

ABSTRACT
A single dominantly expressed allele of major histocompatibility complex class I (MHC I) may be responsible for the duck's high tolerance to highly pathogenic influenza A virus (HP-IAV) compared to the chicken's lower tolerance. In this study, the crystal structures of duck MHC I (Anpl-UAA*01) and duck β2-microglobulin (β2m) with two peptides from the H5N1 strains were determined. Two remarkable features were found to distinguish the Anpl-UAA*01 complex from other known MHC I structures. A disulfide bond formed by Cys95 and Cys112 and connecting the β5 and β6 sheets at the bottom of peptide binding groove (PBG) in Anpl-UAA*01 complex, which can enhance IAV peptide binding, was identified. Moreover, the interface area between duck MHC I and β2m was found to be larger than in other species. In addition, the two IAV peptides that display distinctive conformations in the PBG, B, and F pockets act as the primary anchor sites. Thirty-one IAV peptides were used to verify the peptide binding motif of Anpl-UAA*01, and the results confirmed that the peptide binding motif is similar to that of HLA-A*0201. Based on this motif, approximately 600 peptides from the IAV strains were partially verified as the candidate epitope peptides for Anpl-UAA*01, which is a far greater number than those for chicken BF2*2101 and BF2*0401 molecules. Extensive IAV peptide binding should allow for ducks with this Anpl-UAA*01 haplotype to resist IAV infection.
IMPORTANCE Ducks are natural reservoirs of influenza A virus (IAV) and are more resistant to the IAV than chickens. Both ducks and chickens express only one dominant MHC I locus providing resistance to the virus. To investigate how MHC I provides IAV resistance, crystal structures of the dominantly expressed duck MHC class I (pAnpl-UAA*01) with two IAV peptides were determined. A disulfide bond was identified in the peptide binding groove that can facilitate Anpl-UAA*01 binding to IAV peptides. Anpl-UAA*01 has a much wider recognition spectrum of IAV epitope peptides than do chickens. The IAV peptides bound by Anpl-UAA*01 display distinctive conformations that can help induce an extensive cytotoxic T lymphocyte (CTL) response. In addition, the interface area between the duck MHC I and β2m is larger than in other species. These results indicate that HP-IAV resistance in ducks is due to extensive CTL responses induced by MHC I.
KEYWORDS: crystal structure, duck, MHC class I, influenza A virus, disulfide bond
INTRODUCTION
Influenza A virus (IAV) poses a large threat for both animal and human health and is a growing problem (1–4). Ducks play a pivotal role in its epidemiology because they are natural reservoirs of IAV (5). Although all subtypes of IAV are perpetuated in ducks (6, 7), they typically do not show serious signs of the disease, even that caused by highly pathogenic IAVs (HP-IAVs), such as the H5N1 strain, which is lethal to chickens (8, 9). Since 2002, many Asian lineage H5N1 HP-IAVs have been shown to produce similar symptoms and mortality, although clinical outcomes are also affected by the age and species of the ducks and the strains of IAVs (10, 11). Moreover, ducks have more active and robust cellular immune responses than chickens (12). These findings suggest that at least some species of duck show efficient immune responses against IAV infection.
Studies have indicated that major histocompatibility complex class I (MHC I) plays a role in the anti-IAV response (13–15). MHC I can present viral epitope peptides to specific T cell receptors (TCRs), resulting in the proliferation of cytotoxic T lymphocytes (CTLs) and eventual clearance of the virus from the host (16–18). CTL responses mediated by MHC I play a significant role in primary IAV infection and provide cross-protection against different IAV strains in chickens, mice, and humans (19–23). Suppressive subtractive hybridization libraries have been constructed to enrich differentially expressed genes from the lungs of ducks infected with IAVs of high or low pathogenicity compared to mock-infected controls (13). These data showed that the MHC I gene was upregulated in ducks under both conditions, especially in the highly pathogenic group, in which the MHC I gene was increased by more than 1,000-fold (13). Another study showed that there was an increase in MHC I gene expression in two different duck species after injection with a commercial inactivated vaccine (24). These results suggest that duck MHC I may play a role similar to that of the human HLA against infection.
MHC I molecules are encoded by polymorphic alleles from several loci. These loci make up the peptide binding groove (PBG) and contain epitope peptides against multiple pathogens. In humans and mice, there are three functional MHC I loci that provide polymorphic alleles for peptides (25, 26); however, many birds predominantly express only one MHC class I gene, such as in chickens and ducks (27). Chickens express only one dominant MHC I locus, referred to as BF2, which is adjacent to the TAP gene (28). Although there are five different loci in the duck MHC I genome region (named the UAA to UEA loci), only UAA, which lies adjacent to the polymorphic TAP2 gene, is predominantly expressed for the CTL immune response (29, 30). To date, more than 400 structures of the peptide-MHC I-β2-microglobulin β2m) protein complexes (here called pMHC I) of different species have been determined, and most of them are from human and mouse. Structural studies revealed that the epitope peptides are fixed in the PBG of the MHC I heavy chain by six pockets (A to F) (31). Some pockets, typically the B and F pockets, play a critical role to bind the peptides and determine the peptide binding motif of a certain MHC I molecule. In recent years, pMHC I structures of other species have been solved, while in avian species, only chicken MHC I (BF2) was resolved (32–34). These structures have greatly facilitated the identification of MHC I-restricted CTL epitopes.
Several studies have indicated that “minimal MHC” is related to resistance and the susceptibility of chickens to viruses, such as the Rous sarcoma virus and Marek's disease virus (MDV) (32, 35, 36). Chickens expressing BF2*0401 (B4 haplotype) are susceptible to MDV, and chickens expressing BF2*2101 (B21 haplotype) are resistant to MDV. Crystal studies of BF2 structures have illustrated its resistance or susceptibility to MDV (32). The MHC I complex presents viral peptides to CTLs, depending on the six (A to F) pockets in its peptide binding groove (PBG). The MHC I polymorphisms determine the distinct three-dimensional (3D) structure of the MHC I PBG, and each classical MHC I molecule has a specific peptide binding motif. Crystal studies of B21 have shown that it can remodel its pockets to accommodate different peptides based on its wide binding groove and interplay of two charged residues (Arg9 and Asp24); B21 could bind multiple peptides from MDV and activate extensive T cell repertories to clear MDV (32). In contrast, B4 has a narrow peptide binding groove and strong polar pockets that restrict its peptide binding motif and the presented MDV peptides (33). Therefore, the CTL response to MDV induced by B4 is not strong enough to prevent infection and suggests that resistance is controlled by the type of MHC I and its 3D structure.
MHC I could also play a role in the susceptibility of viruses. For IAV, the higher resistances of some species of duck indicate that their MHC I alleles would present more IAV peptide epitopes than those of chickens (11, 14). Our previous data, as well as a recently published study, confirmed the presence of two or more cysteines, which have never been found in humans or other species, in the PBGs of many duck MHC I alleles (30, 37). To date, little is known about the 3D structure and peptide presentation of duck MHC I. In this study, we determined two crystal structures of the dominantly expressed duck MHC I (Anpl-UAA*01) molecules with two IAV peptides. The crystal structures showed that Anpl-UAA*01 exhibits characteristics consistent with the duck MHC I-peptide-β2m complex (pMHC I) architecture; however, we also identified a disulfide bond in its PBG. Unlike the chicken MHC I, only B and F pockets of Anpl-UAA*01 are the primary anchor sites for peptide binding. The two pockets, whose compositions are similar to those of the B and F pockets of HLA-A2, could accommodate diverse residues and make Anpl-UAA*01 an extensive peptide binding motif. By screening the entire sequences of IAV strains, approximately 600 nonapeptides fit the identified peptide binding motif of Anpl-UAA*01. Our study demonstrates that Anpl-UAA*01 could present more IAV epitope peptides, suggesting that ducks with this MHC I haplotype may be protected from infection.
RESULTS
The Anpl-UAA*01 structure shows an unexposed disulfide bond in the PBG and a large interface between heavy and light (H and L) chains.
Two trimer complexes formed by Anpl-UAA*01, duck β2m (Anpl-β2m), and the MVM9 and RLI9 peptides were crystallized in the P1211 space group at resolutions of 1.71 Å and 2.06 Å, respectively (Table 1). Both of the complexes displayed a canonical pMHC I structure, in which the peptide was located in the platform formed by the α1 and α2 domains of Anpl-UAA*01 above the α3 domain and light-chain Anpl-β2m (Fig. 1A and B). The most remarkable features of the pAnpl-UAA*01 complexes are the two disulfide bonds formed by 4 cysteines in the PBG (Fig. 1C). In other known pMHC I structures, there is only one disulfide bond formed by a pair of cysteines in their PBGs, such as in chicken pBF2*2101 (32) and human pHLA-A*0201 (38) (Fig. 1D and E). These two cysteines have been found only in ducks (Fig. 2). This novel disulfide formed by Cys95 and Cys112 of pAnpl-UAA*01 complexes connects the β5 and β6 sheets at the bottom of the PBG.
TABLE 1.
