

ABSTRACT
Human herpesviruses 6A/B (HHV-6A/B) can integrate their viral genomes in the telomeres of human chromosomes. The viral and cellular factors contributing to HHV-6A/B integration remain largely unknown, mostly due to the lack of efficient and reproducible cell culture models to study HHV-6A/B integration. In this study, we characterized the HHV-6A/B integration efficiencies in several human cell lines using two different approaches. First, after a short-term infection (5 h), cells were processed for single-cell cloning and analyzed for chromosomally integrated HHV-6A/B (ciHHV-6A/B). Second, cells were infected with HHV-6A/B and allowed to grow in bulk for 4 weeks or longer and then analyzed for the presence of ciHHV-6. Using quantitative PCR (qPCR), droplet digital PCR, and fluorescent in situ hybridization, we could demonstrate that HHV-6A/B integrated in most human cell lines tested, including telomerase-positive (HeLa, MCF-7, HCT-116, and HEK293T) and telomerase-negative cell lines (U2OS and GM847). Our results also indicate that inhibition of DNA replication, using phosphonoacetic acid, did not affect HHV-6A/B integration. Certain clones harboring ciHHV-6A/B spontaneously express viral genes and proteins. Treatment of cells with phorbol ester or histone deacetylase inhibitors triggered the expression of many viral genes, including U39, U90, and U100, without the production of infectious virus, suggesting that the tested stimuli were not sufficient to trigger full reactivation. In summary, both integration models yielded comparable results and should enable the identification of viral and cellular factors contributing to HHV-6A/B integration and the screening of drugs influencing viral gene expression, as well as the release of infectious HHV-6A/B from the integrated state.
IMPORTANCE The analysis and understanding of HHV-6A/B genome integration into host DNA is currently limited due to the lack of reproducible and efficient viral integration systems. In the present study, we describe two quantitative cell culture viral integration systems. These systems can be used to define cellular and viral factors that play a role in HHV-6A/B integration. Furthermore, these systems will allow us to decipher the conditions resulting in virus gene expression and excision of the integrated viral genome resulting in reactivation.
KEYWORDS: HHV-6, chromosomal integration, ddPCR, telomere
INTRODUCTION
In the early 1990s, Luppi and colleagues reported the presence of integrated human herpesvirus 6A (HHV-6A) genome in chromosomes of three patients (1). Since then, many other individuals with chromosomally integrated HHV-6A/B (ciHHV-6A/B) were identified (reviewed in references 2 and 3). Daibata and colleagues provided the first conclusive evidence that integrated HHV-6 can be vertically transmitted from parents to their children according to the Mendelian law of chromosome segregation (4, 5). In these individuals, all somatic cells contain a copy of the entire HHV-6 genome integrated in their chromosomes. This condition is referred to as inherited ciHHV-6 (iciHHV-6). In the largest study ever conducted (n = 20,000), the prevalence of iciHHV-6A/B in the province of Quebec (Canada) was found to be 0.6%, 60% of which were iciHHV-6B (6). Comparable results were obtained in different parts of the world, with iciHHV-6A/B prevalence estimates ranging between 0.5% and 2% (reviewed in reference 3). The consequences of harboring an integrated copy of HHV-6A/B in all somatic cells remains poorly understood. Gravel and colleagues recently demonstrated that patients with iciHHV-6A/B are at greater risk of developing angina pectoris than are age-matched controls and independently of other known associated cardiovascular risk factors (6). Additional large-scale studies are required to determine whether iciHHV-6A/B represents an inherited risk factor for the development of other diseases.
Whether HHV-6A/B integration represents a mechanism of viral latency remains a hot research topic. Several studies provided evidence that integrated virus can be excised from chromosomes, resulting in the generation of progeny of infectious virions (7–9). Arbuckle et al. were the first to show that HHV-6A can integrate into cell lines in vitro (7). Although HHV-6A/B integration can occur in several distinct chromosomes, the integration sites are generally near the internal end of the host telomeres (reviewed in references 2 and 3). So far, the factors involved in HHV-6A/B integration remain unknown. Intriguingly, the viral genome harbors telomeric repeats that are identical to the human telomere sequences, suggesting that homologous recombination (HR) events between host and viral telomere sequences could facilitate integration. In support of this, Marek's disease virus (MDV) telomeric repeats are reported to play a role in MDV integration into host chromosomes (10, 11). A recent study also confirmed the importance of viral telomeric sequences for efficient HHV-6A integration (12). Beyond that, it is unclear if these processes require cellular and/or viral proteins. Trempe and colleagues demonstrated that the HHV-6A/B U94 protein possesses some of the biological properties needed for homologous recombination and likely also viral integration (13). However, U94 was recently reported to be dispensable for HHV-6A integration (14).
A prerequisite for the analysis of HHV-6A/B integration mechanisms is a reliable and efficient in vitro experimental system for viral integration. In this study, we describe the development of an HHV-6A/B integration system in several human cell lines. The system can be used to estimate integration frequency as well as to study the spontaneous and chemically induced HHV-6A/B gene expression and production of infectious virions from an integrated state.
RESULTS
HHV-6 chromosomal integration assay using single-cell cloning.
To establish a reliable and efficient in vitro integration system, we tested several human cell lines for their susceptibility to HHV-6A/B chromosomal integration (Table 1). Following infection, cells were seeded at 1 cell/well, and approximately 1 month later, HHV-6A/B DNA was isolated from individual clones and analyzed by quantitative PCR (qPCR) and/or droplet digital PCR (ddPCR). We could detect HHV-6A/B DNA in clones of most human cell lines tested, albeit at various frequencies. The frequency of clones that harbor the virus genome varied between 1% and 22% depending on the cell line and the viral stocks used. The difference between the cell lines could be due to some degree to their susceptibility to HHV-6A/B infection. For U2OS, HeLa, and MCF-7, HHV-6A and HHV-6B were equally efficient at integration. HEK293T cells preferentially supported HHV-6B integration, but only one experiment was performed. Lastly, out of 478 NIH 3T3 (murine fibroblasts) clones tested, none were positive for HHV-6A or HHV-6B, despite intracellular detection of HHV-6 DNA measured 48 h post-HHV-6 exposure (threshold cycle [CT] for HHV-6, 22.5 ± 3.5; CT for GAPDH, 28.6 ± 3.8).
