 50%-60% relative humidity and 12-h light-dark cycles). The study complies with the Institution’s guidelines and the Mexican official regulation (NOM-062-ZOO-1999) regarding technical specifications for production, care and handling of laboratory animals.
Study design
Wistar rats initially weighing 120-150 g were used. Cirrhosis was produced by intraperitoneal (i.p.) administration of CCl4 (400 mg/kg body weight) dissolved in mineral oil three times per week for 8 wk. To determine the capacity of NAR to prevent liver fibrosis, four groups were formed and treated for 8 wk. Group 1 (n = 8) consisted of control animals receiving carboxy methyl-cellulose p.o., the NAR vehicle; group 2 (n = 8) was administered CCl4; group 3 (n = 8) was administered CCl4 plus NAR (100 mg/kg body weight, daily p.o.); group 4 (n = 8) was administered NAR only. All animals were sacrificed after 8 wk under ketamine (100 mg/kg of body weight) and xylazine (8 mg/kg of body weight) anesthesia. Blood was collected by cardiac puncture, and the liver was rapidly removed and properly stored for further analysis.
Serum enzyme activities
The determination of liver damage was performed by measuring the activities of alanine aminotransferase (ALT)[21], alkaline phosphatase (AP)[22], and γ-glutamyl transpeptidase (γ-GTP)[23] in serum samples of blood that were centrifuged at 1300 rpm for 15 min.
Glycogen determination
Small pieces of liver were separated for glycogen determination using the anthrone reagent[24]. Briefly, fresh liver samples (0.5 g) were boiled in 1.5 mL of 30% KOH for 30 min. After cooling, samples were diluted in a volumetric flask of 25 mL. Then, 40 μL (control and NAR groups) or 160 μL (CCl4 or CCl4 + NAR groups) was added to 960 μL and 840 μL of deionized water, respectively. Then, 2 mL of 0.2% anthrone (dissolved in concentrated sulfuric acid) was added. Samples were boiled for 15 min and the absorbance was read at 620 nm. Appropriate glucose standards were prepared.
Assessment of lipid peroxidation
The extent of lipid peroxidation (LPO) was evaluated in liver homogenates via the measurement of malondialdehyde (MDA) formation with the thiobarbituric acid method[25]. Fresh liver samples (0.5 g) were homogenized in 5 mL of deionized water on ice with a polytron homogenizer. Later, 300 μL of 10% liver homogenate with 700 μL of 150 mmol/L Tris-HCl (pH 7.4) and 2 mL of 0.375% thiobarbituric acid (dissolved in 15% trichloroacetic acid) were boiled for 45 min and centrifuged at 3000 rpm for 10 min. The supernatant absorbance was read at 532 nm. The protein concentration was determined via the Bradford method using bovine serum albumin as a standard[26].
Reduced glutathione in liver and blood samples
Fresh liver (0.3 g) and blood (0.3 mL) samples were homogenized in 1.2 mL of precipitating solution (5 mmol/L EDTA in 5% trichloroacetic acid) and centrifuged for 20 min at 12000 rpm. Then, 0.1 mL of the supernatant with 2.1 mL of phosphate solution (0.3 mol/L sodium phosphate dibasic) and 0.25 mL of Ellman’s reagent (40 mg of 5,5’-dithiobis nitrobenzoic acid dissolved in 100 mL of 1% sodium citrate) were mixed. The absorbance was read at 412 nm[27].
Glutathione peroxidase activity in the liver
The method of Lawrence and Burk[28] was used to determine glutathione peroxidase (GPx) activity with cumene hydroperoxide as a substrate. An aliquot of 1.5 mL of the 10% liver homogenate with 75 mmol/L potassium phosphate buffer (pH 7.0) was filtered through muslin cloth and centrifuged at 3000 rpm for 5 min at 4 °C. The reaction mixture contained 200 mL of the homogenate supernatant, 2.0 mL of 75 mmol/L potassium phosphate buffer (pH 7.0), 50 mL of 60 mmol/L glutathione, 0.1 mL of 30 U/mL glutathione reductase, 0.1 mL of 15 mmol/L EDTA, 0.1 mL of 3 mmol/L β-nicotinamide adenine dinucleotide phosphate (NADPH) and 0.3 mL of water. The reaction was started by the addition of 0.1 mL of 45 mmol/L cumene hydroperoxide. Oxidation of NADPH was recorded at 340 nm for 4 min, and the enzymatic activity was calculated as nmol of NADPH oxidized min-1mg-1 of protein using a molar extinction coefficient of 6.22 × 106 M-1 cm-1.
Collagen quantification
Fresh liver samples (100 mg) were placed in ampoules, 2 mL of 6 N HCl was added, and then the samples were sealed and hydrolyzed at 100 °C for 48 h. Next, the samples were evaporated at 50 °C for 24 h and resuspended in 3 mL of sodium acetate-citric buffer, pH 6.0; 0.5 g of activated charcoal was added, and the mixture was stirred vigorously and then centrifuged at 3000 rpm for 15 min. Then, 1 mL of chloramine T was added to 1 mL of the supernatant. The mixture was kept for 20 min at room temperature, and the reaction was stopped by the addition of 0.5 mL of 2 mol/L sodium thiosulfate and 1 mL of 1 N sodium hydroxide. The aqueous layer was transferred into test tubes. The oxidation product from hydroxyproline was converted to a pyrrole by boiling the samples. The pyrrole-containing samples were incubated with Ehrlich’s reagent for 30 min, and the absorbance was read at 560 nm. The recovery of known amounts of standards was carried out on similar liver samples for quantification[29].
Histology
Liver samples were taken from all animals and fixed with 10% formaldehyde in phosphate-buffered saline for 24 h. Tissue samples were then washed with tap water, dehydrated in alcohol and embedded in paraffin. Five-micrometer-thick sections were mounted on silane covered glass slides. Staining was performed with hematoxylin and eosin (H&E) and Masson’s trichrome stain.
Zymography
Proteolytic activity was assayed with gelatin-substrate gels. Liver tissue (0.25 g) was homogenized with 1.7 mL of 1 × PBS. Then, the samples were homogenized on ice with a polytron homogenizer, sonicated and centrifuged at 13000 rpm for 10 min. The supernatant was collected and proteins were quantified via the bicinchoninic acid method (Pierce BCA Protein Assay Prod # 23223 Thermo Scientific). Volumes equivalent to 50 μg of non-heated proteins were mixed with sample buffer (2.5% SDS, 1% sucrose and 4 mg/mL phenol red) without reducing agent and applied to 8% acrylamide gels copolymerized with 1 mg/mL gelatin. After electrophoresis at 72 V for 2 h, the gels were rinsed twice in 2.5% Triton X-100 to remove SDS and then incubated in 50 mmol/L Tris-HCl at pH 7.4 and 5 mmol/L CaCl2 assay buffer at 37 °C for 48 h. The gels were then fixed and stained with 0.25% Coomassie Brilliant Blue G-250 in 10% acetic acid and 30% methanol. Proteolytic activity was detected as clear bands against the background stain of undigested substrate in the gel at the expected location according to the molecular weight of metalloproteinase (MMP)-9 and MMP-2. Images were digitized and then analyzed densitometrically with ImageJ software.
Western blot assays
To carry out western blot assays, lysis buffer (1 mol/L Tris-HCl pH 8, 5 mol/L NaCl, NP40, Triton, 0.5 mol/L EDTA pH 8, 0.1 mol/L PMSF, 0.1 mol/L Na3VO4, 0.1 mol/L NaF) with protease and phosphatase inhibitors (Pierce BCA Protein Assay Prod # 23223 Thermo Scientific), each at a ratio of 1:100, was used to isolate total protein from liver tissue samples. Liver tissue (50 mg) was homogenized with 500 μL of lysis buffer. Then the samples were sonicated and centrifuged at 12000 rpm for 2 min. The supernatant was collected and proteins were quantified via the bicinchoninic acid method.