Data collection and refinement statistics for pAnpl-UAA*01–MVM9/RLI9
| Parameter | Valuea for: |
|
|---|---|---|
| pAnpl-UAA*01–MVM9 | pAnpl-UAA*01–RLI9 | |
| Data collection | ||
| Space group | P1211 | P1211 |
| Cell dimensions | ||
| a, b, c (Å) | 46.27, 64.39, 77.92 | 46.49, 63.54, 79.95 |
| α, β, γ (°) | 90.00, 105.74, 90,00 | 90.00, 106.10, 90.00 |
| Resolution (Å) | 50.00–1.80 (1.86–1.80) | 50.00–2.05 (2.09–2.05) |
| No. of reflections | ||
| Total | 112,298 | 180,543 |
| Unique | 41,752 | 27,433 |
| Rsym or Rmergeb | 0.063 (0.542) | 0.092 (0.568) |
| I/σI | 20.759 (2.667) | 24.100 (3.691) |
| Completeness (%) | 96.2 (98.2) | 99.8 (99.9) |
| Redundancy | 4.5 (4.7) | 4.1 (4.1) |
| Refinement | ||
| Resolution (Å) | 50.00–1.71 | 50.00–2.06 |
| No. of reflections | 40612 | 26122 |
| Rwork/Rfree (%)c | 18.27/21.06 | 20.80/25.33 |
| Root mean square deviation | ||
| Bond length (Å) | 0.019 | 0.006 |
| Bond angle (°) | 1.931 | 1.057 |
| Avg B factor | 32.794 | 33.678 |
| Ramachandran plot quality (%) | ||
| Most favored region | 98.33 | 98.92 |
| Allowed region | 1.67 | 1.08 |
| Disallowed | 0.00 | 0.00 |
Values in parentheses are for the highest-resolution shell.
Rmerge = Σhkl Σi |Ii(hkl) − 〈I(hkl)〉 |/Σhkl Σi Ii(hkl), where Ii(hkl) is the observed intensity and 〈I(hkl)〉 is the average intensity from multiple measurements.
R = Σhkl || Fobs | − k | Fcalc | |Σhkl | Fobs |, where Rfree is calculated for a randomly chosen 5% of reflections and Rwork is calculated for the remaining 95% of reflections used for structure refinement.
FIG 1.

Structural overview and additional disulfide bond of the pAnpl-UAA*01 complex. (A and B) The overall structures of pAnpl-UAA*01–RLI9 (A) and pAnpl-UAA*01–MVM9 (B). pAnpl-UAA*01 chains are shown with distinct colors: for pAnpl-UAA*01–RLI9, H in red and L in olive, and for pAnpl-UAA*01–MVM9, H in marine and L in green). Peptides are shown as spheres using the same colors with their light chains. (C to E) The additional disulfide bond of pAnpl-UAA*01 compared with chicken and human pMHC I complexes. Disulfide bonds are marked with spheres. The additional disulfide bond formed by Cys95-Cys112 of pAnpl-UAA*01 is labeled in a box and amplified to show its position and electronic density map. Both chicken pBF2*2101 (green, Protein Data Bank code 3BEV) and human pHLA-A*0201 (lemon, Protein Data Bank code 3PWN) have just three disulfide bonds distributed in the α2 and α3 domains and β2m chains.
FIG 2.

Structure-based amino acid sequence alignment of Anpl-UAA*01 and representatives of other crystallized MHC I molecules, with the secondary structure elements indicated. Black arrows above the alignment indicate β-strands; cylinders denote α-helices. Green numbers denote residues that form disulfide bonds. Cysteines at positions 95 and 112 are marked by green stars. Conserved residues are highlighted in red. Residues highlighted in green are species-specific amino acids that differ between duck and other animals. Solid blue dots indicate that the residues interact with Anpl-β2m. The total amino acid (AA) identities between Anpl-UAA*01 and the listed MHC I molecules are given at end of each sequence.
The no-peptide refolding product (only Anpl-UAA*01 and Anpl-β2m) was used as a negative control to judge the refolding efficiencies of different peptides; however, a peak (approximately 300 milli-absorbance units [mAU]) eluted at the position representing the pAnpl-UAA*01. The gel filtration and SDS-PAGE results showed that the peak represented the complex of Anpl-UAA*01 and Anpl-β2m (Fig. 3), which is different from results of studies on cattle, pig, chicken, and horse pMHC I refolding (33, 39–41). This result could be due to the Cys95-Cys112 disulfide bond of pAnpl-UAA*01. In order to clarify the impact of this disulfide bond, Cys95 and Cys112 were mutated to alanine (A), alone and together. However, the results for refolding of the mutated Anpl-UAA*01s were similar to those for the wild type (Fig. 3), indicating that the additional disulfide bond is not directly related to the stability of Anpl-UAA*01 without peptide binding.
FIG 3.

Gel filtration and SDS-PAGE results of no-peptide refolding. Anpl-UAA*01 and Anpl-β2m can co-refold in the absence of participation of the peptide, either the wild-type Anpl-UAA*01 heavy chain or the C-A mutant. Different heavy chains and Anpl-β2m are marked in the corresponding positions. Target, co-refolding products eluted via gel filtration. M, protein ladder. (A) Co-refolding results for the wild-type Anpl-UAA*01 and Anpl-β2m. (B) Co-refolding results for the C95A-C112A double mutant heavy chain and Anpl-β2m. (C) Co-refolding results for the C95A mutant heavy chain and Anpl-β2m. (D) Co-refolding results for the C112A mutant heavy chain and Anpl-β2m.
The formation of a complex without the peptide may be due to the strong binding affinity between H and L chains. There are a total of 18 hydrogen bonds and salt bridges between Anpl-UAA*01 and Anpl-β2m (Fig. 4A), which is greater than the numbers found in the pMHC I structures of chickens (13) (pBF2*0401, PDB code 4E0R) and pigs (16) (pSLA-1*0401, PDB code 3QQ3). In addition, the size of the interface between Anpl-UAA*01 and Anpl-β2m is 1,554.2 Å2, which is the largest among the known pMHC I structures of different species (Fig. 4B), including human (PDB code 3PWN), monkey (PDB code 1ZVS), mouse (PDB code 3TID), rat (PDB code 1ED3), swine (PDB code 3QQ3), cattle (PDB code 3PWU), horse (PDB code 4ZUV), and chicken (PDB code 3BEV) (33, 38–44). The interactions and interface area between Anpl-UAA*01 and Anpl-β2m likely contribute to the stability of the complex without binding peptides.
FIG 4.

Strong binding between duck H and L chains. (A) Interactions between H and L chains of pAnpl-UAA*01. pAnpl-UAA*01 is shown in red, with the interactive residues shown as sticks and colored according to the atom type (blue, N; red, O). The interactive residues on the L chain are colored white. The hydrogen bonds and salt bridges are shown as green dashed lines. All of the interactions and interactive residues are magnified within a blue dotted box and labeled. (B) Interface areas of the L chains of pAnpl-UAA*01 and pMHC I structures from the other known species. All the L chains are surface representations with different colors. The white areas indicate the interactive regions contacting H chains. Detailed interface areas are shown under each structure. The structures are as follows: chicken (BF2*2101, PDB code 3BEV), human (HLA-A2, PDB code 3PWN), bovine (BoLA-N*01801, PDB code 3PWU), swine (SLA-1*0401, PDB code 3QQ3), horse (Eqca-N*00602, PDB code 4ZUV), rat (RT1-Aa, PDB code 1ED3), macaque (Mamu-A*01, PDB code 1ZVS), and mouse (H-2 Kb, PDB code 3TID).
The distinct peptide conformations presented by the Anpl-UAA*01 molecule.
The conformations of IAV-MVM9 and IAV-RLI9 presented by Anpl-UAA*01 are distinct, especially in the middle region from the residue at position 3 (P3) to that at P6 (Fig. 5). The IAV-RLI9 peptide is clear on the electronic density map and adopts an “M” overall conformation, which is common to other pMHC I structures. The B factors of the IAV-RLI9 middle part are relatively higher than at the N and C termini, indicating that P4 to P6 are more flexible than other residues (Fig. 5A). In IAV-MVM9, the electronic density map is missing at P3 and P4, which means that these two residues are quite flexible (Fig. 5B). The side chain orientations of P5 are obviously different between the IAV-MVM9 and IAV-RLI9 peptides. In IAV-MVM9, the side chain of P5 stretches to the α2 helix but in RLI9, its side chain stretches upward and most parts are exposed from the PBG (Fig. 5C).
FIG 5.

Divergent presentations of RLI9/MVM9 peptides presented by Anpl-UAA*01. (A and B) Electron densities and overall conformations of the structurally defined peptides RLI9 and MVM9. Simulated annealing omit maps (CNS) calculated for the two peptides are shown in blue at a contour of 1.5, and the coloration of the two peptides is according to the isotropic B factors. (C) General side chain orientations and the different interfacing areas of RLI9/MVM9 presented by Anpl-UAA*01 PBG, as viewed in profile from the peptide N terminus toward the C terminus. Black arrows indicate the directions in which the residues point: up is toward the T cell receptor, down is toward the floor of the peptide binding groove, left is toward the α1 helix domain, and right is toward the α2 helix domain. Pockets accommodating each residue are listed under the corresponding anchors within the peptide binding groove. ASA, accessible surface area of each residue; BSA, buried surface areas of the residues.
The conformations of the MVM9 and RLI9 peptides were significantly distinct when the two pAnpl-UAA*01 structures were superimposed (Fig. 6A and B); their deviation is focused mainly at the center, where the distance between the Cα atoms of MVM9 and RLI9 P5 residues can reach 4.4 Å. The N and C termini of the two peptides matched well. Structural analysis showed that the N termini of the MVM9 and RLI9 peptides (P1 and P2 residues) form the same hydrogen bonds with the PBG (Fig. 6C). From the P3 to P7 residues, there are no hydrogen bonds between RLI9 and the PBG. Although the P3 and P4 residues are missing in MVM9, three hydrogen bonds were found between P5, P7, and the PBG (Fig. 6D). These distinct peptide conformations of this part should be related to the differences in hydrogen bonds found here. In the C termini, the hydrogen bonds between the main chain of peptides and the PBG are the same; the only difference is that the Lys142 of pAnpl-UAA*01 forms 3 hydrogen bonds with the side chains of the P8 and P9 residues in the RLI9 peptide but does not form a bond with the MVM9 peptide (Fig. 6E).