TABLE 1.
HHV-6 integration frequency in various cell lines
| Cell line | Phenotype | Virus | Integration frequency (%) | No. of clones analyzed | No. of expt |
|---|---|---|---|---|---|
| U2OS | ALT+ | HHV-6A | 22.4 ± 10.4 | 294 | 4 |
| U2OS | ALT+ | HHV-6B | 14.2 ± 11.6 | 550 | 8 |
| U2OS | ALT+ | HHV-6A WTR | 4.9 ± 2.4 | 185 | 2 |
| GM847 | ALT+ | HHV-6B | 3.4 | 87 | 1 |
| HCT-116 | TELO+ | HHV-6B | 12.2 | 107 | 1 |
| HEK293T | TELO+ | HHV-6A | 1.1 | 91 | 1 |
| HEK293T | TELO+ | HHV-6B | 19.4 | 98 | 1 |
| HELA | TELO+ | HHV-6A | 3.4 ± 1.1 | 507 | 4 |
| HELA | TELO+ | HHV-6B | 5.8 | 86 | 1 |
| MCF-7 | TELO+ | HHV-6A | 4.1 ± 3.9 | 231 | 2 |
| MCF-7 | TELO+ | HHV-6B | 10.4 ± 7.8 | 231 | 2 |
| NIH 3T3 | TELO+ | HHV-6A | 0 | 235 | 1 |
| NIH 3T3 | TELO+ | HHV-6B | 0 | 243 | 1 |
As U2OS cells reliably facilitated the maintenance of the ciHHV-6A/B genome, they were studied in greater detail. Twenty individual U2OS clones positive for ciHHV-6 (10 ciHHV-6A and 10 ciHHV-6B) were analyzed by ddPCR to determine the number of integrated viral copies/cell. A representative ddPCR result from a U2OS clone, BP6, containing integrated HHV-6B is presented in Fig. 1A and B. The DNA sample is partitioned into approximately 20,000 droplets and subjected to PCR amplification using primer pairs and probes specific for HHV-6 and the RPP30 single-copy cellular gene. After PCR, the samples were analyzed, the number of droplets positive for HHV-6 (blue), RPP30 (green), or HHV-6 and RPP30 (orange) was determined, and the number of HHV-6 copy/cell was calculated. DNA from clonal cell lines without HHV-6A/B integration and DNA from known iciHHV-6A+/B+ individuals (6) were used as negative and positive controls, respectively (Fig. 1C). Samples from iciHHV-6A+/B+ individuals had a median number of HHV-6 copies/cell of 1. U2OS cells with integrated virus have median copies/cell of 1.4 for HHV-6A and 0.95 for HHV-6B. The range of DNA copy/cell varied between 0.2 and 4.3 (Fig. 1C). To confirm that the virus genome is integrated into the host chromosomes, we performed fluorescent in situ hybridization (FISH) on several clonal cell lines. FISH analyses confirmed that the virus genome is indeed localized at the ends of metaphase chromosomes. A representative result of HHV-6 integrated in the telomeric region of cellular chromosomes is presented in Fig. 1D.
FIG 1.

Characterization of clones with integrated HHV-6. (A and B) DNA samples from U2OS and a U2OS-BP6 clone containing ciHHV-6B were analyzed by ddPCR. After PCR, the content of droplets was analyzed for the presence of HHV-6 (blue), cellular gene RPP30 (green), HHV-6+ RPP30 (orange), or no DNA (gray). (C) The average number of HHV-6 copies/cell from U2OS clones with ciHHV-6A and ciHHV-6B was determined by ddPCR. DNA from cell clones without HHV-6 integration and DNA samples from known iciHHV-6A+/B+ subjects were used as negative and positive controls (neg and pos ctl), respectively. (D) FISH analysis of a U2OS clone containing ciHHV-6A. Telomeres (TMR) and HHV-6A were visualized using Cy3-labeled telomeric probe (red) and HHV-6A using Alexa-488 (green)-labeled probe. An enlarged portion of the image is presented on the right.
HHV-6 chromosomal integration assay using bulk cultures.
Although very useful, the single-cell cloning procedure is labor-intensive. Furthermore, not all cell types can be easily cloned. To find an alternative assay that would bypass these issues, we studied the HHV-6 integration frequency in bulk cultures using ddPCR. HHV-6A- and HHV-6B-infected U2OS cells were expanded and passaged once a week for several weeks. At regular intervals, aliquots of cells were analyzed to determine the HHV-6 DNA copy number relative to RPP30, a single-copy gene. The frequency of cells containing integrated HHV-6 was estimated assuming a single integrated HHV-6/cell and calculated with the following formula: (number of HHV-6 copies)/(number of RPP30 copies/2 copies per cell) × 100. As presented in Fig. 2A, the percentage of ciHHV-6A+/B+ cells remained relatively constant over 150 days, suggesting that the virus is stably integrated. The second assumption is that cells, with or without ciHHV-6A/B, proliferate at a similar rate. In support of this, the proliferation rates of U2OS cells and U2OS-AP3 containing ciHHV-6A were similar (Fig. 2B). To validate the frequency of ciHHV-6A+/B+ U2OS cells in bulk cultures, an aliquot of cells was taken from the bulk cultures at day 108 and processed for single-cell cloning. After an additional month, the HHV-6A/B copy number from each clone was determined by ddPCR and compared to that of cells from the bulk culture. Cells with >0.5 copies of HHV-6A/B per cell were considered ciHHV-6A+/B+. As presented in Table 2, the estimated frequencies of ciHHV-6A+/B+ U2OS cells from the bulk culture were comparable to those in clones (P > 0.05).
FIG 2.