Volumes equivalent to 50 and 250 μg of protein were transferred onto 15, 12 and 10% polyacrylamide and electrophoresis was carried out. Separated proteins were transferred onto an Inmuno-BlotTM PVDF membrane (BIO-RAD, Hercules, CA, United States). Next, blots were blocked with 7% skim milk and 0.05% Tween-20 for 2 h at room temperature and independently incubated overnight at 4 °C with specific primary antibodies (Table 1). The following day, membranes were washed with TBS-tween-20 and then exposed to a secondary peroxidase-labeled antibody α-mouse (62-6520 Invitrogen) or α-rabbit (31460 Thermo Fisher Scientific) in the blocking solution for 2 h at room temperature. Blots were then washed with TBS-tween-20, and protein development was performed with the western lightningTM Plus-ECL Enhanced Chemiluminescence detection system (NEN Life Sciences Products, Elmer LAS Inc., Boston, MA, United States). β-actin was used as a control to normalize cytokine protein expression levels. Images were digitized and then analyzed densitometrically with ImageJ software.
Table 1.
Antibodies employed for this research
| Protein | Company | Catalog number | Dilution |
| TGF-β | Millipore | MAB1032 | 1:500 |
| α-SMA | Sigma Aldrich | A5691 | 1:500 |
| CTGF | Santa Cruz Biotechnology | SC-14939 | 1:500 |
| Col-1 | Sigma Aldrich | C-2456 | 1:500 |
| MMP-13 | Millipore | MAB13426 | 1:500 |
| NF-κB (p65) | Millipore | MAB3026 | 1:500 |
| IL-1 | Millipore | AB1832P | 1:500 |
| IL-10 | Invitrogen | ARC9102 | 1:500 |
| Smad3 | Abcam | Ab65847 | 1:500 |
| pSmad3L | Abcam | Ab63403 | 1:250 |
| Smad7 | Abcam | Ab90086 | 1:500 |
| JNK | Cell Signaling | 9252 | 1:500 |
| pJNK | Abcam | Ab131499 | 1:500 |
Statistical analysis
All data are expressed as the mean values ± SE. Comparisons were carried out via the analysis of one way variance followed by Tukey’s test, as appropriate, using the Graph Pad Prism software. Differences were considered statistically significant when P was < 0.05.
RESULTS
NAR prevented necrosis and cholestasis and improved liver biosynthetic capacity in CCl4 -treated rats
As shown in Figure 2, chronic administration of CCl4 significantly increased the serum activity of ALT, a hepatocyte necrosis indicator[30]. After CCl4 administration, AP and γ-GTP (two markers of cholestasis[30]) serum activity significantly increased compared to the levels in the control group. NAR administration completely prevented the increase in ALT, AP and GTP activity, suggesting that NAR is able to prevent necrosis and cholestasis caused by CCl4 administration.
Figure 2.
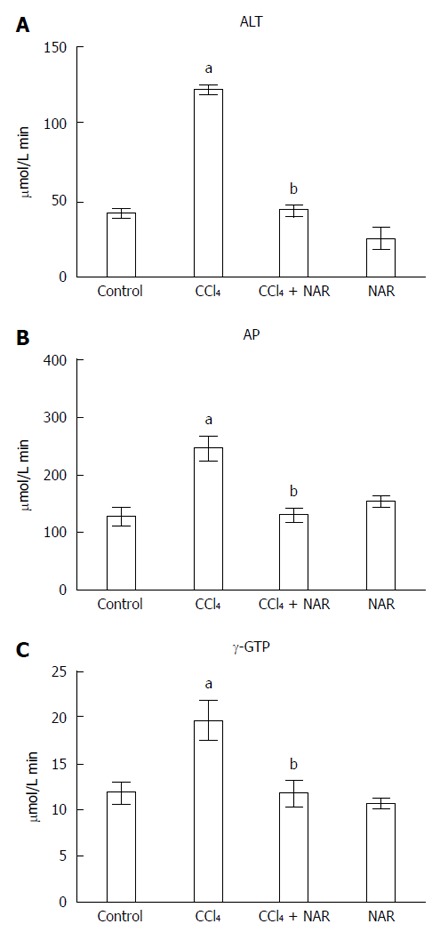
Naringenin prevents necrosis and cholestasis in CCl4-treated rats. Alanine aminotransferase (ALT) (A); alkaline phosphatase (AP) (B) and γ-glutamyl transpeptidase (γ-GTP) (C) activities were determined in serum from control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). Values represent the mean of experiments performed in duplicate assay ± SE (n = 8). aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
Glycogen measurement is used to determine the biosynthetic capacity and proper function of the liver[30]. Cirrhotic livers had significantly lower glycogen levels; however, NAR completely prevented the depletion of hepatic glycogen (Figure 3).
Figure 3.
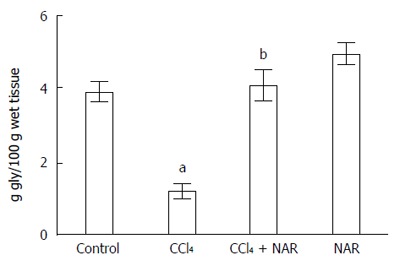
Naringenin prevents glycogen depletion in CCl4-treated rats. Liver glycogen content determined in control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). Each bar represents the mean value of experiments performed in duplicate assay ± SE (n = 8). aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
NAR prevented the oxidative stress caused by chronic liver damage
One of the main products of LPO is MDA, which is utilized to measure oxidative stress in tissues[31]. As expected, the induction of cirrhosis triggered LPO, since MDA levels were significantly elevated compared to the control group. Interestingly, NAR prevented the increase in MDA levels (Figure 4A).
Figure 4.
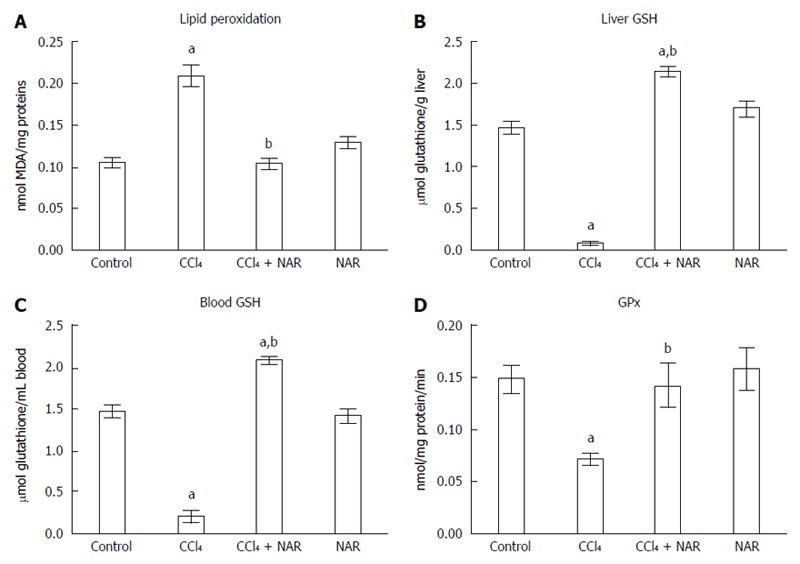
Naringenin prevents oxidative stress in CCl4-treated rats. Lipid peroxidation (A), reduced glutathione (GSH) determinations in liver (B) and blood (C) and GPx activity (D) from control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). Each bar represents the mean value of experiments performed in duplicate assay ± SE (n = 8). aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
Reduced glutathione (GSH) is one of the most important endogenous antioxidants[32]. CCl4-treated rats exhibited low liver and blood GSH levels compared with the control group, but NAR completely prevented this decrease and even raised GSH level above that of the control group (Figure 4B and C).
One of the most important antioxidant enzymes is GPx, which utilizes GSH to detoxify H2O2[33]. GPx activity was significantly decreased by the chronic administration of CCl4, while NAR coadministration partially prevented this effect (Figure 4D).
Together, these results show that NAR prevents oxidative stress during experimental liver cirrhosis at several levels.