FIG 6.

Comparisons of dissimilar conformations and interactions of RLI9/MVM9 peptides. (A and B) Superposition of RLI9 (olive) and MVM9 (green) presented by Anpl-UAA*01. To conveniently observe the different conformations of the two peptides, the side chains of residues are presented. Both the side (A) and top (B) views of the peptide alignments demonstrate that most conformational distinctions are located in the central region of the peptides, with the farthest distance between the backbone carbon atoms on P4 of the two peptides marked using red dashes. (C to E) Hydrogen bond comparisons of RLI9 and MVM9 peptides interacting with their PBGs. The interacting residues on PBG are shown (pAnpl-UAA*01–RLI9, red; pAnpl-UAA*01–MVM9, blue), and the hydrogen bonds are represented as dashed lines with the corresponding colors. (C) Similar interactions at the P1 and P2 positions of RLI9/MVM9 peptides with the residues in the A pocket of Anpl-UAA*01. (D) Different conformations and hydrogen bonds between the central region of peptides and Anpl-UAA*01. Three hydrogen bonds are formed in pAnpl-UAA*01–MVM9, and no hydrogen bonds are found in pAnpl-UAA*01–RLI9. (E) Interactions between the C-terminal residues of the RLI9/MVM9 peptides and their PBGs. Three more hydrogen bonds of RLI9 with the Lys142 residues of Anpl-UAA*01 are observed.
These data suggest that the conformation of the middle section of the peptide presented by Anpl-UAA*01 is flexible and is determined by the peptide's specific amino acid sequence.
Anpl-UAA*01 selects peptides by relying on the B and F pockets.
The compositions of the six pockets of Anpl-UAA*01 are shown in Fig. 7, and the interactions between peptides and these pockets are listed in Table 2. The RLI9 peptide was selected to illustrate the interactions with the six pockets in Fig. 6 because of its completeness.
FIG 7.

Composition of the pockets of Anpl-UAA*01. Pockets are shown as surface representations in light pink. Residues comprising these pockets (light pink) and the bound peptide RLI9 (C, olive; N, blue; O, red) are labeled. The hydrogen bonds between RLI9 and pockets are shown as red dashed lines. (A) Pocket A with the P1-Arg. (B) Pocket B with the P2-Leu. (C) Pocket C with the P6-Ser. The additional disulfide bond are shown as thick sticks and colored by atom type (C, red; N, blue; O, red; S, yellow). (D) Pocket D with the P3-Ile. (E) Pocket E with the P7-Ile. (F) Pocket F with the PC-Ile.
TABLE 2.
Interactions between the RLI9/MVM9 peptide and Anpl-UAA*01
| Complex | Hydrogen bonds and salt bridges |
van der Waals contact residuesa | |||
|---|---|---|---|---|---|
| Peptide |
Heavy chain |
||||
| Residue | Atom | Residue | Atom | ||
| pAnpl-UAA*01–RLI9 | P1-Arg | N | Tyr168 | OH | Leu5, Tyr7, Tyr58, Glu62, Tyr156, Thr160, Trp164, Tyr168 (59) |
| N | Tyr7 | OH | |||
| O | Tyr156 | OH | |||
| P2-Leu | N | Glu62 | OE1 | Tyr7, Tyr9, Thr24, Met43, Glu62, Thr65, Ser66, Tyr156 (46) | |
| P3-Ile | Thr65, His97, Trp153, Tyr156 (42) | ||||
| P4-Gln | Arg61, Thr65 (18) | ||||
| P5-Asn | (0) | ||||
| P6-Ser | Asn69, Ile72 (5) | ||||
| P7-Ile | Trp143, Lys142, Glu146, Val149, Ile72 (21) | ||||
| P8-Thr | OG1 | Lys142 | NZ | Ile72, Lys142, Asn76, Val75, Trp143 (31) | |
| O | Lys142 | NZ | |||
| O | Trp143 | NE1 | |||
| P9-Ile | N | Asn76 | OD1 | Lys142, Trp143, Asn76, Thr139, Phe119, Thr79, Ala80, Arg83, Trp93 (69) | |
| OXT | Arg83 | NH2 (S) | |||
| OXT | Thr139 | OG1 | |||
| O | Lys142 | NZ (S) | |||
| Total | 11 | 291 | |||
| pAnpl-UAA*01–MVM9 | P1-Met | N | Tyr7 | OH | Leu5, Tyr7, Tyr58, Arg61, Glu62, Tyr156, Trp164, Tyr168 (56) |
| O | Tyr156 | OH | |||
| N | Tyr168 | OH | |||
| P2-Val | N | Glu62 | OE1 | Tyr7, Tyr9, Glu62, Thr65, Asn69, His97, Tyr156 (37) | |
| P5-Leu | O | Trp153 | NE1 | Gln152, Trp153, Tyr156 (33) | |
| P6-Ile | Tyr9, Asn69, Ile72, Phe73, Trp153 (26) | ||||
| P7-Arg | O | Asn76 | ND2 | Ile72, Asn76, Trp143, Glu146, Val149, Gln152 (31) | |
| NH2 | Glu146 | OE2 (S) | |||
| P8-Met | O | Trp143 | NE1 | Ile72, Val75, Asn76, Lys142, Trp143 (22) | |
| P9-Ile | N | Asn76 | OD1 | Asn76, Thr79, Ala80, Arg83, Trp93, Phe119, Thr139, Lys142, Trp143 (70) | |
| OXT | Arg83 | NH2 (S) | |||
| OXT | Thr139 | OG1 | |||
| Total | 11 | 275 | |||
Numbers in parentheses are the amounts of van der Waals force.
The A pocket of Anpl-UAA*01, composed of Leu5, Tyr7, Tyr58, Glu62, Tyr156, Thr160, Cys161, Trp164, and Tyr168, fixes P1-Arg by hydrogen bonds and strong van der Waals forces (VDWs) (Fig. 7A; Table 2). As in the known pMHC I structures, P1-Arg forms the hydrogen bonds with the A pocket by its main-chain atoms, and its side chain stretches upward out of the A pocket (45, 46). Therefore, although the binding between the A pocket and P1 residue is strong, the A pocket dose not play a restrictive role in the PBG of Anpl-UAA*01.
The B pocket is a primary anchor site and plays a restrictive role in peptide binding. The B pocket of Anpl-UAA*01 accommodates P2-Leu (Fig. 7B) The charged Glu62, which is on the top of the B pocket, can form a hydrogen bond with the main chain of P2-Leu. The side chain of the P2 residue inserts into the B pocket and is fixed by the VDWs provided by the surrounding residues (Table 2).
The C, D, and E pockets usually connect with residues in the middle part of the binding peptides. The amino acid compositions of these three pockets of Anpl-UAA*01 are shown in Fig. 7C to E. The C and D pockets can interact with the side chains of the P3 and P6 residues, respectively, but the E pocket can only interact with the main chain of the P7 residue, as the orientation of the P7 side chain faces upward (Table 2).
The additional disulfide bond formed by Cys95 and Cys112 is in the C pocket of Anpl-UAA*01. The impact of this disulfide bond on peptide binding was checked by the refolding of MVM9 and RLI9 peptides with the C95A, C112A, and C95A-C112A double mutant heavy chains. We found that that the mutant H chains can still form a complex with peptides, but the refolding is worse than that of the wild-type H chain, especially for the C112A mutant H chain (Fig. 8A and B). The reduced refolding efficiencies of the mutants indicated that the disulfide bound in the C pocket helps with the peptide binding of Anpl-UAA*01; however, the distances between the peptide residues and disulfide bond are over 5.0 Å, which means that their direct interactions are negligible (Fig. 6C). Previous studies have suggested that Cys95 and Cys112 do not meet the preferred geometry to form the disulfide bond (30), and our pAnpl-UAA*01 structures show that the distance between Cys95 and Cys112 is less than that for the corresponding positions in HLA-A2 and B21 (Fig. 8C). To some extent, the additional disulfide bond of Anpl-UAA*01 alters the bottom of the C pocket and strengthens the stability. Improved peptide binding efficiency may strengthen the stability of the C pocket with the additional disulfide bound.
FIG 8.

Mutants of Anpl-UAA*01 influence the in vitro refolding efficiency with L chain and RLI9/MVM9 peptides. The refolding products were analyzed by chromatography on a Superdex 200 10/300 GL column (GE Healthcare). Curves in different colors indicate different H chains. (A) Refolding results for the wild-type Anpl-UAA*01 and three mutants with the MVM9 peptide. (B) Refolding results with the RLI9 peptide. (C) The bottom β sheet of Anpl-UAA*01 is more compact than those in B21 and HLA-A2. All the structures are in ribbon models. The distances between C95 and C112 in Anpl-UAA*01 and their corresponding positions in B21 and HLA-A2 are labeled by dashed lines.
The F pocket is the most important anchor site at the C terminus of PBG. The F pocket of Anpl-UAA*01 is composed of Asn76, Thr79, Ala80, Arg83, Tyr84, Trp93, Phe119, and Ala135, and it has a strong binding affinity with P9-I (Fig. 7F). The main chain of P9-I can form 2 salt bridges with Arg83 and Lys142 and 2 hydrogen bonds with Asn76 and Thr139 in the F pocket. The side chain of P9-I inserts into the F pocket and is fixed by the strong hydrophobic forces (Table 2).