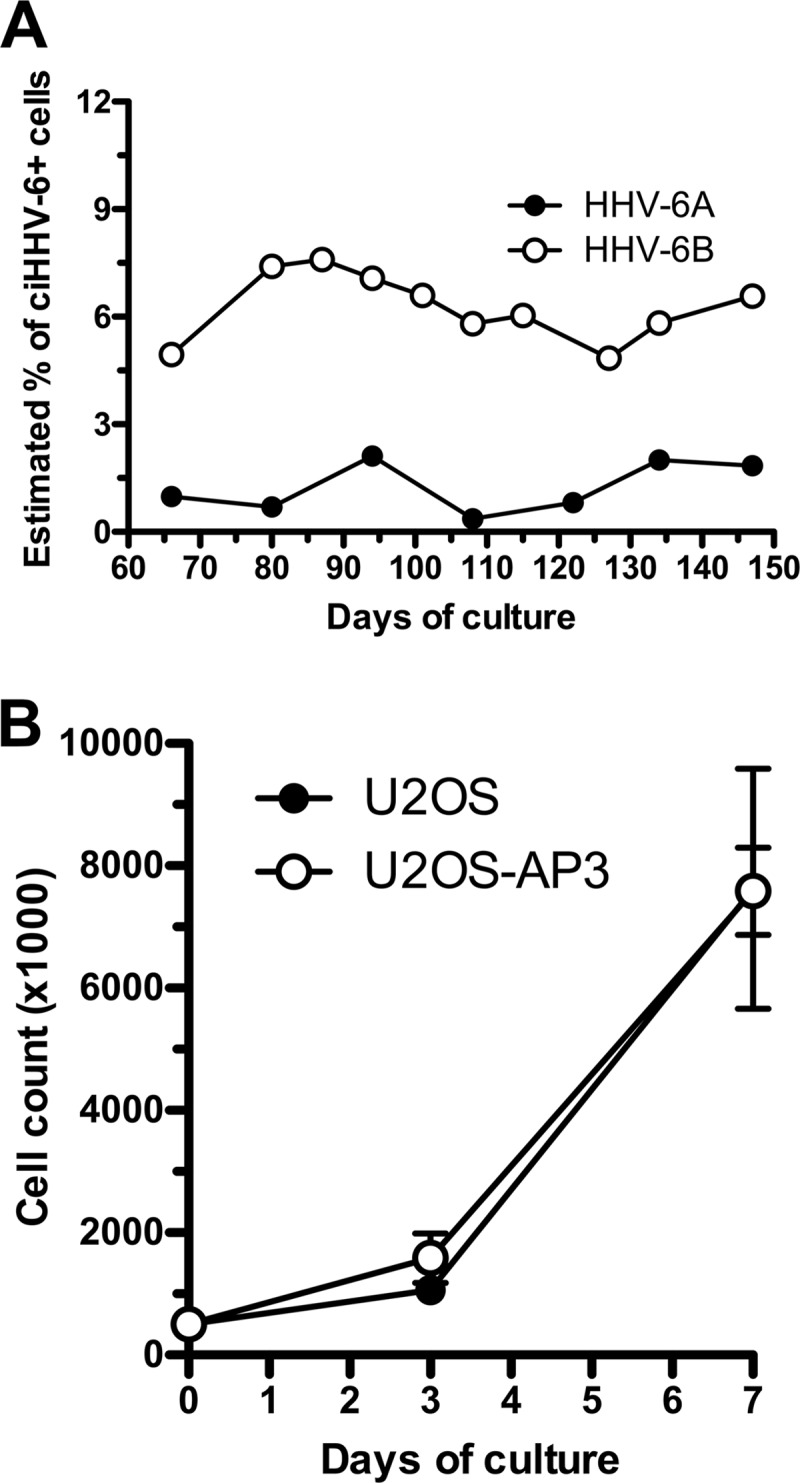
Maintenance and growth of cells containing ciHHV-6 over time. (A) U2OS cells were infected with HHV-6A or HHV-6B and cultured for 150 days. At various times, DNA was extracted and analyzed by ddPCR for estimation of ciHHV-6 frequency. (B) U2OS and U2OS-AP3 cells (containing ciHHV-6A) were analyzed for their growth rates. Cells were plated on day 0, and on days 3 and 7 the number of cells was determined. Results are expressed as mean cell number ± standard deviations (SD) from triplicate cultures.
TABLE 2.
Comparative analysis of bulk culture and single-cell cloning assays
| Cell line and virus | Bulk assay |
Single cell cloning |
P valueb | ||||
|---|---|---|---|---|---|---|---|
| No. of copies of: |
% ciHHV-6+ | No. of ciHHV-6+ clonesa | Total no. of clones analyzed | % ciHHV-6+ | |||
| U65/U66 | RPP30 | ||||||
| U2OS-HHV-6A | 400 | 43,800 | 1.8 | 0 | 87 | 0.0 | 0.4 |
| U2OS-HHV-6B | 1,460 | 44,400 | 6.6 | 6 | 107 | 5.6 | 0.8 |
| MCF7-HHV-6A | 404 | 32,000 | 2.5 | 2 | 144 | 1.4 | 0.7 |
| MCF7-HHV-6B | 386 | 30,740 | 2.5 | 7 | 143 | 4.9 | 0.7 |
For single-cell cloning analysis, only clones with >0.5 copies of HHV-6/cell were considered positive.
P values were determined using chi-square analysis.
Using a second cell line, a complementary approach was taken to compare the single-cell cloning and bulk culture assays. MCF-7 cells were infected with HHV-6A or HHV-6B for a few hours. An aliquot of the cells was used for single-cell cloning, and the remaining cells were cultured in bulk and passaged weekly. After approximately 1 month, clones and bulk cultures were analyzed by ddPCR. As presented in Table 2, the frequency of ciHHV-6A+/B+ MCF-7 cells in both assays were equivalent (P > 0.05).
These results suggest that the estimation of HHV-6A/B chromosomal integration frequencies using bulk cultures and single-cell cloning procedures are equivalent.
Spontaneous and induced expression of ciHHV-6 genes.
To determine if ciHHV-6A+ clones express viral genes, 11 ciHHV-6A+ clones were analyzed for the expression of immediate early (IE; U90) and late (U100) gene expression. As presented in Fig. 3A, some clones spontaneously expressed U90 and U100. Gene expression varied greatly between clones, with 1- to 2-log variations observed. To confirm that mRNAs were translated into proteins, we monitored the spontaneous expression of the IE1 protein, encoded by U90, in several clones. In some clones, such as AP3, we could easily detect IE1 expression in 10 to 20% of cells (Fig. 3B). For others (the majority), such as BP6, IE1 was undetectable.