Inflammation during liver injury was prevented by NAR administration
NF-κB is a key protein in inflammation, since it regulates interleukin expression, including IL-1 and IL-10[34,35]. Baseline values of NF-κB, IL-1 and IL-10 were increased after CCl4 treatment, and the expression of NF-κB increased almost 3-fold compared to the control group. The levels of IL-1 and IL-10 increased by 3.8- and 1.8-fold, respectively, compared to the control group. NAR prevented inflammation and necrosis in CCl4-treated rats by maintaining normal NF-κB, IL-1 and IL-10 levels (Figure 5).
Figure 5.
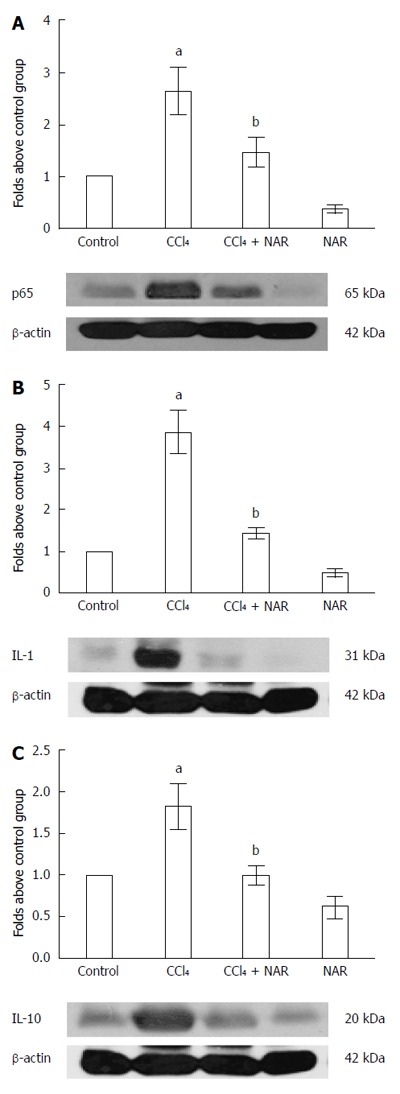
Naringenin prevents inflammation in CCl4-treated rats. The NF-κB (A), IL-1 (B) and IL-10 (C) protein levels in samples of liver tissue were determined by western blot analysis from control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). β-actin was used as a control. Values are expressed as fold increase of relative IOD normalized to the control group values (control = 1). Each bar represents the mean value of three rats ± SE. aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
Collagen accumulation was prevented by NAR in CCl4-induced experimental cirrhosis
Collagen quantification provides information about the balance between ECM synthesis and degradation[3,4]. Rats treated with CCl4 showed a nearly 4-fold increment in collagen content compared to the control group. This effect was completely prevented by NAR (Figure 6).
Figure 6.
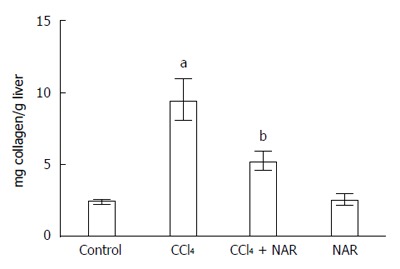
Naringenin prevents collagen deposition in CCl4-treated rats. Liver collagen content determined in control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). Each bar represents the mean value of experiments performed in duplicate assay ± SE (n = 8). aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
The general appearance of the livers at the macroscopic and microscopic level can be seen in Figure 7. Figure 7A shows a normal liver from the control group. CCl4 treatment produced macro nodular fibrosis (Figure 7B) that was prevented by NAR administration (Figure 7C). NAR-treated rats had livers that were macroscopically similar to those of control animals (Figure 7D).
Figure 7.
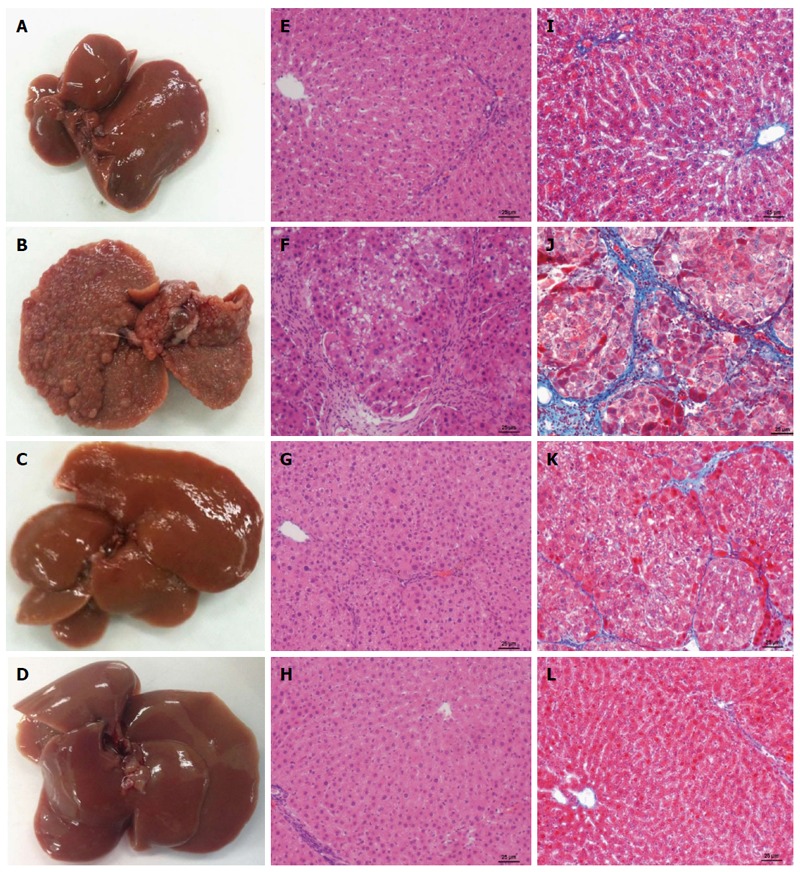
Naringenin effect on macroscopic and microscopic hepatic architecture in CCl4-treated rats. Macroscopic aspect of livers in control rats (A), carbon tetrachloride (CCl4)-treated rats (B), CCl4 plus naringenin rats (CCl4 + NAR) (C), and NAR alone rats (D). Hematoxylin and Eosin stain in livers of control rats (E), CCl4-treated rats (F), CCl4 + NAR rats (G), and rats administered with NAR alone (H). Masson’s trichromic staining in livers of control rats (I), CCl4-treated rats (J), CCl4 + NAR (K), and NAR alone rats (L). Bar scale = 50 μm. Magnification × 100.
H&E staining is shown in Figure 7E-H. Figure 7E, the control group, shows no alterations of the hepatic parenchyma. Figure 7F corresponds to a representative liver section of chronic CCl4-induced liver injury; in this case, the tissue shows liver parenchymal disruption, steatosis, hyperchromatic nuclear hepatocytes, and atypical pleomorphic nuclei. Fibrosis was decreased by the administration of NAR (Figure 7G). NAR treatment of control rats produced no effect on liver histology (Figure 7H).
Masson’s stained liver slices are shown in Figure 7I-L. Figure 7J presents a sample of a liver from a cirrhotic rat; a large amounts of collagen around fibrotic nodules was detected. The distortion of the parenchyma is evident when compared with a normal control liver (Figure 7I). NAR treatment prevented collagen accumulation and the formation of regenerative nodules (Figure 7K), and NAR alone produced no effect (Figure 7L).
NAR preserved the normal activity of MMP-9 and MMP-2 in experimental liver cirrhosis
MMPs are enzymes that are responsible for ECM degradation, and among the most important are MMP-9 and MMP-2[36]. As seen in Figure 8, during normal conditions, MMP-9 and MMP-2 exhibit basal activity. However, after CCl4 administration, MMP-9 and MMP-2 activities increased 3.7- and 5.4-fold compared to the control group, respectively. NAR treatment effectively maintained basal MMP activity levels in animals with experimental cirrhosis induced by CCl4.
Figure 8.