In order to determine the primary anchor residues of Anpl-UAA*01-presenting peptides and the vital restriction pockets for peptide binding, the RLI9 peptide was mutated by alanine scanning and circular dichroism (CD) spectral analysis was used to test the stabilities of Anpl-UAA*01 complexes with these mutant peptides (Fig. 9). The wild-type RLI9 peptide was used as a control, and its midpoint transition temperature (Tm) value was 43.3°C. Among all of the alanine mutant peptides, only the Tm values of the P2-Ala and P9-Ala mutant peptides were significantly lower than that of the wild-type RLI9 peptide, indicating that the side chains of P2 and P9 play key roles in RLI9 peptide binding and that these two residues are the primary anchor residues. The pockets (B and F) at the two termini of the Anpl-UAA*01 PBG anchor the peptides and determine the peptide binding motif of Anpl-UAA*01.
FIG 9.

P2 and P9 of the peptide are critical for binding to Anpl-UAA*01. The denaturation and elution curves of the complexes with the different peptides are indicated in different colors. (A) Thermostabilities of Anpl-UAA*01 with the RLI9 peptide and its substitutions (residues at each position in RLI9 are replaced with alanine). Circular dichroism spectropolarimetry was utilized to assess the thermostabilities of purified pAnpl-UAA*01 complexes. Denaturation was monitored at 218 nm as the temperature was ramped up from 25 to 75°C at 1°C/min. Shown here are the data for fitting to the denaturation curves using the Origin 9.1 program (OriginLab).The Tms of different peptides are indicated by the gray line at 50% fraction unfolded. (B and C) Peptide-induced assembly and stabilization assay of Anpl-UAA*01 and Anpl-β2m with RLI9 and its mutants by in vitro refolding. (B) Gel filtration chromatograms of the refolded products obtained using a Superdex 200 10/300 GL column (GE Healthcare). The aggregated H chain, the correctly refolded pAnpl-UAA*01 complex (∼45 kDa), and the extra β2m are indicated. The refolding efficiencies are represented by the heights of pAnpl-UAA*01 complex peaks. A higher peak indicates a better efficiency of the peptide to help the MHC renature. (C) Results of further stabilization assays of the refolded products tested by anion exchange. A higher peak here also indicates better stability. With peptides P2-A and P9-A, the refolded complex proteins dissociated at the corresponding eluting NaCl concentration (9% to 11%), implying poor stabilities.
The peptide binding motif of Anpl-UAA*01 is most similar to that of HLA-A2.
The peptide binding manner of pAnpl-UAA*01 is similar to those of some mammalian pMHCs but not similar to that of chicken pMHC I; only the B and F pockets are the key anchor caves, similar to what is observed in human MHC I (also named human lymphocyte antigen [HLA]). By comparison with the solved structures in the Protein Data Bank (PDB), the peptide conformation and binding manner of pAnpl-UAA*01 were found to be most similar to those of HLA-A2 (Fig. 10). In the B pockets of Anpl-UAA*01 and HLA-A2, the side chains of P2-Leu residues have similar orientations and are fixed in a similar way by the residues at the same positions in the two pockets (Fig. 10A). A similar situation was also found in the two F pockets, where the hydrogen bonds fixing P9-I are almost the same (Fig. 10B). The peptide presentations and pocket compositions make the peptide conformations of pAnpl-UAA*01 and pHLA-A2 almost identical (Fig. 10C and D).
FIG 10.

Alignments of B pocket, F pocket, and binding peptides indicate similar peptide binding motifs of Anpl-UAA*01 with HLA-A*0201 (PDB code 3PWN). Residues making up these pockets and binding peptides are as follows: for pAnpl-UAA*01, C, red; N, blue; O, red; and for pHLA-A*0201, C, lemon; N, blue; O, red). (A and B) B and F pocket alignments of Anpl-UAA*01 with HLA-A*0201. The same compositions of P2-Leu and P9-Ile in both RLI9/HuDG2L peptides are shown using the same color as their H chains. Hydrogen bonds formed by P2/P9 and their H chains are represented as dashed lines with each corresponding color. (C and D) Side and top views of the RLI9 peptide alignment with HuDG2L peptide presented by HLA-A*02. Both of the peptides' main chains are shown as bold sticks. Shared P2-Leu and P9-Ile in RLI9/HuDG2L peptides are colored black in their sequences in the upper left corner. (E and F) Residues fitting the B and F pockets of Anpl-UAA*01 and HLA-A*0201 can mostly overlap. Residues within the solid red circle are motifs for Anpl-UAA*01, and those within the circle are motifs for HLA-A*0201.
Previous studies have shown that the B pocket of HLA-A2 prefers hydrophobic anchor residues, such as I, L, V, A, and F, and can also accommodate the same polar residues, such as T. The most frequently occurring anchor residues in the F pocket of HLA-A2 are I, V, and L. The two peptides presented by Anpl-UAA*01, MVM9 and RLI9, perfectly fit the peptide binding motif of HLA-A2. Eighteen additional peptides from IAV and 11 mutant peptides were used to verify the peptide binding motif of Anpl-UAA*01 (Table 3). The results confirmed that the peptide binding motifs of these two MHC I molecules from different species are mostly overlapping (Fig. 10E and F). In brief, the nonapeptide binding motif of Anpl-UAA*01 is x-(A, V, I, L, M, T, S, or F)-x-x-x-x-x-x-(A, V, I, L, M, or F).
TABLE 3.
Predicted peptides used in this study and their binding to Anpl-MHC I, evaluated by in vitro refolding
| Name | Derived protein | Position | Sequence | % randoma | Stability with Anpl-MHC Ib |
|---|---|---|---|---|---|
| IAY9 | Nucleocapsid protein (IAV) | 45–53 | IAYERMCNI | 0.185 | ++ |
| RTS9 | Nucleocapsid protein (IAV) | 22–30 | RTSDMRTEI | 0.168 | ++ |
| MVM9 | Nucleocapsid protein (IAV) | 17–25 | MVMELIRMI | 0.172 | ++ |
| RLI9 | Nucleocapsid protein (IAV) | 55–63 | RLIQNSITI | 0.245 | ++ |
| WMA9 | Nucleocapsid protein (IAV) | 68–76 | WMACHSAAF | 0.213 | ++ |
| IFL9 | Nucleocapsid protein (IAV) | 18–26 | IFLARSALI | 0.125 | ++ |
| VSG9 | Nucleocapsid protein (IAV) | 28–36 | VSGIGRFYI | 0.134 | ++ |
| ATA9 | Nucleocapsid protein (IAV) | 53–61 | ATAGLTHLM | 0.200 | +/− |
| SLAP5 | Hemagglutinin (IAV) | 25–33 | TSADQQSLY | 0.100 | +/− |
| SLAP9 | Polymerase PA (IAV) | 6–14 | GTFDLGGLY | 0.041 | +/− |
| AI-4 | Hemagglutinin (IAV) | 442–450 | VAMENQHTI | 0.387 | ++ |
| EN5 | Gag protein (IAV) | 226–234 | MTARFIRGL | 0.130 | ++ |
| KMN-P9I | Hemagglutinin (IAV) | 402–410 | KMNTQFIAI | 0.207 | ++ |
| NS1-VKN | Nonstructural protein 1 (IAV) | 174–182 | VKNAVGVLI | 0.054 | +/− |
| PA-ENK | Polymerase PA (IAV) | 613–621 | ENKSETWPI | 0.035 | +/− |
| PA-KTN | Polymerase PA (IAV) | 497–505 | KTNLYGFII | 0.150 | ++ |
| HA-ITN | Hemagglutinin (IAV) | 393–401 | ITNKVNSVI | 0.162 | ++ |
| HA-YIN | Hemagglutinin (IAV) | 182–190 | YINDKGKEV | 0.073 | +/− |
| PA-RRN | Polymerase PA (IAV) | 442–450 | RRNYFTAEV | 0.059 | +/− |
| PB1-KM | Polymerase PB1 (IAV) | 531–539 | KNNMINNDL | 0.046 | +/− |
| RLI9-P1A | Mutant RLI9 | ALIQNSITI | ++ | ||
| RLI9-P2A | Mutant RLI9 | RAIQNSITI | +/− | ||
| RLI9-P3A | Mutant RLI9 | RLAQNSITI | ++ | ||
| RLI9-P4A | Mutant RLI9 | RLIANSITI | ++ | ||
| RLI9-P5A | Mutant RLI9 | RLIQASITI | ++ | ||
| RLI9-P6A | Mutant RLI9 | RLIQNAITI | ++ | ||
| RLI9-P7A | Mutant RLI9 | RLIQNSATI | ++ | ||
| RLI9-P8A | Mutant RLI9 | RLIQNSIAI | ++ | ||
| RLI9-P9A | Mutant RLI9 | RLIQNSITA | +/− | ||
| RLI9-P9V | Mutant RLI9 | RLIQNSITV | +/− | ||
| RLI9-P9L | Mutant RLI9 | RLIQNSITL | ++ |
Base value for estimating the binding affinities of peptides with the NetMHCpan 3.0 server (http://www.cbs.dtu.dk/services/NetMHCpan/): rank threshold for strongly binding peptides, 0.100; rank threshold for weakly binding peptides, 1.000.
++, peptide binds strongly and can tolerate anion-exchange chromatography; +/−, peptide binds Anpl-MHC I but cannot tolerate anion-exchange chromatography.
Many IAV peptides match the motif of pAnpl-UAA*01.