FIG 3.
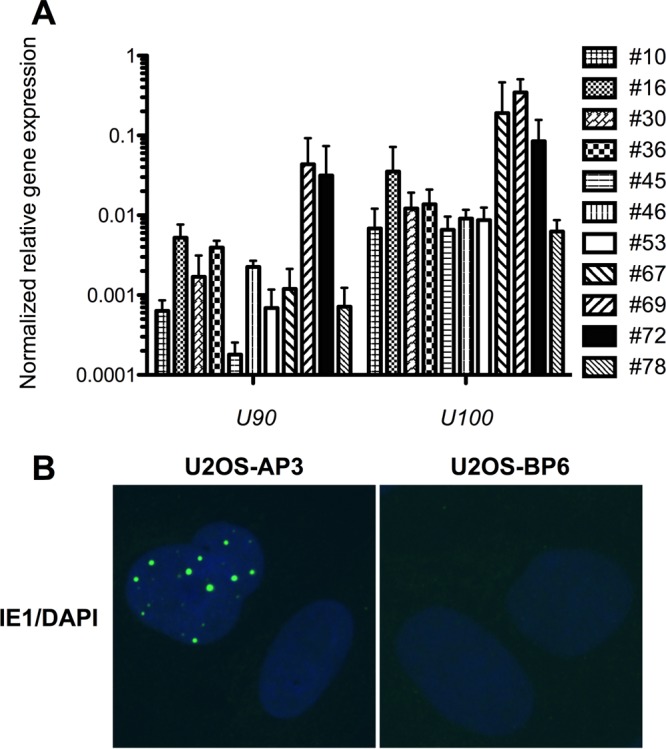
Spontaneous HHV-6 gene expression from ciHHV-6A/B U2OS clones. (A) Eleven U2OS clones containing ciHHV-6A were analyzed for the spontaneous expression of IE and U100 by RT-qPCR. Results are expressed as mean ratios ± SD of viral genes relative to GAPDH mRNA levels. (B) U2OS-AP3 (ciHHV-6A+) and U2OS-BP6 (ciHHV-6B+) were analyzed for the spontaneous expression of IE1 protein by IFA using purified anti-IE1 rabbit IgG coupled to Alexa 488. Nuclei were stained blue using 4′,6-diamidino-2-phenylindole (DAPI).
To determine if expression of genes from the integrated virus can be induced, U2OS (Fig. 4A to F) and MCF-7 (Fig. 4G to I) clones with integrated virus were stimulated for 8 to 96 h with a TNT cocktail (tetradecanoylphorbol-13-acetate [TPA], sodium butyrate [NaBy], trichostatin A [TSA], and hydrocortisone) or suberoylanilide hydroxamic acid (SAHA) and processed for reverse transcription-qPCR (RT-qPCR) analyses. Upon stimulation, certain genes, such as U90 (immediate early), U39 (early [E]), and U100 (late), were efficiently activated. Others, such as U54 and U94, were either undetectable or minimally induced upon stimulation (data not shown). These results indicate a great heterogeneity between ciHHV-6A/B clones in terms of spontaneous and inducible expression of viral genes. However, the possibility that expression originates from an episome in a cell with a silent integration cannot be excluded.
FIG 4.

Spontaneous and induced HHV-6 gene expression from ciHHV-6A+/B+ U2OS and MCF-7 clones. (A to F) The expression of HHV-6A/B U90, U39, and U100 genes from ciHHV-6A+ (AP3 and WTR-BAC#25) and ciHHV-6B+ (BP6) U2OS clones was determined by RT-qPCR at the indicated times following TNT (TPA, NaBy, TSA, and hydrocortisone) or SAHA stimulation. Results are shown as mean fold induction ± SD relative to untreated (resting) cells. (G to I) Expression of U90, U39, and U100 from ciHHV-6A+ (MCF7-66A) and ciHHV-6B+ (MCF-7 32B) MCF-7 clones was determined by RT-qPCR at the indicated times following TNT or SAHA stimulation. Results are shown as mean fold induction ± SD relative to untreated (resting) cells. NS, not stimulated. (J) GFP expression in resting and TSA-activated U2OS clone 9 containing integrated WTR-BAC HHV-6A. Results are representative of a minimum of three independent experiments performed in triplicate. *, P < 0.05; **, P < 0.01.
We next studied whether cells harboring the integrated wild-type HHV-6A bacterial artificial chromosome (WTR-BAC) would spontaneously express green fluorescent protein (GFP) upon stimulation with TSA, as the WTR-BAC encodes a GFP gene under the control of the major immediate early promoter of human cytomegalovirus. Under resting conditions, GFP was expressed only at a very low level in about 1% of cells, while GFP expression was upregulated and detectable in 8% of the cells upon TSA treatment (Fig. 4J). As for viral genes, levels of spontaneous GFP expression varied greatly between clones. In some clones, viral genes/GFP expression could be readily induced by stimulation, while others were not responsive (data not shown).
HHV-6A and HHV-6B double integration.
We next determined if ciHHV-6+ cells are resistant or permissive to de novo integration with the other HHV-6 species. Clone AP3 with ciHHV-6A and clone BP6 with ciHHV-6B were infected with HHV-6B and HHV-6A, respectively, and processed for single-cell cloning. Approximately 100 AP3 and BP6 clones were analyzed by qPCR for de novo integration with the other HHV-6 species. HHV-6B could integrate in 9% of AP3 clones containing ciHHV-6A. Similarly, HHV-6A could integrate in 38% of BP6 clones. To confirm the presence of two viral genomes in these cell lines, we performed FISH analyses. Two signals could be detected in metaphase chromosomes and interphase nuclei (Fig. 5).
FIG 5.