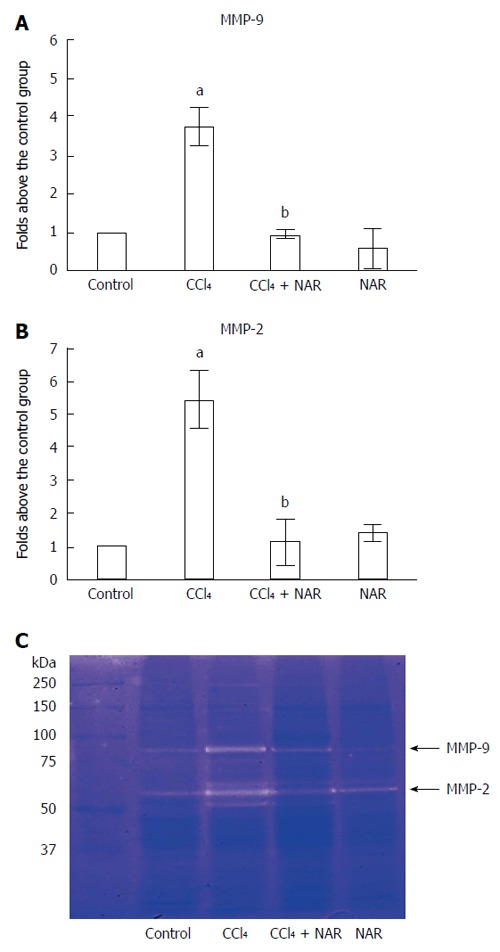
Naringenin prevents elevation of metalloproteinase-9 and 2 activities in CCl4-treated rats. Matrix metalloproteinase (MMP)-9 (A) and MMP-2 (B) activities was analyzed by zymography using gelatin-substrate gels (C). Liver samples for control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR) were analyzed. Values are expressed as average of relative IOD, normalized to the control group values (control = 1). Each bar represents the mean value of three rats ± SE. aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
NAR blocked HSC transdifferentiation and Col-1 synthesis by inhibiting profibrogenic proteins
It is well known that TGF-β induces HSC transdifferentiation, as well as α-SMA, Col-1 and connective tissue growth factor (CTGF) expression in HSCs[1,37]. In this study, under normal conditions, basal TGF-β, α-SMA, CTGF and Col-1 protein levels were observed; however, experimental fibrosis induced by CCl4 administration increased the expression of such proteins several-fold compared to the control group. NAR administration completely prevented the elevation of TGF-β, α-SMA, CTGF and Col-1 levels during CCl4 administration. Unexpectedly, NAR alone reduced α-SMA and Col-1 levels to below the control group (Figure 9A-D).
Figure 9.
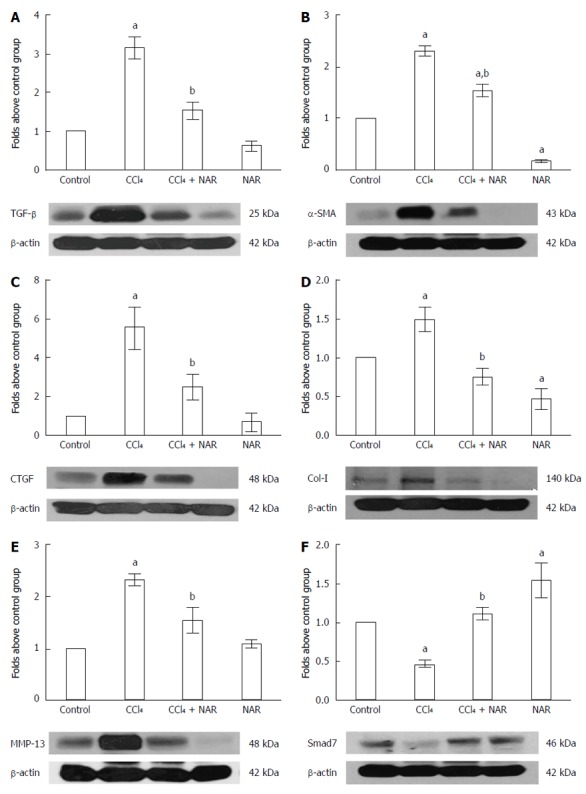
Naringenin prevents elevation of TGF-β, α-SMA, CTGF, MMP-13, and Col-1 protein levels, and preserves Smad7 protein levels in CCl4-treated rats. The TGF-β (A), α-SMA (B), CTGF (C), MMP-13 (D), Col-1 (E) and Smad7 (F) protein levels in samples of liver tissue were determined by western blot analysis from control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). β-actin was used as a control. Values are expressed as fold increase of relative IOD normalized to the control group values (control = 1). Each bar represents the mean value of three rats ± SE. aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
MMP-13 is involved in the migration and proliferation of HSCs and the activation of TGF-β[38]. As seen in Figure 9E, during normal conditions MMP-13 depicts basal protein levels; however, due to CCl4 administration, the MMP-13 level increased 2.3 fold above the control group. NAR treatment effectively maintained basal MMP-13 protein levels in animals with experimental cirrhosis induced by CCl4.
Since Smad7 inhibits the TGF-β signaling pathway by TGF-β receptor ubiquitination[39], we evaluated Smad7 to determine its participation in the mechanism of action of NAR. Smad7 levels were significantly decreased by CCl4 administration, but this effect was prevented by NAR treatment. Interestingly, NAR administration by itself increased Smad7 protein expression above control values (Figure 9F).
NAR inhibited HSC proliferation by blocking the JNK-pSmad3L pathway
Smad3 is normally activated by TGF-β, but JNK, via the linker phosphorylation pathway, also enables Smad3 to induce HSC proliferation[9,10]. Our results confirmed that animals treated with CCl4 over 8 wk showed a significant increase in the expression of pJNK, pSmad3L and Smad3 compared with basal conditions. Interestingly, NAR administration prevented JNK activation, the elevation of Smad3 protein levels and phosphorylation in the linker region (Figure 10).
Figure 10.
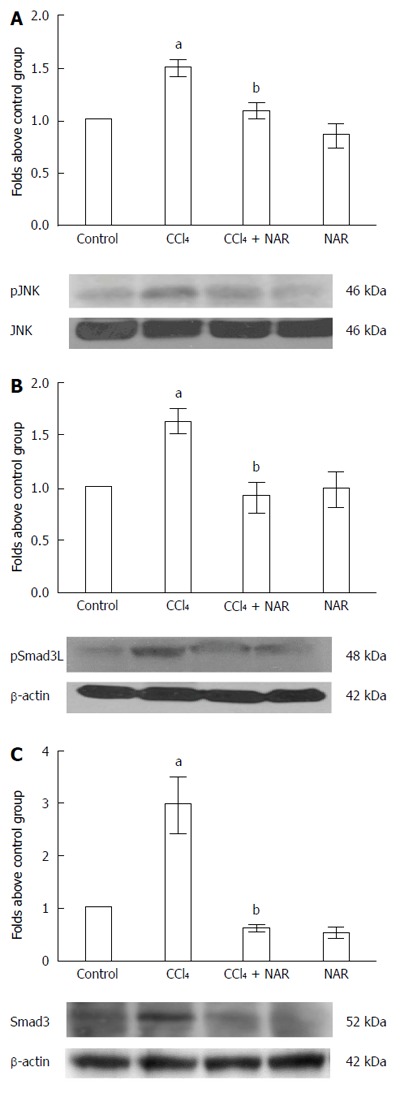
Naringenin prevents Smad3 linker phosphorylation by JNK inhibition in CCl4-treated rats. pJNK (A) and pSmad3L protein activation (B) and Smad3 protein levels (C) in samples of liver tissue were determined by western blot analysis from control rats, carbon tetrachloride (CCl4)-treated rats, CCl4 plus naringenin rats (CCl4 + NAR), and rats administered with NAR alone (NAR). β-actin was used as a control. Values are expressed as fold increase of relative IOD normalized to the control group values (control = 1). Each bar represents the mean value of three rats ± SE. aP < 0.05 vs control group; bP < 0.05 vs CCl4 group.