Using the motif of Anpl-UAA*01 that we identified, the proteomes of several IAV strains were screened to identify the epitope peptides that could be presented by Anpl-UAA*01, including the H1N1, H3N2, H3N8, H4N6, H5N1, H7N9, and H9N2 subtypes (47–53). There were approximately 600 candidate nonapeptides from different IAV strains matching the peptide binding motif of Anpl-UAA*01 (Fig. 11). Most selected peptides also fit the motif of HLA-A2, and 11 nonapeptides were proven to activate HLA-A2-restricted CTL responses efficiently according to IEDB data (http://www.iedb.org/) (54). The in vitro refolding results for 31 peptides confirmed that the predicted IAV epitopes could be bound by Anpl-UAA*01 (Table 3).
FIG 11.
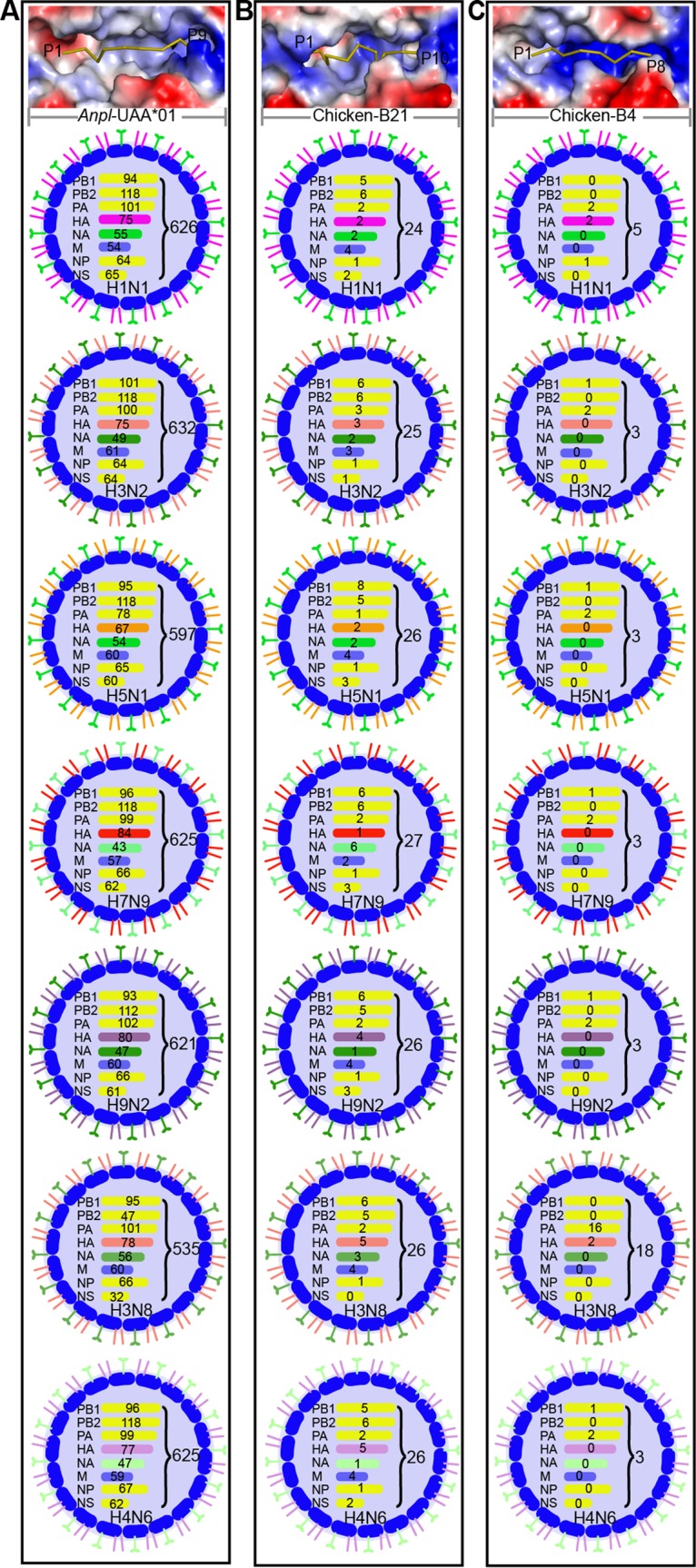
Peptide predictions from different influenza virus subtypes according to the distinct binding motifs of Anpl-UAA*01, BF2*0401, and BF2*2101. Genome-wide scanning results of peptides matching the motifs of Anpl-UAA*01 (A), BF2*0401 (B), and BF2*2101 (C) are as follows: Anpl-UAA*01, x-(A, V, I, L, M, T, S, or F)-x-x-x-x-x-x-(A, V, I, L, M, or F); BF2*0401: x-(D, E)-x-x-(D, E)-x-x-E; BF2*2101, x-(H, K, or R)-x-x-x-x-x-(E or D)-x-(A V, L, I, F, or W), x-(E or D)-x-x-x-x-x-L-x-(A, V, L, I, F, or W). The numbers of peptides derived from the PB1, PB2, PA, HA, NA, M, NP, and NS proteins of each IAV subtype are marked, as well as the total numbers. Vacuum electrostatic surface potential presentations (red, negative; blue, positive; gray, neutral) and predominant lengths of binding peptides of PBGs in Anpl-UAA*01, BF2*0401, and BF2*2101 are shown below. The peptides are shown as ribbons (olive) with their lengths labeled: nonamers for Anpl-UAA*01, octamers for B4, and 10-mers for B21.
Based on the motifs of chicken B4 and B21, the longer peptides from IAV strains were also screened. There are fewer than 20 octapeptides for B4 and 24 to 27 decapeptides for B21 (Fig. 11), which are much less than the numbers of peptides found in Anpl-UAA*01.
DISCUSSION
The most distinguishable feature of Anpl-UAA*01 was the additional disulfide bond in the PBG. The mutant experiments showed that this disulfide bond located in the C pocket can increase the efficiency of peptide binding to pAnpl-UAA*01. Although all of the mutants could still form complexes with the RLI9/MVM9 peptide and β2m, their refolding efficiencies were significantly lower than that of the wild type (Fig. 8A and B). Without the peptide, this disulfide bond did not increase the refolding production of the complex of Anpl-UAA*01 and β2m (Fig. 3). These data indicate that this additional disulfide bond can improve peptide binding to Anpl-UAA*01 and lead to the increased formation of a trimer complex; however, no significant contacts between the presented peptide and disulfide bond were identified. We also discovered that the additional disulfide bond makes the bottom β sheet of the C pocket of Anpl-UAA*01 more compact than in other MHC I molecules. The refolding effect of the C112A mutant indicated that the free side chain of Cys95 could disturb peptide binding in the C pocket region (Fig. 8A and B). Therefore, we hypothesize that this additional disulfide bond provides a more stable C pocket, which helps with peptide presentation of Anpl-UAA*01.
Another characteristic of the Anpl-UAA*01 structure is the intensive interaction between its H and L chains. In comparison with other known pMHC I structures, pAnpl-UAA*01 has the greatest interface area and many interchain bonds (Fig. 5). The interface area in ducks and chickens is significantly larger than that in mammals. Compared with BF2*2101 (32), pAnpl-UAA*01 had a 100-Å2-larger interface area and 4 more hydrogen bonds; this relatively stronger interaction could lead to the formation of complexes containing only Anpl-UAA*01 and β2m through in vitro refolding, which has never been identified in pMHC I structures of other species (33, 39–41).
Anpl-UAA*01 is similar to human HLA in peptide presentation (but not to chicken BF2), although the amino acid identities among Anpl-UAA*01 and BF2 molecules are much higher than those between Anpl-UAA*01 and HLA (>60% versus <50%, respectively). There are only two primary anchor pockets in pAnpl-UAA*01, the B and F pockets, which is common in HLA molecules (Fig. 10A and B). Using structural analysis and mutant peptide refolding tests, we found that the peptide binding motif of pAnpl-UAA*01 overlaps with HLA-A*0201 to a large degree. The B pocket of Anpl-UAA*01 can accommodate extensive uncharged residues, and the F pocket can accommodate multiple hydrophobic residues (Fig. 10E). However, BF2 exhibits peptide-presenting strategies that differ from those of HLA. B4 has a narrow and highly charged PBG, which limits the binding peptides that must fit its B, C, and F pockets together (33). B21 has a large central cavity and flexible Arg9, which make its binding motif promiscuous with three anchor residues (32). The moderate limits of the peptide binding motif suggest that ducks can accommodate more peptides from pathogens than chickens.
The role of MHC I mediating the CTL response in clearance of IAV has been confirmed in human and mouse. Although substantial data on anti-IAV CTL responses in duck and chicken are scarce, some evidence indicates that duck MHC genes respond to infection with IAV (13, 55). The studies showed that duck MHC locus genes (MHC I/II and TAP) are overexpressed after infection with IAV, especially the duck MHC I gene, and under the condition of HP-IAV infection its expression could be increased by about 1,000 times in the lung (13). Unlike mammals, both ducks and chickens have a “minimal MHC” region with a limited set of genes and express only one dominant MHC I allele. The “minimal MHC” in chickens is critical to defense against a particular pathogen because it is completely dependent on whether or not it can load peptides from that pathogen. The best illustrated example is that chickens of the BF2*2101 genotype could defend against MDV, while chickens of the BF2*0401 genotype could not (32, 33, 56). The difference in duck versus chicken responses to IAV might also relate to the different peptide loading abilities of duck and chicken MHC I. Approximately 600 nonapeptides from different IAV strains matching the motif of pAnpl-UAA*01 were identified in this study, which is much higher than the numbers of BF2*2101 and BF2*0401 from chicken (Fig. 11; see Data Set S1 in the supplemental material). Therefore, the duck Anpl-UAA*01 should exhibit stronger resistance to IAV strains than chicken BF2*2101 and BF2*0401 (57, 58). In addition, the Anpl-UAA*01 structure showed that it can present IAV peptides in distinct conformations, which is believed to be useful for activating the T cell repertoire and inducing stronger CTL responses. Our study illustrates the structural basis of duck MHC I and provides a novel approach for explaining why ducks are more resistant to HP-IAV.