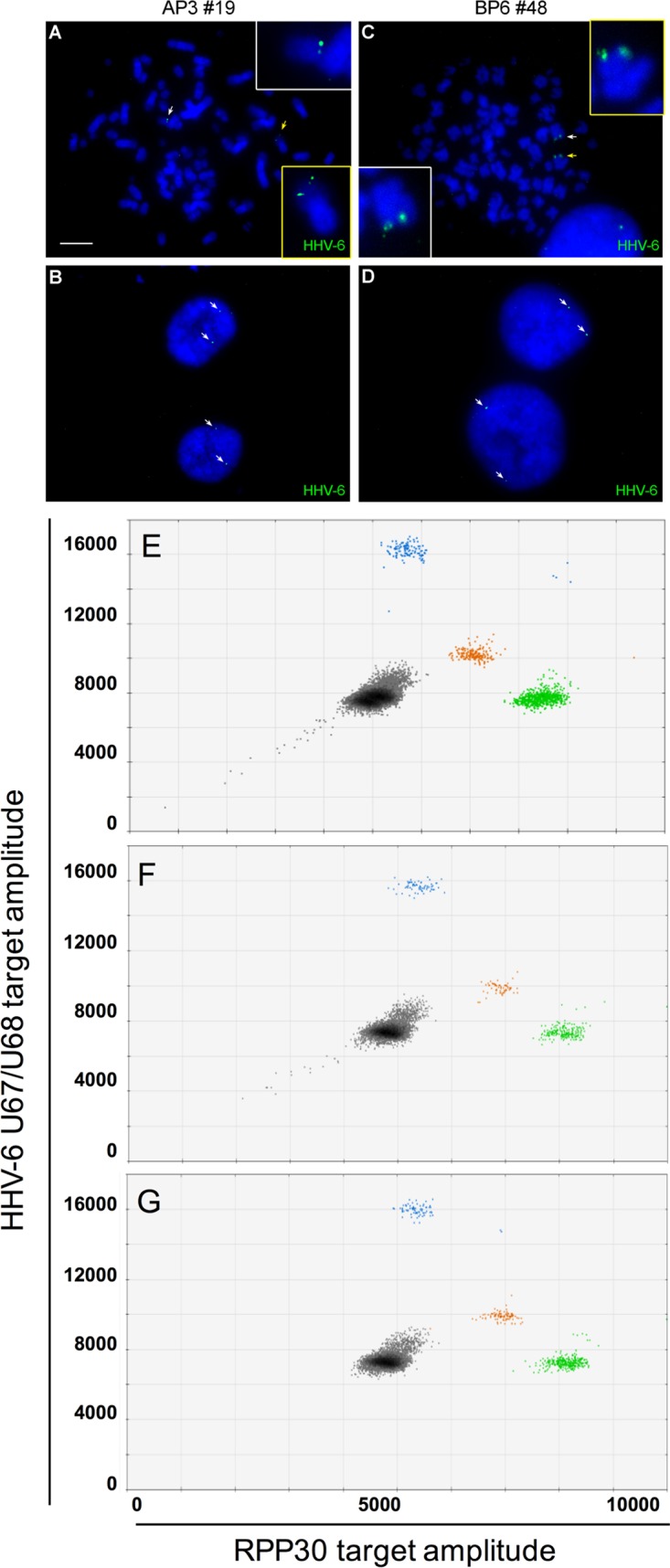
Generation of HHV-6A and HHV-6B doubly integrated U2OS cells. U2OS-AP3 (ciHHV-6A+) and U2OS-BP6 (ciHHV-6B+) were superinfected with HHV-6B and HHV-6A, respectively, and submitted to single-cell cloning. Clones positive for two viruses were identified by FISH and ddPCR. Metaphase spreads (A and C) and interphase nuclei (B and D) of AP3#19 (A and B) and BP6#48 (C and D) showing the presence of two distinct HHV-6 hybridization signals (arrows). (E and G) Droplet digital PCR analysis of cell lines containing integrated HHV-6A and integrated HHV-6B. (E) ciHHV-6A+ and ciHHV-6B+ cell lines assayed together to produce a droplet plot with multiple clusters. Gray droplets are negative for template. Blue droplets are positive for HHV-6A. Orange droplets are positive for HHV-6B. Green droplets are positive for RPP30. (F) ddPCR analysis of the U2OS AP3 superinfected with HHV-6B to generate clone 19 containing both HHV-6 species. On average, this clone contained 2.9 HHV-6A and 2.2 HHV-6B genomes/cell. (G) ddPCR analysis of the U2OS BP6 clone superinfected with HHV-6A, resulting in clone 48 containing both HHV-6 species. On average, this clone contained 0.6 HHV-6A and 0.6 HHV-6B genomes/cell.
Since FISH does not allow discrimination between HHV-6A and HHV-6B, we used a modified ddPCR approach that allowed the simultaneous differentiation of HHV-6A and HHV-6B (15). To validate the assay, DNA from two clones, one ciHHV-6A+ and one ciHHV-6B+, were mixed and analyzed by ddPCR. As shown in Fig. 5E, HHV-6A (blue), HHV-6B (orange), and the RPP30 (green) cellular gene could be detected and easily discriminated. The same ddPCR assay was used to analyze the U2OS-AP3 clone superinfected with HHV-6B (Fig. 5F) and clone U2OS-BP6 superinfected with HHV-6A (Fig. 5G). As shown, in both doubly integrated clones, DNA corresponding to HHV-6A and HHV-6B was detected. The number of HHV-6A and HHV-6B integrated genomes varied between 0.6 and 2.9 copies/cell depending on the clone analyzed. These results indicate that cells containing ciHHV-6 are permissive to additional integration events.
Role of viral and cellular factors in integration.
Lastly, we set out to identify cellular and viral factors that could be involved in HHV-6 chromosomal integration. One factor that could aid in the integration process is the telomerase complex. Our observation that HHV-6A/B integrates efficiently in telomerase-negative cells such as U2OS and GM847 cells suggests that the telomerase activity is dispensable for HHV-6 integration (Table 1). Another potential cellular factor is p53, a crucial player in the DNA repair pathway that could affect the integration of HHV-6A/B. We infected wild-type and p53−/− HCT-116 cells with HHV-6B and determined the integration frequency using the single-cell cloning procedure. Integration was similar (P = 0.39) in the absence of p53 compared to the parental cell line, suggesting that p53 does not play a major role in HHV-6A/B integration (Table 3).
TABLE 3.