DISCUSSION
The aim of this study was to evaluate the hepatoprotective effect of NAR in a model of CCl4-induced chronic damage and to investigate whether the beneficial effects of NAR are associated with its antioxidant properties, and/or the disruption of NF-κB, and/or the canonical or non-canonical TGF-β pathways. The results show that the flavonoid is able to prevent necrosis, cholestasis, glycogen stores depletion and oxidative stress induced by chronic CCl4 administration. NAR preserved liver function, normal collagen levels and MMP basal activity during CCl4 administration. In addition, NAR prevented HSC transdifferentiation and Col-1 synthesis by inhibiting profibrogenic proteins such as TGF-β and CTGF. NAR also preserved Smad7 protein levels, which decreased during liver injury. Regarding the non-canonical Smad pathway, the flavonoid prevented the activation of JNK and Smad3 phosphorylation in the linker region. Finally, NAR showed anti-inflammatory properties because it maintained normal levels of NF-κB, IL-1 and IL-10 during liver injury. In summary, NAR prevented CCl4 -cirrhosis by means of its antioxidant, anti-inflammatory, immunomodulatory and antifibrotic properties.
NAR protects the liver via an antioxidant mechanism
The antinecrotic and anticholestatic effects of NAR may be associated with the ability of the flavonoid to prevent membrane damage due to its antioxidant properties. The toxic effect of CCl4 is dependent on its degraded metabolites, trichloromethyl and trichloromethyl peroxyl radicals, which are formed by the liver microsomal enzyme CYP2E1. These molecules are unstable radicals and exhibit strong binding affinity to protein and lipids of cell membranes by abstracting a hydrogen atom from an unsaturated lipid, thereby triggering LPO and thus causing liver damage[40,41].
NAR was able to prevent LPO due to its molecular structure; perhaps its hydroxyl groups facilitate its adherence to lipid bilayer polar groups, and its nonpolar nucleus may interact with hydrophobic tails of phospholipids, thereby reducing the deleterious effects of free radicals on membranes[12,42,43].
On the other hand, GSH is one of the main components of the endogenous antioxidant system; it scavenges hydroxyl and peroxynitrite radicals and is a GPx cofactor, among other important activities[32]. Chronic CCl4 administration decreased GSH levels; however, NAR was could preserve normal GSH levels in the liver and blood. In addition to its antioxidant properties as a free radical scavenger, NAR may also act though Nrf2 activation and induce endogenous antioxidant enzymes. In fact, NAR upregulates the expression of the enzyme glutamate-cysteine ligase, the rate limiting enzyme involved in de novo GSH synthesis[44-48], as well as glutathione reductase, which catalyzes the reduction of oxidized glutathione to the reduced form[15,43]. GPx detoxifies H2O2 by reducing it to water and oxygen, utilizing two molecules of GSH to form GSSG in the reduction of a molecule of H2O2[49]. In agreement with previous reports[13,15,50-53], CCl4 decreased GPx activity in this study. NAR cotreatment partially prevented the decrement in GPx activity, probably by up regulating its expression through Nrf2 modulation[15,40,43,44-47,50], providing another mechanism to fight oxidative stress induced by CCl4.
NAR prevents hepatic necrosis by blocking the NF-κB pathway
Rats treated with CCl4 showed increased NF-κB, IL-1 and IL-10 protein levels, but concomitant NAR administration with CCl4 prevented this increase. NAR inhibits NF-κB via the downregulation of TLR4 and TLR2 mRNA and protein levels and the decreased translocation and DNA binding of NF-κB[54-56]. This leads to the inhibition of the expression of NF-κB dependent interleukins, such as IL-1 and IL-10, and thus, necrosis is prevented.
NAR preserves MMP-9 and MMP-2 activity in CCl4-treated rats
During fibrosis, activated HSCs and Kupffer cells express MMP-9 and MMP-2; these enzymes lead to TGF-β activation by cleaving TGF-β from its reservoir on ECM, thus enhancing HSC invasive activity[38,53,57,58]. CCl4 chronic administration increased MMP-9 and MMP-2, while concomitant NAR administration maintained the normal activity of MMPs. In agreement with these findings, several reports indicate that NAR reduces both protein and mRNA levels of MMP-9 and MMP-2, thereby reducing the activity of these MMPs[59-61]. Therefore, it seems likely that the antifibrotic effect of NAR may be due, in part, to the downregulation of MMP-9 and MMP-2.
NAR inhibits the TGF-β-Smad3 pathway, leading to the downregulation of α-SMA, CTGF and Col-I
Through pSmad3C, TGF-β induces the expression of α-SMA, CTGF and Col-1 in activated HSCs; α-SMA is a highly specific transdifferentiation marker that is closely related to HSC contractile and migration capacities. CTGF amplifies the profibrogenic action of TGF-β, and Col-I is one of the main types of ECM collagen[4,37,62,63]. In this study, CCl4 administration increased TGF-β, α-SMA, CTGF and Col-I protein levels; importantly, NAR preserved the normal levels of these proteins.
The possible mechanism by which NAR inhibits the TGF-β-Smad3 pathway are (1) a reduction in tissue TGF-β levels; (2) a decrease in Smad3 mRNA and protein levels, with consequent reduction in Smad3 phosphorylation, thus preventing the nuclear translocation of pSmad3C; and (3) a reduction in the binding of TGF-β to its specific receptor, TβRII, leading to the inhibition of Smad3 phosphorylation[8,20,59,64-66]. In agreement with the results that were obtained, it has been reported that the inhibition of the TGF-β-Smad3 pathway by NAR results in decreased α-SMA, CTGF and Col-I mRNA and protein levels[8,19,37,67], therefore providing a suitable mechanism to further explain the antifibrotic properties of NAR.
NAR downregulates the profibrogenic TGF-β pathway by preserving Smad7 protein levels
Contrary to Smad3, Smad7 exerts an inhibitory effect on the TGF-β pathway by activating TβRI degradation[39]. As reported by others[68,69], CCl4 administration reduced Smad7 protein levels, but NAR was able to prevent this event. Lou et al[59], reported that NAR is able to preserve Smad7 mRNA levels in pancreatic cells treated with TGF-β. The prevention of Smad7 diminution by NAR constitutes another valuable mechanism by which the TGF-β pathway is downregulated to prevent fibrosis.
NAR blocks the profibrogenic action of TGF-β by downregulating MMP-13
MMP-13 is expressed by HSCs, Kupffer cells, and perisinusoidal cells; it releases ECM-bound cytokines such as TGF-β, leading to HSC proliferation and migration[38]. CCl4-induced liver damage increased protein levels of MMP-13; however, NAR maintained normal MMP-13 levels during liver damage. MMP-13 can be upregulated by IL-1, NF-κB and JNK[38,70]; however, these proteins were also downregulated by NAR. By downregulating MMP-13, NAR may prevent ECM deposition in rats treated with CCl4.
NAR also prevents fibrosis by blocking the non-canonical TGF-β pathway
After CCl4 administration, the activation of JNK was elevated, as were Smad3 protein levels and phosphorylation in the linker region of this protein. During CCl4 administration, pSmad3L has been found in the nucleus of HSCs. The phosphorylation of Smad3 in the linker region is catalyzed by pJNK, resulting in a rapid translocation of pSmad3L to the nucleus and the stimulation of expression of c-myc, an important HSC proliferation inducer[9,71,72], which leads to increased ECM production. NAR was capable of preventing the increase in Smad3 protein levels and JNK phosphorylation, and therefore, pSmad3L formation was decreased, leading to the inhibition of ECM deposition.