MATERIALS AND METHODS
Prediction and synthesis of IAV-derived peptides.
All nonamer peptides that potentially bound to Anpl-UAA*01 were predicted using the NetMHCpan 3.0 server (http://www.cbs.dtu.dk/services/NetMHCpan/) (59, 60). The 31 peptides used in this study (Table 3) were synthesized and purified to 90% by reverse-phase high-performance liquid chromatography (HPLC) and mass spectrometry (SciLight Biotechnology). These peptides were stored in lyophilized aliquots at −80°C after synthesis and dissolved in dimethyl sulfoxide (DMSO) before use.
Protein preparation.
DNA fragments encoding extracellular domains of Anpl-UAA*01 (GenBank accession no. AB115245, residues 1 to 270 of the mature protein with EcoRI and HindIII restriction sites) (37) and Anpl-β2-microglobulin (Anpl-β2m) (GenBank accession no. AB246408, residues 1 to 101 of the mature protein) were synthesized by Shanghai Invitrogen Life Technologies and then cloned into pET21a(+) vectors (Novagen) and expressed in Escherichia coli BL21(DE3). The recombinant Anpl-UAA*01 and β2m were expressed in inclusion bodies and purified as previously described (61, 62). Finally, the Anpl-UAA*01 heavy chain and Anpl-β2m bodies were separately dissolved in 6 M guanidinium chloride buffer to a protein concentration of 30 mg/ml.
Assembly of the pAnpl-UAA*01 complex.
To assemble the pAnpl-UAA*01 complex, the peptide, Anpl-UAA*01, and Anpl-β2m inclusion bodies were refolded (in a 1:1:1 molar ratio) according to the gradual dilution method that we described previously (39, 63). After a 24-h refolding step at 277 K, the remaining soluble portion of the complex was concentrated and purified using a Superdex 200 16/60 column (GE Healthcare), followed by Resource Q anion-exchange chromatography (GE Healthcare). Purified proteins were buffer exchanged with 10 mM Tris-HCl and 50 mM NaCl at a pH of 8.0.
Crystallization and data collection.
The purified pAnpl-UAA*01 complex was ultimately concentrated to 10 mg/ml. After mixing with reservoir buffer at a 1:1 ratio, the purified protein was crystallized using the sitting-drop vapor diffusion technique at 277 K. Index, Crystal Screen I/II, and Crystal Screen Cryo I/II kits (Hampton Research, Riverside, CA) were used to screen for optimal crystal growth conditions. After several days, crystals (pAnpl-UAA*01–MVM9 and pAnpl-UAA*01–RLI9) were observed with solutions NO.7 from the Crystal Screen Cryo II kit (8% polyethylene glycol 1000, 8% [wt/vol] polyethylene glycol 8000, and 20% [vol/vol] glycerol) and NO.43 from the Crystal Screen Cryo I kit (24% [wt/vol] polyethylene glycol 1500 and 20% [vol/vol] glycerol), respectively. Diffraction data for pAnpl-UAA*01 crystals were collected to resolutions of 1.71 Å (pAnpl-UAA*01–MVM9) and 2.06 Å (pAnpl-UAA*01–RLI9) at the Shanghai Synchrotron Radiation Facility (SSRF) using beamline BL17U at a wavelength of 1.5418 Å (Shanghai, China) (64).The crystals were first soaked in reservoir solution containing 25% glycerol as a cryoprotectant and were then flash-cooled in a stream of gaseous nitrogen at 100 K (65). The collected intensities were indexed, integrated, corrected for absorption, scaled, and merged using the HKL2000 package (66).
Structure determination and refinement.
The crystals of pAnpl-UAA*01–MVM9/RLI9 all belong to the P1211 space group, and their structures were solved by molecular replacement using Molrep and Phaser in the CCP4 package, with the chicken BF2*0401 structure (PDB code 4E0R) as the search model (67–69). Extensive model building was performed by hand with COOT (70), and restrained refinement was performed using REFMAC5. Additional rounds of refinement were performed using the Phenix refine program implemented in the PHENIX package (71) together with isotropic atomic displacement parameter (ADP) refinement and bulk solvent modeling. The stereochemical quality of the final model was assessed with the PROCHECK program (72). Detailed information about collection and refinement is shown in Table 1.
Structural analysis and generation of illustrations.
Peptide-contacting residues were identified using the program CONTACT and were defined as residues containing an atom within 3.3 Å of the target partner (69). Structural illustrations and electron density-related figures were generated using the PyMOL molecular graphics system (http://www.pymol.org/). Solvent-accessible surface areas and the B factor were calculated with CCP4.
Preparation of Cys-to-Ala mutants of pAnpl MHC I.
To investigate the function of the additional disulfide bond in Anpl-UAA*01, Cys95 and Cys112 were mutated to Ala by overlap PCR (the primers used for Cys95-to-Ala mutation were 5′-GGCAGGCGATGCATGGCTGTG-3′ and 5′-GCCATGCATCGCCTGCCATG-3′ and those for Cys112-to-Ala mutation were 5′-CAACAAGCGGGCTATGATGGG-3′ and 5′-CCCATCATAGCCCGCTTGTTG-3′, where the underlined sequences mutated the codon encoding Ala). These mutants were inserted into the pET21a vector and expressed in BL21(DE3) cells. The mutants were termed Anpl-UAA*01-C95A, Anpl-UAA*01-C112A, and Anpl-UAA*01-C95A-C112A, respectively. Recombinant mutants were expressed as inclusion bodies and further purified as described above. The mutant inclusion bodies were refolded with Anpl-β2m using the in vitro gradual dilution method as described above (61, 62). In addition, all of the mutant pAnpl-UAA*01 complexes formed by refolding were further purified by gel filtration and anion-exchange chromatography as described above (39, 63).
CD spectra and thermal unfolding.
Circular dichroism (CD) experiments for the pAnpl-UAA*01 with parental RLI9 or mutant peptides were performed on a Jasco J-810 spectropolarimeter equipped with a water-circulating cell holder. The CD spectra were collected at a protein concentration of 8 mΜ in pH 8.0 Tris buffer (20 mM Tris and 50 mM NaCl), using a 1-mm-optical-path-length cuvette with ellipticity at 218 nm. Thermal denaturation curves were determined as the temperature was raised from 25 to 80°C at a linear rate of 1°C/min. The temperature of the sample solution was directly measured with a thermistor. The fraction of unfolded protein was calculated from the mean residue ellipticity (θ) using the standard method. The unfolded fraction (%) was expressed as (θ − θN)/(θU − θN), where θN and θU are the mean residue ellipticity values in the fully folded and fully unfolded states, respectively. The midpoint transition temperature (Tm) was determined by fitting data to the denaturation curves using the Origin 9.1 program (OriginLab) (73).
Accession number(s).
The coordinates and structure factors for pAnpl-UAA*01–RLI9 and pAnpl-UAA*01–MVM9 have been deposited in the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) under accession numbers 5GJX and 5GJY, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported financially by grants from the National Basic Research Program of China (973 Program) (2013CB835302, http://program.most.gov.cn), the 13th Five-Year National Key Research project (2016YFD0500106, http://program.most.gov.cn/), and the National Natural Science Foundation of China (NSFC) (31572493, http://www.nsfc.gov.cn).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We thank the staff at the Shanghai Synchrotron Radiation Facility of China (SSRF) for their technical assistance during data collection.
We declare no conflict of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.02511-16.