Effects of p53 deletion or PAA treatment on HHV-6 integration
| Cell line | Virus | Condition | No. of ciHHV-6+ clones/total no. of clones tested (%) | P value |
|---|---|---|---|---|
| HCT-116 | HHV-6B | p53+/+ | 13/107 (12.2) | |
| HCT-116 | HHV-6B | p53−/− | 12/71 (16.9) | 0.39 |
| U2OS | HHV-6A | Untreated | 66/294 (22.4) | |
| U2OS | HHV-6A | PAA | 17/70 (24.3) | 0.74 |
| U2OS | HHV-6B | Untreated | 53/543 (9.8) | |
| U2OS | HHV-6B | PAA | 7/74 (9.5) | 1.00 |
To determine if HHV-6 integration requires replication of the HHV-6 genome, cells were treated with the well-characterized and highly effective viral DNA polymerase inhibitor phosphonoacetic acid (PAA) prior to HHV-6 infection and throughout the assay. The efficiency of PAA at inhibiting HHV-6 replication was confirmed using HHV-6-infected T cells as previously described (16 and data not shown). Integration frequencies of HHV-6A and HHV-6B upon inhibition of the viral polymerase were comparable to those for untreated cultures (Table 3), indicating that integration into host chromosomes is independent of viral replication and late gene expression.
DISCUSSION
As far as we know, HHV-6A and HHV-6B are the only human herpesviruses that can efficiently and reproducibly integrate their genomes into host chromosomes. Whether this represents a bona fide state of latency remains to be fully addressed. Recent studies provided evidence that integrated HHV-6A can be excised in vivo, resulting in viral reactivation and pathogenesis (8, 9). The mechanisms by which HHV-6A and HHV-6B integrate into the telomeric/subtelomeric junction of host chromosomes remain unclear. Analysis of the viral genome of iciHHV-6A+/B+ individuals indicates that all HHV-6A/B open reading frames are largely intact, with no gross rearrangements (17). However, the pac1 sequence from DRL and the pac2 sequence from DRR are lost during the integration process (17). This is unlikely to have major impacts on the ability of HHV-6A/B to generate infectious virions, considering that invasion of the telomeric 3′ overhang into the internal HHV-6A/B DRR-T1 could facilitate the release of a complete viral genome from the chromosome with a single reconstituted DR (17).
Until now, analysis of the HHV-6A/B integration mechanism was hampered by the lack of a robust and well-defined in vitro integration model. In a previous report, Arbuckle et al. demonstrated that HHV-6A integrates into HEK293 and SupT1 cells (7). However, the low integration efficiency in these cells does not allow for a quantitative assessment of integration. No studies were conducted with HHV-6B. Our goal was to develop a cell culture system that would enable the study of integration by both HHV-6 species and allow the identification of cellular and viral factors involved in this process.
Several immortalized cell lines were tested for their susceptibility to HHV-6A and HHV-6B integration. HHV-6A/B can integrate into most cell lines tested, independent of their telomerase activity. No integration events could be detected using the murine NIH 3T3 cells, suggesting that the murine cellular environment is not suitable for HHV-6A/B integration despite the report indicating expression of several viral transcripts (18). U2OS, MCF-7, and HeLa cells proved to be susceptible to HHV-6A and HHV-6B integration. Clones containing less than 1 HHV-6 DNA copy/cell are likely oligoclonal, contaminated by cells lacking integrated virus. Alternatively, integrated virus may be lost in some cells during growth of the cellular clone. Intriguingly, a recent study demonstrated that the integrated HHV-6 can be lost, presumably via telomere-loop formation and excision of the viral genome (7, 17, 19). Therefore, a similar scenario is possible in our system, considering that U2OS cells have highly dynamic telomere trimming mechanisms involving telomere loop formation (20, 21). Cells with >1 HHV-6 copy/cell either represent clones with multiple integration events or the integrated viral genome could have been copied from one telomere to another by an ALT mechanism occurring in both primary and cancerous cells (22).
A fundamental question remaining is whether the presence or expression of specific HHV-6A/B genes/proteins is required for virus integration. Interestingly, restriction of viral gene expression to immediate early (IE) and early (E) genes using the viral DNA polymerase inhibitor PAA did not affect integration frequencies of HHV-6A and HHV-6B (P > 0.05). These results indicate that HHV-6A and HHV-6B integration is independent of viral DNA replication, and that expression of IE and/or E genes is sufficient for integration to occur. Considering that there are more than 30 IE and E genes (23), identification of the viral proteins involved in the integration process will not be a trivial undertaking but should be an important emphasis for future work.
At least two hypotheses have been proposed that could explain how HHV-6A/B integration into the telomere region of host chromosomes might occur (2, 3). The first is based on a role for the U94 recombinase (13), and the second involves homologous recombination (HR) events between viral and cellular telomeric repeats. The expression kinetics of U94 have been classified as those of either an IE (24, 25) or an E (23) gene. Biochemical data suggest that the U94 protein could facilitate integration (2, 3, 13). However, despite some growth defects, an HHV-6A U94 deletion mutant proved as efficient as WT HHV-6A at integrating host chromosomes, indicating that U94 is dispensable for HHV-6A integration (14). The quest for other viral candidate proteins involved in integration is ongoing. On the cellular side, a key protein for homologous recombination is Rad51 (26). Intriguingly, using the Rad51 RI-1 inhibitor (27), Wallaschek et al. recently reported that the Rad51 recombinase is dispensable for HHV-6A and HHV-6B integration in U2OS cells (14). Our results also suggest that abrogation of the p53 protein, which is involved in DNA repair mechanisms and interacts with the HHV-6 U14 and U19 proteins (28, 29), does not affect HHV-6 chromosomal integration. Thus, our results indicate that HHV-6 U94 and the cellular p53 and Rad51 proteins are dispensable for HHV-6 integration, suggesting that other viral or cellular recombinases can complement their functions.