Figure 11 summarizes the mechanisms by which NAR acts to prevent liver oxidative stress, necrosis and fibrosis induced by chronic CCl4 intoxication; (1) during fibrosis, TGF-β is a major HSC transdifferentiation inductor, so the levels are increased. NAR administration maintained basal levels of TGF-β; (2) normally, latent TGF-β is anchored to the ECM, but during liver fibrogenesis, the activation of metalloproteinases such as MMP-2, MMP-9 and MMP-13 leads to TGF-β activation by cleaving it from its reservoir. NAR effectively preserved basal MMP activity levels and prevented TGF-β activation; (3) TGF-β binds to the TGF-β type II receptor (TβRII), which recruits and phosphorylates the TGF-β type I receptor (TβRI), which in turn phosphorylates Smad3 in its C-terminal region, leading to pSmad3C formation. Smad7 inhibits the TGF-β signaling pathway via TβRI ubiquitination and posterior degradation in the proteasome. NAR increased Smad7 expression, therefore downregulating the TGF-β pathway; (4) it is known that PDGF activates JNK, and in turn, this kinase phosphorylates Smad3 in the linker domain to generate pSmad3L. NAR administration prevented JNK activation and Smad3 phosphorylation; (5) Smad3 is indispensable for the canonical and non-canonical pathways and in liver samples from cirrhotic rats Smad3 levels are increased; however, NAR maintained basal Smad3 levels; (6) NF-κB regulates the expression of interleukins such as IL-1 and IL-10, while NAR prevented inflammation by downregulating the expression of NF-κB; and (7) one of the most important antioxidant enzymes is GPx, which utilizes GSH to detoxify H2O2; during cirrhosis, this activity decreased, and NAR partially prevented this effect and blocked LPO.
Figure 11.
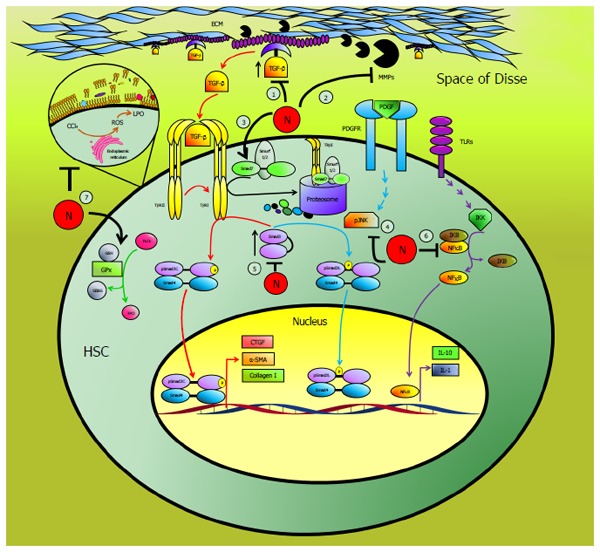
Schematic representation of the antifibrotic effect of naringenin. Naringenin (N) may act at 7 levels to prevent liver fibrosis: (1) by maintaining basal TGF-β levels, thus, preventing hepatic stellate cells activation (HSC), (2) preserving normal metalloproteinases (MMPs) activity blocking TGF-β liberation from extracellular matrix (ECM), (3) increasing the inhibitory protein Smad7, (4) by blocking the activation of JNK, (5) preserving Smad3 levels within control values, (6) blocking the proinflammatory factor NF-κB, (7) by counteracting oxidative stress, preserving reduced glutathione (GSH) levels, glutathione peroxidase (GPx) activity and lipid peroxidation (LPO) induced by reactive oxygen species (ROS), within normal levels.
In conclusion, our results demonstrate, for the first time, that NAR completely prevents CCl4-induced liver fibrosis in rats, not only because of its antioxidant properties but also via its effects as an immunomodulator and a downregulator of several profibrogenic pathways. The present results not only provide important information about the mechanism of the antifibrotic actions of NAR but also suggest that this flavonoid may be utilized in patients with fibrosis previous clinical and toxicological evaluation.
ACKNOWLEDGMENTS
The authors thank Ramón Hernández Guadarrama, Benjamín Salinas Hernández, Laura Dayana Buendia Montaño, Paula Vergara, Ma. Teresa García Camacho, Silvia Galindo, Angélica Silva Olivares, Rafael Leyva, Benjamín E. Chávez, and Ricardo Gaxiola for excellent technical assistance. The authors also acknowledge the Animal Lab Facility, UPEAL-Cinvestav.
COMMENTS
Background
Liver fibrosis results from chronic liver damage and is characterized by extracellular matrix (ECM) protein deposition. The mechanisms by which the ECM is induced by hepatic stellate cells includes oxidative stress and involves canonical and non-canonical TGF-β pathways and increased metalloproteinases. Naringenin (NAR) has shown some hepatoprotective properties; however, the capacity of NAR to prevent liver fibrosis has not yet been evaluated.
Research frontiers
NAR is a flavonoid that is widely distributed in nature and possesses antioxidant, anti-inflammatory and immunomodulatory properties that may be very useful to prevent hepatic fibrosis.
Innovations and breakthroughs
Oxidative stress, NF-κB activity, TGF-β-Smad3 and metalloprotease activity lead to necrosis and fibrosis. NAR may inhibit hepatic necrosis and fibrosis by blocking free radicals and inhibiting these proinflammatory and profibrotic pathways.
Applications
The current results indicate that the antinecrotic, anti-inflammatory and antifibrotic effects of NAR may be due to its ability to directly and indirectly fight free radicals and to inhibit NF-κB, IL-1, IL-10, and TGF-β-Smad3 and metalloproteases activity.
Terminology
NAR is the natural flavonoid aglycone of naringin; it is a flavanone with a stereogenic center at C2. It has two enantiomers, namely, (R)-NAR and (S)-NAR, and both NAR enantiomers are present in natural sources.
Peer-review
The paper by Hernández-Aquino et al investigated the hepato-protective and anti-fibrotic effects of NAR using a CCl4-induced liver fibrosis model. The authors found NAR protects liver functions in the CCl4-treated livers, and reduces levels of oxidative stress, fibrosis, and inflammation. This is an interesting paper which has implications for possible mechanisms underlying hepato-protective and anti-fibrotic effects of NAR. The manuscript was well written and the data support their claims.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Mexico
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Institutional animal care and use committee statement: The study complies with the Institution’s guidelines and the Mexican official regulation (NOM-062-ZOO-1999).
Conflict-of-interest statement: The authors declare no conflicts of interest.
Data sharing statement: No additional data are available.