REFERENCES
- 1.Kong W, Wang F, Dong B, Ou C, Meng D, Liu J, Fan ZC. 2015. Novel reassortant influenza viruses between pandemic (H1N1) 2009 and other influenza viruses pose a risk to public health. Microb Pathog 89:62–72. doi: 10.1016/j.micpath.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Del Rio C, Franco-Paredes C. 2009. The perennial threat of influenza pandemics. Arch Med Res 40:641–642. doi: 10.1016/j.arcmed.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Yen HL, Webster RG. 2009. Pandemic influenza as a current threat. Curr Top Microbiol Immunol 333:3–24. doi: 10.1007/978-3-540-92165-3_1. [DOI] [PubMed] [Google Scholar]
- 4.Capua I, Alexander DJ. 2007. Animal and human health implications of avian influenza infections. Biosci Rep 27:359–372. doi: 10.1007/s10540-007-9057-9. [DOI] [PubMed] [Google Scholar]
- 5.Huang YH, Li YR, Burt DW, Chen HL, Zhang Y, Qian WB, Kim H, Gan SQ, Zhao YQ, Li JW, Yi K, Feng HP, Zhu PY, Li B, Liu QY, Fairley S, Magor KE, Du ZL, Hu XX, Goodman L, Tafer H, Vignal A, Lee T, Kim KW, Sheng ZY, An Y, Searle S, Herrero J, Groenen MAM, Crooijmans RPMA, Faraut T, Cai QL, Webster RG, Aldridge JR, Warren WC, Bartschat S, Kehr S, Marz M, Stadler PF, Smith J, Kraus RHS, Zhao YF, Ren LM, Fei J, Morisson M, Kaiser P, Griffin DK, Rao M, Pitel F, Wang J, Li N. 2013. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet 45:776. doi: 10.1038/ng.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinshaw VS, Webster RG, Turner B. 1980. The perpetuation of orthomyxoviruses and paramyxoviruses in Canadian waterfowl. Can J Microbiol 26:622–629. doi: 10.1139/m80-108. [DOI] [PubMed] [Google Scholar]
- 7.Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992. Evolution and ecology of influenza A viruses. Microbiol Rev 56:152–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JK, Negovetich NJ, Forrest HL, Webster RG. 2009. Ducks: the “Trojan horses” of H5N1 influenza. Influenza Other Respir Viruses 3:121–128. doi: 10.1111/j.1750-2659.2009.00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guan XT, Li YB, Liu JL, Chai HL, Wang YL, Hua YP. 2014. Evaluation of specific pathogen-free ducks infected with the highly pathogenic avian influenza virus H5N1 subtype derived from wild birds. Pakistan J Zool 46:625–631. [Google Scholar]
- 10.Pantin-Jackwood MJ, Suarez DL. 2013. Vaccination of domestic ducks against H5N1 HPAI: a review. Virus Res 178:21–34. doi: 10.1016/j.virusres.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Bingham J, Green DJ, Lowther S, Klippel J, Burggraaf S, Anderson DE, Wibawa H, Hoa DM, Long NT, Vu PP, Middleton DJ, Daniels PW. 2009. Infection studies with two highly pathogenic avian influenza strains (Vietnamese and Indonesian) in Pekin ducks (Anas platyrhynchos), with particular reference to clinical disease, tissue tropism and viral shedding. Avian Pathol 38:267–278. doi: 10.1080/03079450903055371. [DOI] [PubMed] [Google Scholar]
- 12.Huang Z, Fang D, Lv P, Bian X, Ruan X, Yan Y, Zhou J. 2012. Differential cellular immune responses between chickens and ducks to H9N2 avian influenza virus infection. Vet Immunol Immunopathol 150:169–180. doi: 10.1016/j.vetimm.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 13.Vanderven HA, Petkau K, Ryan-Jean KE, Aldridge JR Jr, Webster RG, Magor KE. 2012. Avian influenza rapidly induces antiviral genes in duck lung and intestine. Mol Immunol 51:316–324. doi: 10.1016/j.molimm.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang QL, Luo J, Zhou K, Dong JX, He HX. 2011. Immune-related gene expression in response to H5N1 avian influenza virus infection in chicken and duck embryonic fibroblasts. Mol Immunol 48:924–930. doi: 10.1016/j.molimm.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 15.Adams SC, Xing Z, Li J, Cardona CJ. 2009. Immune-related gene expression in response to H11N9 low pathogenic avian influenza virus infection in chicken and Pekin duck peripheral blood mononuclear cells. Mol Immunol 46:1744–1749. doi: 10.1016/j.molimm.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 16.McMichael AJ, Gotch FM, Noble GR, Beare PA. 1983. Cytotoxic T-cell immunity to influenza. N Engl J Med 309:13–17. doi: 10.1056/NEJM198307073090103. [DOI] [PubMed] [Google Scholar]
- 17.Kane K, Clark WR. 1984. The role of class I MHC products in polyclonal activation of CTL function. J Immunol 133:2857–2863. [PubMed] [Google Scholar]
- 18.Jondal M, Schirmbeck R, Reimann J. 1996. MHC class I-restricted CTL responses to exogenous antigens. Immunity 5:295–302. doi: 10.1016/S1074-7613(00)80255-1. [DOI] [PubMed] [Google Scholar]
- 19.Bender BS, Croghan T, Zhang L, Small PA Jr. 1992. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 175:1143–1145. doi: 10.1084/jem.175.4.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langley WA, Bradley KC, Li ZN, Talekar GR, Galloway SE, Steinhauer DA. 2010. The effects of preexisting immunity to influenza on responses to influenza vectors in mice. Vaccine 28:6305–6313. doi: 10.1016/j.vaccine.2010.06.112. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Wu B, Zhang S, Tan S, Sun Y, Chen Z, Qin Y, Sun M, Shi G, Wu Y, Sun M, Liu N, Ning K, Ma Y, Gao B, Yan J, Zhu F, Wang H, Gao GF. 2013. Conserved epitopes dominate cross-CD8+ T-cell responses against influenza A H1N1 virus among Asian populations. Eur J Immunol 43:2055–2069. doi: 10.1002/eji.201343417. [DOI] [PubMed] [Google Scholar]
- 22.Sridhar S, Begom S, Bermingham A, Ziegler T, Roberts KL, Barclay WS, Openshaw P, Lalvani A. 2012. Predominance of heterosubtypic IFN-gamma-only-secreting effector memory T cells in pandemic H1N1 naive adults. Eur J Immunol 42:2913–2924. doi: 10.1002/eji.201242504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yap KL, Ada GL. 1978. Cytotoxic T cells in the lungs of mice infected with an influenza A virus. Scand J Immunol 7:73–80. doi: 10.1111/j.1365-3083.1978.tb00428.x. [DOI] [PubMed] [Google Scholar]
- 24.Cagle C, To TL, Nguyen T, Wasilenko J, Adams SC, Cardona CJ, Spackman E, Suarez DL, Pantin-Jackwood MJ. 2011. Pekin and Muscovy ducks respond differently to vaccination with a H5N1 highly pathogenic avian influenza (HPAI) commercial inactivated vaccine. Vaccine 29:6549–6557. doi: 10.1016/j.vaccine.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 25.McKenzie LM, Pecon-Slattery J, Carrington M, O'Brien SJ. 1999. Taxonomic hierarchy of HLA class I allele sequences. Genes Immun 1:120–129. doi: 10.1038/sj.gene.6363648. [DOI] [PubMed] [Google Scholar]
- 26.Rawle FC, Knowles BB, Ricciardi RP, Brahmacheri V, Duerksen-Hughes P, Wold WS, Gooding LR. 1991. Specificity of the mouse cytotoxic T lymphocyte response to adenovirus 5. E1A is immunodominant in H-2b, but not in H-2d or H-2k mice. J Immunol 146:3977–3984. [PubMed] [Google Scholar]
- 27.Kaufman J. 1999. Co-evolving genes in MHC haplotypes: the “rule” for nonmammalian vertebrates? Immunogenetics 50:228–236. doi: 10.1007/s002510050597. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman J, Milne S, Gobel TW, Walker BA, Jacob JP, Auffray C, Zoorob R, Beck S. 1999. The chicken B locus is a minimal essential major histocompatibility complex. Nature 401:923–925. doi: 10.1038/44856. [DOI] [PubMed] [Google Scholar]
- 29.Moon DA, Veniamin SM, Parks-Dely JA, Magor KE. 2005. The MHC of the duck (Anas platyrhynchos) contains five differentially expressed class I genes. J Immunol 175:6702–6712. doi: 10.4049/jimmunol.175.10.6702. [DOI] [PubMed] [Google Scholar]
- 30.Fleming-Canepa X, Jensen SM, Mesa CM, Diaz-Satizabal L, Roth AJ, Parks-Dely JA, Moon DA, Wong JP, Evseev D, Gossen DA, Tetrault DG, Magor KE. 2016. Extensive allelic diversity of MHC class I in wild mallard ducks. J Immunol doi: 10.4049/jimmunol.1502450. [DOI] [PubMed] [Google Scholar]
- 31.Saper MA, Bjorkman PJ, Wiley DC. 1991. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 A resolution. J Mol Biol 219:277–319. doi: 10.1016/0022-2836(91)90567-P. [DOI] [PubMed] [Google Scholar]
- 32.Koch M, Camp S, Collen T, Avila D, Salomonsen J, Wallny HJ, van Hateren A, Hunt L, Jacob JP, Johnston F, Marston DA, Shaw I, Dunbar PR, Cerundolo V, Jones EY, Kaufman J. 2007. Structures of an MHC class I molecule from B21 chickens illustrate promiscuous peptide binding. Immunity 27:885–899. doi: 10.1016/j.immuni.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 33.Zhang J, Chen Y, Qi J, Gao F, Liu Y, Liu J, Zhou X, Kaufman J, Xia C, Gao GF. 2012. Narrow groove and restricted anchors of MHC class I molecule BF2*0401 plus peptide transporter restriction can explain disease susceptibility of B4 chickens. J Immunol 189:4478–4487. doi: 10.4049/jimmunol.1200885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chappell P, Meziane el K, Harrison M, Magiera L, Hermann C, Mears L, Wrobel AG, Durant C, Nielsen LL, Buus S, Ternette N, Mwangi W, Butter C, Nair V, Ahyee T, Duggleby R, Madrigal A, Roversi P, Lea SM, Kaufman J. 2015. Expression levels of MHC class I molecules are inversely correlated with promiscuity of peptide binding. eLife 4:e05345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaufman J, Volk H, Wallny HJ. 1995. A “minimal essential Mhc” and an “unrecognized Mhc”: two extremes in selection for polymorphism. Immunol Rev 143:63–88. doi: 10.1111/j.1600-065X.1995.tb00670.x. [DOI] [PubMed] [Google Scholar]