The fate of the integrated HHV-6 genome has gained a lot of attention not only for an unconventional mode of herpesvirus latency but also for medical reasons. Several studies have reported spontaneous expression of HHV-6A/B mRNA and/or protein in freshly isolated leukocytes from iciHHV-6A+/B+ individuals (30–32). In addition, Endo et al. provided conclusive evidence that ciHHV-6 can reactivate from the integration state in vivo and cause disease (8). Furthermore, expression of viral proteins, even in the absence of infectious virion production, has potential pathogenic effects, such as destruction of cells/tissue expressing viral antigens (reviewed in reference 3). This phenomenon could be of importance in, for example, solid-organ transplantation. Organ rejection could be caused by immune responses against HHV-6A/B proteins expressed in cells from iciHHV-6A+/B+ organ donors that previously would have been attributed to histocompatibility differences. Analyses of several U2OS and MCF7 clones carrying ciHHV-6A/B indicate that viral gene expression is largely dependent on the cellular clone studied, suggesting that there are differences in the level of epigenetic silencing. Certain clones spontaneously express viral genes while others do not. Similarly, induction of viral gene expression with activators such as phorbol ester and histone deacetylase (HDAC) inhibitors is also clone dependent. Intriguingly, the clinically used HDAC inhibitor SAHA efficiently activated HHV-6 gene expression. Whether this effect is also induced in iciHHV-6A+/B+ patients treated with SAHA remains to be investigated. In certain clones, such as AP3, a variety of genes were readily activated, while modulation of viral gene expression was minimal in the BP6 cell line. Similar observations were made in cell lines harboring the HHV-6A WTR, in which GFP expression could be induced by the treatment of cells with phorbol ester and HDAC inhibitors. However, GFP expression may or may not correlate with the expression of HHV-6 genes, as it is driven by the very strong human cytomegalovirus (HCMV) IE promoter. Differences in viral gene expression modulation could be affected by the chromosome integrated, epigenetics, and the degree of chromatin condensation. These results indicate that our conditions were not adequate to induce HHV-6 reactivation, reflected by the lack of infectious virus particle production. Viral preparations contain a significant number of noninfectious viral particles. Under our conditions, the number of infectious virus particles is typically 100× to 1,000× lower than the number of DNase-protected viral genome equivalents. These defective particles likely enter cells and possibly integrate their genomes into host chromosomes. Since we mostly studied cellular clones, many of them could contain a defective virus that is unable to reactivate.
In summary, we established an efficient, reproducible, and quantitative model system to study HHV-6A/B integration. Both models proved equivalent at determining the integration frequency. The single-cell cloning procedure has the advantage that it allows the study of individual clones, which could prove useful in certain situations, such as the study of preferred sites of integration. On the other hand, the bulk approach is less tedious and easily adapted to study the impact of drugs on the integration process. Both systems can be exploited to identify viral and cellular proteins involved in the integration process using recombinant viruses, short hairpin RNA (shRNA), or CRISPR-Cas9 technologies. In addition, they also allow the testing of clinically approved drugs, such as SAHA, for their potential effects on the expression of viral genes from the integrated HHV-6A/B genome. Further studies are needed to identify the optimal conditions that can promote complete viral reactivation, which has been observed in vivo.
MATERIALS AND METHODS
Cell lines and viruses.
Molt3, JJHan, and HSB-2 cells (American Type Culture Collection [ATCC], Manassas, VA, USA) were cultured in RPMI 1640 (Corning Cellgro, Manassas, VA, USA) supplemented with 10% Nu serum (Corning Cellgro), 10 mM HEPES, and 5 μg/ml plasmocin (Invivogen, San Diego, CA, USA). U2OS and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Corning Cellgro) supplemented with 10% Nu serum (Corning Cellgro), nonessential amino acids (Corning Cellgro), HEPES, sodium pyruvate (Wisent Inc., St-Bruno, QC, Canada), and 5 μg/ml plasmocin (Invivogen). HCT-116, GM847, HEK293T, MCF-7, and NIH 3T3 cells (all from ATCC, except for HCT-116) were cultured in the same medium but supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA, USA) instead of Nu serum. HCT-116 and HCT-116 (p53−/−) were kindly provided by Bert Vogelstein (33). HHV-6A (U1102 and GS) and HHV-6B (Z29) were propagated on HSB-2 and Molt3 cells, respectively, as described previously (16). Virus reconstituted from the wild-type HHV-6A (WTR-HHV-6A) bacterial artificial chromosome (BAC) (34) was propagated on JJHan cells as previously described (12).
Single-cell cloning integration assay.
Ten thousand cells/well (U2OS, HCT-116, HeLa, GM847, MCF-7, NIH 3T3, and HEK293T) were seeded in 48-well plates. The next day, the cells were infected with either HHV-6A, HHV-6B, or WTR-HHV-6A at a multiplicity of infection (MOI) of 1 for 5 h at 37°C. Cells were washed 3× with phosphate-buffered saline (PBS) to remove unadsorbed virions prior to the addition of fresh culture medium. The next day, cells were harvested, counted, and plated at a density of 1 cell/well in 96-well flat-bottom plates (typically 2 to 3 plates/condition). After 10 to 14 days, wells containing only a single clone were identified. Cell clones were propagated for an additional 3 weeks and harvested, and a portion of the cells was used for DNA extraction and qPCR or droplet digital PCR (ddPCR) analysis (see below). HHV-6-positive clones were expanded and frozen for future use. For qPCR, crude DNA extraction was made. Cells were centrifuged and resuspended in 20 μl of buffer (10 mM Tris-HCl, pH 8.4, 50 mM KCl, 2 mM MgCl2, 0.45% NP-40, 0.45% Tween 20, and 60 μg/ml of proteinase K). Cells were incubated at 55°C for 1 h, followed by 95°C for 10 min. The lysates were kept on ice (or frozen) until used. For ddPCR, DNA was isolated using the QiaAMP blood extraction kit as described by the manufacturer (Qiagen Inc., Toronto, ON, Canada).
qPCR analysis.
Four microliters (approximately 50 ng) of the DNA was mixed with primers and probes in a final volume of 10 μl, mixed with an equal volume of 2× Rotor-Gene multiplex PCR kit (Qiagen), and subjected to 40 cycles of amplification. The conditions for detection of HHV-6A/B were described previously (6, 35). Primers and probe used are listed in Table 4. DNA isolated from known iciHHV-6A+, iciHHV-6B+, and iciHHV-6− individuals were used as positive and negative controls in every run. A standard curve generated from DNA isolated from an iciHHV-6+ individual was used to estimate the number of HHV-6 copies. Samples with an HHV-6 copy number of ≥0.5 per cell were considered positive and confirmed by ddPCR to determine the exact HHV-6 copy numbers.