Peer-review started: November 18, 2016
First decision: March 3, 2017
Article in press: June 1, 2017
P- Reviewer: Dang SS, Ro SW S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pellicoro A, Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Fibrogenesis Tissue Repair. 2012;5:S26. doi: 10.1186/1755-1536-5-S1-S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiskirchen R, Tacke F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig Dis. 2016;34:410–422. doi: 10.1159/000444556. [DOI] [PubMed] [Google Scholar]
- 4.Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22 Suppl 1:S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 5.Pollard TD, Earnshaw WC, Lippincott-Schwartz J. Cell Biology. 2th edition. Philadelphia: Elsevier; 2008. pp. 433–435. [Google Scholar]
- 6.Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, Ten Dijke P. TGF-β signalling and liver disease. FEBS J. 2016;283:2219–2232. doi: 10.1111/febs.13665. [DOI] [PubMed] [Google Scholar]
- 7.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19–27. doi: 10.1016/j.cytogfr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Liu X, Wang W, Hu H, Tang N, Zhang C, Liang W, Wang M. Smad3 specific inhibitor, naringenin, decreases the expression of extracellular matrix induced by TGF-beta1 in cultured rat hepatic stellate cells. Pharm Res. 2006;23:82–89. doi: 10.1007/s11095-005-9043-5. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki K. Smad phospho-isoforms direct context-dependent TGF-β signaling. Cytokine Growth Factor Rev. 2013;24:385–399. doi: 10.1016/j.cytogfr.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Murata M, Yamaguchi T, Matsuzaki K, Okazaki K. Reversible Human TGF-β Signal Shifting between Tumor Suppression and Fibro-Carcinogenesis: Implications of Smad Phospho-Isoforms for Hepatic Epithelial-Mesenchymal Transitions. J Clin Med. 2016;5 doi: 10.3390/jcm5010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erlund I. Review of the flavonoids quercetin, hesperetin, and naringenin. Dietary sources, bioactivities, bioavailability, and epidemiology. Nutr Res. 2004;24:851–874. [Google Scholar]
- 12.Jayaraman J, Veerappan M, Namasivayam N. Potential beneficial effect of naringenin on lipid peroxidation and antioxidant status in rats with ethanol-induced hepatotoxicity. J Pharm Pharmacol. 2009;61:1383–1390. doi: 10.1211/jpp/61.10.0016. [DOI] [PubMed] [Google Scholar]
- 13.Yen FL, Wu TH, Lin LT, Cham TM, Lin CC. Naringenin-loaded nanoparticles improve the physicochemical properties and the hepatoprotective effects of naringenin in orally-administered rats with CCl(4)-induced acute liver failure. Pharm Res. 2009;26:893–902. doi: 10.1007/s11095-008-9791-0. [DOI] [PubMed] [Google Scholar]
- 14.Jayaraman J, Jesudoss VA, Menon VP, Namasivayam N. Anti-inflammatory role of naringenin in rats with ethanol induced liver injury. Toxicol Mech Methods. 2012;22:568–576. doi: 10.3109/15376516.2012.707255. [DOI] [PubMed] [Google Scholar]
- 15.Esmaeili MA, Alilou M. Naringenin attenuates CCl4 -induced hepatic inflammation by the activation of an Nrf2-mediated pathway in rats. Clin Exp Pharmacol Physiol. 2014;41:416–422. doi: 10.1111/1440-1681.12230. [DOI] [PubMed] [Google Scholar]
- 16.Pinho-Ribeiro FA, Zarpelon AC, Mizokami SS, Borghi SM, Bordignon J, Silva RL, Cunha TM, Alves-Filho JC, Cunha FQ, Casagrande R, et al. The citrus flavonone naringenin reduces lipopolysaccharide-induced inflammatory pain and leukocyte recruitment by inhibiting NF-κB activation. J Nutr Biochem. 2016;33:8–14. doi: 10.1016/j.jnutbio.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Ozkaya A, Sahin Z, Dag U, Ozkaraca M. Effects of Naringenin on Oxidative Stress and Histopathological Changes in the Liver of Lead Acetate Administered Rats. J Biochem Mol Toxicol. 2016;30:243–248. doi: 10.1002/jbt.21785. [DOI] [PubMed] [Google Scholar]
- 18.Arul D, Subramanian P. Inhibitory effect of naringenin (citrus flavonone) on N-nitrosodiethylamine induced hepatocarcinogenesis in rats. Biochem Biophys Res Commun. 2013;434:203–209. doi: 10.1016/j.bbrc.2013.03.039. [DOI] [PubMed] [Google Scholar]
- 19.Lee MH, Yoon S, Moon JO. The flavonoid naringenin inhibits dimethylnitrosamine-induced liver damage in rats. Biol Pharm Bull. 2004;27:72–76. doi: 10.1248/bpb.27.72. [DOI] [PubMed] [Google Scholar]
- 20.Du G, Jin L, Han X, Song Z, Zhang H, Liang W. Naringenin: a potential immunomodulator for inhibiting lung fibrosis and metastasis. Cancer Res. 2009;69:3205–3212. doi: 10.1158/0008-5472.CAN-08-3393. [DOI] [PubMed] [Google Scholar]
- 21.Reitman S, Frankel S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am J Clin Pathol. 1957;28:56–63. doi: 10.1093/ajcp/28.1.56. [DOI] [PubMed] [Google Scholar]
- 22.Bergmeyer HU, Grabl M, Walter HE. Enzymes. In: Bergmeyer J, Grabl M, editors. Methods of Enzymatic Analysis. Weinheim: Verlag - Chemie; 1983. pp. 269–270. [Google Scholar]
- 23.Glossmann H, Neville DM. gamma-Glutamyltransferase in kidney brush border membranes. FEBS Lett. 1972;19:340–344. doi: 10.1016/0014-5793(72)80075-9. [DOI] [PubMed] [Google Scholar]
- 24.Seifter S, Dayton S. The estimation of glycogen with the anthrone reagent. Arch Biochem. 1950;25:191–200. [PubMed] [Google Scholar]
- 25.Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 26.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Sedlak J, Lindsay RH. Estimation of total, protein-bound, and nonprotein sulfhydryl groups in tissue with Ellman’s reagent. Anal Biochem. 1968;25:192–205. doi: 10.1016/0003-2697(68)90092-4. [DOI] [PubMed] [Google Scholar]
- 28.Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun. 1976;71:952–958. doi: 10.1016/0006-291x(76)90747-6. [DOI] [PubMed] [Google Scholar]
- 29.Prockop DJ, Udenfriend S. A specific method for the analysis of hydroxyproline in tissues and urine. Anal Biochem. 1960;1:228–239. doi: 10.1016/0003-2697(60)90050-6. [DOI] [PubMed] [Google Scholar]
- 30.Rosen HR, Keeffe EB. Evaluation of abnormal liver enzymes, use of liver test, and the serology of viral hepatitis. In: Bacon BR, Di Bisceglie AM, editors. Liver Disease Diagnosis and Management. Philadelphia: Churchill; 2000. pp. 24–35. [Google Scholar]
- 31.Manibusan MK, Odin M, Eastmond DA. Postulated carbon tetrachloride mode of action: a review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2007;25:185–209. doi: 10.1080/10590500701569398. [DOI] [PubMed] [Google Scholar]
- 32.Arauz J, Ramos-Tovar E, Muriel P. Redox state and methods to evaluate oxidative stress in liver damage: From bench to bedside. Ann Hepatol. 2016;15:160–173. doi: 10.5604/16652681.1193701. [DOI] [PubMed] [Google Scholar]
- 33.Carillon J, Rouanet JM, Cristol JP, Brion R. Superoxide dismutase administration, a potential therapy against oxidative stress related diseases: several routes of supplementation and proposal of an original mechanism of action. Pharm Res. 2013;30:2718–2728. doi: 10.1007/s11095-013-1113-5. [DOI] [PubMed] [Google Scholar]
- 34.Kawai T, Akira S. TLR signaling. Semin Immunol. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 35.Muriel P. NF-kappaB in liver diseases: a target for drug therapy. J Appl Toxicol. 2009;29:91–100. doi: 10.1002/jat.1393. [DOI] [PubMed] [Google Scholar]
- 36.Vandooren J, Geurts N, Martens E, Van den Steen PE, Opdenakker G. Zymography methods for visualizing hydrolytic enzymes. Nat Methods. 2013;10:211–220. doi: 10.1038/nmeth.2371. [DOI] [PubMed] [Google Scholar]
- 37.Arauz J, Moreno MG, Cortés-Reynosa P, Salazar EP, Muriel P. Coffee attenuates fibrosis by decreasing the expression of TGF-β and CTGF in a murine model of liver damage. J Appl Toxicol. 2013;33:970–979. doi: 10.1002/jat.2788. [DOI] [PubMed] [Google Scholar]
- 38.Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46:955–975. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Imamura T, Oshima Y, Hikita A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. J Biochem. 2013;154:481–489. doi: 10.1093/jb/mvt097. [DOI] [PubMed] [Google Scholar]
- 40.Weber LW, Boll M, Stampfl A. Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol. 2003;33:105–136. doi: 10.1080/713611034. [DOI] [PubMed] [Google Scholar]