- 36.Wallny HJ, Avila D, Hunt LG, Powell TJ, Riegert P, Salomonsen J, Skjodt K, Vainio O, Vilbois F, Wiles MV, Kaufman J. 2006. Peptide motifs of the single dominantly expressed class I molecule explain the striking MHC-determined response to Rous sarcoma virus in chickens. Proc Natl Acad Sci U S A 103:1434–1439. doi: 10.1073/pnas.0507386103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia C, Lin CY, Xu GX, Hu TJ, Yang TY. 2004. cDNA cloning and genomic structure of the duck (Anas platyrhynchos) MHC class I gene. Immunogenetics 56:304–309. doi: 10.1007/s00251-004-0685-y. [DOI] [PubMed] [Google Scholar]
- 38.Borbulevych OY, Piepenbrink KH, Baker BM. 2011. Conformational melding permits a conserved binding geometry in TCR recognition of foreign and self molecular mimics. J Immunol 186:2950–2958. doi: 10.4049/jimmunol.1003150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Liu J, Qi J, Gao F, Li Q, Li X, Zhang N, Xia C, Gao GF. 2011. Two distinct conformations of a rinderpest virus epitope presented by bovine major histocompatibility complex class I N*01801: a host strategy to present featured peptides. J Virol 85:6038–6048. doi: 10.1128/JVI.00030-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang N, Qi J, Feng S, Gao F, Liu J, Pan X, Chen R, Li Q, Chen Z, Li X, Xia C, Gao GF. 2011. Crystal structure of swine major histocompatibility complex class I SLA-1 0401 and identification of 2009 pandemic swine-origin influenza A H1N1 virus cytotoxic T lymphocyte epitope peptides. J Virol 85:11709–11724. doi: 10.1128/JVI.05040-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yao S, Liu J, Qi J, Chen R, Zhang N, Liu Y, Wang J, Wu Y, Gao GF, Xia C. 2016. Structural illumination of equine MHC class I molecules highlights unconventional epitope presentation manner that is evolved in equine leukocyte antigen alleles. J Immunol doi: 10.4049/jimmunol.1501352. [DOI] [PubMed] [Google Scholar]
- 42.Chu F, Lou Z, Chen YW, Liu Y, Gao B, Zong L, Khan AH, Bell JI, Rao Z, Gao GF. 2007. First glimpse of the peptide presentation by rhesus macaque MHC class I: crystal structures of Mamu-A*01 complexed with two immunogenic SIV epitopes and insights into CTL escape. J Immunol 178:944–952. doi: 10.4049/jimmunol.178.2.944. [DOI] [PubMed] [Google Scholar]
- 43.Shen ZT, Nguyen TT, Daniels KA, Welsh RM, Stern LJ. 2013. Disparate epitopes mediating protective heterologous immunity to unrelated viruses share peptide-MHC structural features recognized by cross-reactive T cells. J Immunol 191:5139–5152. doi: 10.4049/jimmunol.1300852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Speir JA, Stevens J, Joly E, Butcher GW, Wilson IA. 2001. Two different, highly exposed, bulged structures for an unusually long peptide bound to rat MHC class I RT1-Aa. Immunity 14:81–92. doi: 10.1016/S1074-7613(01)00091-7. [DOI] [PubMed] [Google Scholar]
- 45.Mohammed F, Cobbold M, Zarling AL, Salim M, Barrett-Wilt GA, Shabanowitz J, Hunt DF, Engelhard VH, Willcox BE. 2008. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat Immunol 9:1236–1243. doi: 10.1038/ni.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petersen J, Wurzbacher SJ, Williamson NA, Ramarathinam SH, Reid HH, Nair AKN, Zhao AY, Nastovska R, Rudge G, Rossjohn J, Purcell AW. 2009. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc Natl Acad Sci U S A 106:2776–2781. doi: 10.1073/pnas.0812901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. doi: 10.1038/nature08157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russell CA, Jones TC, Barr IG, Cox NJ, Garten RJ, Gregory V, Gust ID, Hampson AW, Hay AJ, Hurt AC, de Jong JC, Kelso A, Klimov AI, Kageyama T, Komadina N, Lapedes AS, Lin YP, Mosterin A, Obuchi M, Odagiri T, Osterhaus AD, Rimmelzwaan GF, Shaw MW, Skepner E, Stohr K, Tashiro M, Fouchier RA, Smith DJ. 2008. The global circulation of seasonal influenza A (H3N2) viruses. Science 320:340–346. doi: 10.1126/science.1154137. [DOI] [PubMed] [Google Scholar]
- 49.Subbarao K, Klimov A, Katz J, Regnery H, Lim W, Hall H, Perdue M, Swayne D, Bender C, Huang J, Hemphill M, Rowe T, Shaw M, Xu X, Fukuda K, Cox N. 1998. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science 279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- 50.Chen Y, Liang W, Yang S, Wu N, Gao H, Sheng J, Yao H, Wo J, Fang Q, Cui D, Li Y, Yao X, Zhang Y, Wu H, Zheng S, Diao H, Xia S, Zhang Y, Chan KH, Tsoi HW, Teng JL, Song W, Wang P, Lau SY, Zheng M, Chan JF, To KK, Chen H, Li L, Yuen KY. 2013. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet 381:1916–1925. doi: 10.1016/S0140-6736(13)60903-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guan Y, Shortridge KF, Krauss S, Webster RG. 1999. Molecular characterization of H9N2 influenza viruses: were they the donors of the “internal” genes of H5N1 viruses in Hong Kong? Proc Natl Acad Sci U S A 96:9363–9367. doi: 10.1073/pnas.96.16.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferreira HL, Vangeluwe D, Van Borm S, Poncin O, Dumont N, Ozhelvaci O, Munir M, van den Berg T, Lambrecht B. 2015. Differential viral fitness between H1N1 and H3N8 avian influenza viruses isolated from mallards (Anas platyrhynchos). Avian Dis 59:498–507. doi: 10.1637/11074-033015-Reg. [DOI] [PubMed] [Google Scholar]
- 53.VanDalen KK, Franklin AB, Mooers NL, Sullivan HJ, Shriner SA. 2010. Shedding light on avian influenza H4N6 infection in mallards: modes of transmission and implications for surveillance. PLoS One 5:e12851. doi: 10.1371/journal.pone.0012851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, Wheeler DK, Gabbard JL, Hix D, Sette A, Peters B. 2015. The immune epitope database (IEDB) 3.0. Nucleic Acids Res 43:D405–D412. doi: 10.1093/nar/gku938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacDonald MR, Veniamin SM, Guo X, Xia J, Moon DA, Magor KE. 2007. Genomics of antiviral defenses in the duck, a natural host of influenza and hepatitis B viruses. Cytogenet Genome Res 117:195–206. doi: 10.1159/000103180. [DOI] [PubMed] [Google Scholar]
- 56.Bacon LD. 1987. Influence of the major histocompatibility complex on disease resistance and productivity. Poult Sci 66:802–811. doi: 10.3382/ps.0660802. [DOI] [PubMed] [Google Scholar]
- 57.Kishida N, Sakoda Y, Isoda N, Matsuda K, Eto M, Sunaga Y, Umemura T, Kida H. 2005. Pathogenicity of H5 influenza viruses for ducks. Arch Virol 150:1383–1392. doi: 10.1007/s00705-004-0473-x. [DOI] [PubMed] [Google Scholar]
- 58.Jeong OM, Kim MC, Kim MJ, Kang HM, Kim HR, Kim YJ, Joh SJ, Kwon JH, Lee YJ. 2009. Experimental infection of chickens, ducks and quails with the highly pathogenic H5N1 avian influenza virus. J Vet Sci 10:53–60. doi: 10.4142/jvs.2009.10.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. 2009. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nielsen M, Andreatta M. 2016. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 8:33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen W, Gao F, Chu F, Zhang J, Gao GF, Xia C. 2010. Crystal structure of a bony fish beta2-microglobulin: insights into the evolutionary origin of immunoglobulin superfamily constant molecules. J Biol Chem 285:22505–22512. doi: 10.1074/jbc.M109.095000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou M, Xu Y, Lou Z, Cole DK, Li X, Liu Y, Tien P, Rao Z, Gao GF. 2004. Complex assembly, crystallization and preliminary X-ray crystallographic studies of MHC H-2Kd complexed with an HBV-core nonapeptide. Acta Crystallogr D Biol Crystallogr 60:1473–1475. doi: 10.1107/S0907444904013587. [DOI] [PubMed] [Google Scholar]
- 63.Garboczi DN, Hung DT, Wiley DC. 1992. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A 89:3429–3433. doi: 10.1073/pnas.89.8.3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang QS, Yu F, Huang S, Sun B, Zhang KH, Liu K, Wang ZJ, Xu CY, Wang SS, Yang LF, Pan QY, Li L, Zhou H, Cui Y, Xu Q, Earnest T, He JH. 2015. The macromolecular crystallography beamline of SSRF. Nucl Sci Tech 26:12–17. doi: 10.13538/j.1001-8042/nst.26.010102. [DOI] [Google Scholar]
- 65.Harp JM, Timm DE, Bunick GJ. 1998. Macromolecular crystal annealing: overcoming increased mosaicity associated with cryocrystallography. Acta Crystallogr D Biol Crystallogr 54:622–628. doi: 10.1107/S0907444997019008. [DOI] [PubMed] [Google Scholar]
- 66.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr A 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 67.Lebedev AA, Vagin AA, Murshudov GN. 2008. Model preparation in MOLREP and examples of model improvement using X-ray data. Acta Crystallogr D Biol Crystallogr 64:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Collaborative Computational Project. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 70.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 71.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 72.Laskowski RA, Moss DS, Thornton JM. 1993. Main-chain bond lengths and bond angles in protein structures. J Mol Biol 231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 73.Tobita T, Oda M, Morii H, Kuroda M, Yoshino A, Azuma T, Kozono H. 2003. A role for the P1 anchor residue in the thermal stability of MHC class II molecule I-Ab. Immunol Lett 85:47–52. doi: 10.1016/S0165-2478(02)00206-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.