- 41.Muriel P, Ramos-Tovar E, Montes-Páez G, Buendía-Montaño LD. Experimental models of liver damage mediated by oxidative stress. In: Muriel P, editor. Liver pathophysiology; therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 529–546. [Google Scholar]
- 42.Hernández-Aquino E, Muriel P. Naringenin and the liver. In: Muriel P, editor. Liver pathophysiology; therapies & antioxidants. Waltham, MA: Elsevier; 2017. pp. 633–651. [Google Scholar]
- 43.Jayaraman J, Namasivayam N. Naringenin modulates circulatory lipid peroxidation, anti-oxidant status and hepatic alcohol metabolizing enzymes in rats with ethanol induced liver injury. Fundam Clin Pharmacol. 2011;25:682–689. doi: 10.1111/j.1472-8206.2010.00899.x. [DOI] [PubMed] [Google Scholar]
- 44.Gopinath K, Sudhandiran G. Naringin modulates oxidative stress and inflammation in 3-nitropropionic acid-induced neurodegeneration through the activation of nuclear factor-erythroid 2-related factor-2 signalling pathway. Neuroscience. 2012;227:134–143. doi: 10.1016/j.neuroscience.2012.07.060. [DOI] [PubMed] [Google Scholar]
- 45.Lou H, Jing X, Wei X, Shi H, Ren D, Zhang X. Naringenin protects against 6-OHDA-induced neurotoxicity via activation of the Nrf2/ARE signaling pathway. Neuropharmacology. 2014;79:380–388. doi: 10.1016/j.neuropharm.2013.11.026. [DOI] [PubMed] [Google Scholar]
- 46.Podder B, Song HY, Kim YS. Naringenin exerts cytoprotective effect against paraquat-induced toxicity in human bronchial epithelial BEAS-2B cells through NRF2 activation. J Microbiol Biotechnol. 2014;24:605–613. doi: 10.4014/jmb.1402.02001. [DOI] [PubMed] [Google Scholar]
- 47.Ramprasath T, Senthamizharasi M, Vasudevan V, Sasikumar S, Yuvaraj S, Selvam GS. Naringenin confers protection against oxidative stress through upregulation of Nrf2 target genes in cardiomyoblast cells. J Physiol Biochem. 2014;70:407–415. doi: 10.1007/s13105-014-0318-3. [DOI] [PubMed] [Google Scholar]
- 48.Han X, Pan J, Ren D, Cheng Y, Fan P, Lou H. Naringenin-7-O-glucoside protects against doxorubicin-induced toxicity in H9c2 cardiomyocytes by induction of endogenous antioxidant enzymes. Food Chem Toxicol. 2008;46:3140–3146. doi: 10.1016/j.fct.2008.06.086. [DOI] [PubMed] [Google Scholar]
- 49.Davies KJ. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life. 2000;50:279–289. doi: 10.1080/713803728. [DOI] [PubMed] [Google Scholar]
- 50.Dong D, Xu L, Yin L, Qi Y, Peng J. Naringin prevents carbon tetrachloride-induced acute liver injury in mice. J Funct Foods. 2015;12:179–191. [Google Scholar]
- 51.Maiti K, Mukherjee K, Gantait A, Saha BP, Mukherjee PK. Enhanced therapeutic potential of naringenin-phospholipid complex in rats. J Pharm Pharmacol. 2006;58:1227–1233. doi: 10.1211/jpp.58.9.0009. [DOI] [PubMed] [Google Scholar]
- 52.Hermenean A, Ardelean A, Stan M, Herman H, Mihali CV, Costache M, Dinischiotu A. Protective effects of naringenin on carbon tetrachloride-induced acute nephrotoxicity in mouse kidney. Chem Biol Interact. 2013;205:138–147. doi: 10.1016/j.cbi.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 53.Hermenean A, Ardelean A, Stan M, Hadaruga N, Mihali CV, Costache M, Dinischiotu A. Antioxidant and hepatoprotective effects of naringenin and its β-cyclodextrin formulation in mice intoxicated with carbon tetrachloride: a comparative study. J Med Food. 2014;17:670–677. doi: 10.1089/jmf.2013.0007. [DOI] [PubMed] [Google Scholar]
- 54.Dou W, Zhang J, Sun A, Zhang E, Ding L, Mukherjee S, Wei X, Chou G, Wang ZT, Mani S. Protective effect of naringenin against experimental colitis via suppression of Toll-like receptor 4/NF-κB signalling. Br J Nutr. 2013;110:599–608. doi: 10.1017/S0007114512005594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yilma AN, Singh SR, Morici L, Dennis VA. Flavonoid naringenin: a potential immunomodulator for Chlamydia trachomatis inflammation. Mediators Inflamm. 2013;2013:102457. doi: 10.1155/2013/102457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yoshida H, Watanabe W, Oomagari H, Tsuruta E, Shida M, Kurokawa M. Citrus flavonoid naringenin inhibits TLR2 expression in adipocytes. J Nutr Biochem. 2013;24:1276–1284. doi: 10.1016/j.jnutbio.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 57.Olaso E, Ikeda K, Eng FJ, Xu L, Wang LH, Lin HC, Friedman SL. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest. 2001;108:1369–1378. doi: 10.1172/JCI12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayashi H, Sakai T. Biological Significance of Local TGF-β Activation in Liver Diseases. Front Physiol. 2012;3:12. doi: 10.3389/fphys.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lou C, Zhang F, Yang M, Zhao J, Zeng W, Fang X, Zhang Y, Zhang C, Liang W. Naringenin decreases invasiveness and metastasis by inhibiting TGF-β-induced epithelial to mesenchymal transition in pancreatic cancer cells. PLoS One. 2012;7:e50956. doi: 10.1371/journal.pone.0050956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yen HR, Liu CJ, Yeh CC. Naringenin suppresses TPA-induced tumor invasion by suppressing multiple signal transduction pathways in human hepatocellular carcinoma cells. Chem Biol Interact. 2015;235:1–9. doi: 10.1016/j.cbi.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 61.Sun Y, Gu J. Study on effect of naringenin in inhibiting migration and invasion of breast cancer cells and its molecular mechanism. Zhongguo Zhong Yao Za Zhi. 2015;40:1144–1150. [PubMed] [Google Scholar]
- 62.Inagaki Y, Mamura M, Kanamaru Y, Greenwel P, Nemoto T, Takehara K, Ten Dijke P, Nakao A. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol. 2001;187:117–123. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1059>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 63.Holmes A, Abraham DJ, Sa S, Shiwen X, Black CM, Leask A. CTGF and SMADs, maintenance of scleroderma phenotype is independent of SMAD signaling. J Biol Chem. 2001;276:10594–10601. doi: 10.1074/jbc.M010149200. [DOI] [PubMed] [Google Scholar]
- 64.Martinez RM, Pinho-Ribeiro FA, Steffen VS, Caviglione CV, Vignoli JA, Barbosa DS, Baracat MM, Georgetti SR, Verri WA, Casagrande R. Naringenin Inhibits UVB Irradiation-Induced Inflammation and Oxidative Stress in the Skin of Hairless Mice. J Nat Prod. 2015;78:1647–1655. doi: 10.1021/acs.jnatprod.5b00198. [DOI] [PubMed] [Google Scholar]
- 65.Meng XM, Zhang Y, Huang XR, Ren GL, Li J, Lan HY. Treatment of renal fibrosis by rebalancing TGF-β/Smad signaling with the combination of asiatic acid and naringenin. Oncotarget. 2015;6:36984–36997. doi: 10.18632/oncotarget.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang Y, Xu Y, Xia T, Chen F, Zhang C, Liang W, Lai L, Fang X. A single-molecule study of the inhibition effect of Naringenin on transforming growth factor-β ligand-receptor binding. Chem Commun (Camb) 2011;47:5440–5442. doi: 10.1039/c1cc10778j. [DOI] [PubMed] [Google Scholar]
- 67.Jung JW, Park IH, Cho JS, Lee HM. Naringenin inhibits extracellular matrix production via extracellular signal-regulated kinase pathways in nasal polyp-derived fibroblasts. Phytother Res. 2013;27:463–467. doi: 10.1002/ptr.4735. [DOI] [PubMed] [Google Scholar]
- 68.Bai G, Yan G, Wang G, Wan P, Zhang R. Anti-hepatic fibrosis effects of a novel turtle shell decoction by inhibiting hepatic stellate cell proliferation and blocking TGF-β1/Smad signaling pathway in rats. Oncol Rep. 2016;36:2902–2910. doi: 10.3892/or.2016.5078. [DOI] [PubMed] [Google Scholar]
- 69.Wu FR, Jiang L, He XL, Zhu PL, Li J. Effect of hesperidin on TGF-beta1/Smad signaling pathway in HSC. Zhongguo Zhong Yao Za Zhi. 2015;40:2639–2643. [PubMed] [Google Scholar]
- 70.Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4:157–164. doi: 10.1186/ar401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshida K, Matsuzaki K, Mori S, Tahashi Y, Yamagata H, Furukawa F, Seki T, Nishizawa M, Fujisawa J, Okazaki K. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005;166:1029–1039. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshida K, Matsuzaki K. Differential Regulation of TGF-β/Smad Signaling in Hepatic Stellate Cells between Acute and Chronic Liver Injuries. Front Physiol. 2012;3:53. doi: 10.3389/fphys.2012.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]