TABLE 4.
DNA primers and probes used in qPCR and ddPCR assays
| Name and purpose | Sequenceb (5′→3′) |
|---|---|
| qPCR and ddPCR | |
| U65-U66 | |
| Fwd | GACAATCACATGCCTGGATAATG |
| Rev | TGTAAGCGTGTGGTAATGGACTAA |
| Probe | FAM-AGCAGCTGG-ZEN-CGAAAAGTGCTGTGC-IABkFQ |
| GAPDH | |
| Fwd | GTCCCTCAATATGGTCCTGTC |
| Rev | TTCTCCATGGTGGTGAAGAC |
| Probe | HEX-CGACGTACT-ZEN-CAGCGCCAGCATC-IABkFQ |
| U67 | |
| Fwd | GTTAGGATATACCGATGTGCGTGAT |
| Rev | TACAGATACGGAGGCAATAGATTTG |
| Probe | FAM-TCCGAAACAACTGTCTGACTGGCAAAA-BHQ1 |
| U67-U68 | |
| Fwd | TTCCGGTATATGACCTTCGTAAGC |
| Rev | GATGTCTCACCTCCAAATCTTTAGAAAT |
| HHV-6A probe | FAM-ACATTATATGTCGAACTTGACACTACCTTCCG-BHQ1 |
| HHV-6B probea | HEX-CATTATATATCGAATCTGACGCTACCTTCCG-BHQ1 |
| RPP30 | |
| Fwd | GATTTGGACCTGCGAGCG |
| Rev | GCGGCTGTCTCCACAAGT |
| Probe | HEX-TCTGACCTG-ZEN-AAGGCTCTGCGCG-IABkFQ |
| cDNA analysis | |
| IE | |
| Fwd | GGCGGTGTCTSAATTTGCATC |
| Rev | CAYTGGATCGGGAYGGTAGTYTT |
| Probe | FAM-ACCCTCTGGAAACAACATGGRATCCAA-BHQ1 |
| GAPDH | |
| Fwd | TTCACACCCATGACGAACAT |
| Rev | AATCCCATCACCATCTTCCAG |
| Probe | HEX-CGACGTACT-ZEN-CAGCGCCAGCATC-IABkFQ |
| U39 | |
| Fwd | TCCTGGCTGTCTTTTTGATG |
| Rev | CCCTCCGACATCTTTGCATT |
| U100A | |
| Fwd | ACCTGTCTCTGCTTTATGGCGCTT |
| Rev | TGGACAGAAASSTATCGCCGTCAT |
| U100B | |
| Fwd | ACTTCTCTCTGCTATATGGCGCTT |
| Rev | TGGACAAAAAGCTATCGCCGTCAT |
HHV-6B probe was labeled with FAM or HEX (see the text for details).
Nucleotide codes for cDNA analysis primers and probes were the following: R, G or A; Y, T or C; S, G or C.
Determination of HHV-6 copy number by ddPCR.
The ddPCR assay used to determine the number of HHV-6 copies per cell was performed as described previously (36).
Detection of HHV-6 in culture supernatant.
To measure the production of extracellular virions, 180 μl of culture supernatant first was mixed with 20 μl of 10× DNase I buffer (100 mM Tris-HCl, pH 7.5, 25 mM MgCl2, and 5 mM CaCl2) and 5 U of DNase I (Sigma-Aldrich, Mississauga, ON, Canada) to digest nonencapsidated HHV-6 DNA, and the mixture was incubated at 37°C for 30 min. After adding 7.5 μl of 200 mM EDTA, the samples were heated at 70°C for 10 min to inactivate DNase I. DNA was then extracted using a QIAamp blood extraction kit as described by the manufacturer (Qiagen).
Bulk culture integration assay.
Cells were plated as described above. The next day, the cells were infected with either HHV-6A, HHV-6B, or WTR-HHV-6A at an MOI of 1 for 5 h at 37°C and then washed 3× with PBS to remove unadsorbed virions prior to the addition of fresh culture medium. Upon confluence, cells were passaged into the well of a 6-well plate for a few days and further expanded into a 25-cm2 flask. Cells were then passaged once a week for several weeks until analyzed by ddPCR.
Validation experiments.
To compare the single-cell cloning and bulk approaches, two experiments were conducted. First, U2OS cells were infected with HHV-6A/B and allowed to proceed as bulk cultures for several weeks. On day 108 postinfection, a portion of the cells was harvested and seeded at one cell/well in 96-well plates. Thirty-three days later, on day 141 (postinfection), cells from the bulk culture or individual clones were analyzed by ddPCR for HHV-6 DNA. Second, MCF-7 cells were infected with HHV-6A/B. After infection (5 h), cells were either allowed to grow in bulk or processed for single-cell cloning. Thirty days postinfection, DNA from the bulk cultures and individual clones were isolated and analyzed by ddPCR.
Cell proliferation.
U2OS and U2OS-AP3 clones were seeded at 500,000 cells/75-cm3 flask in triplicate. On days 3 and 7, cells were collected and counted.
Detection of cells with HHV-6A/B double integration.
To detect cointegration of HHV-6A and HHV-6B in cell clones, a recently described ddPCR assay was used (15). The assay consists of a single primer pair that amplifies the U67/U68 region in both HHV-6A and HHV-6B and two probes that discriminate between the two viruses (Table 4). The probe for HHV-6A was labeled with 6-carboxyfluorescein (FAM), and another, specific for HHV-6B, was labeled at a 50:50 HEX/FAM ratio. The primers and probes to detect the RNase reference gene RPP30 were previously described (36) (Table 4).
FISH.
Cell clones positive for HHV-6 were analyzed by fluorescent in situ hybridization (FISH) as described previously (37).
Treatment of HHV-6A/B cell lines and RT-qPCR.
Clonal cell lines were treated with either phosphonoacetic acid (PAA) (100 μg/ml), an inhibitor of the HHV-6 DNA polymerase, trichostatin A (TSA) (10 ng/ml), suberoylanilide hydroxamic acid (SAHA; also known as vorinostat) (1 μM), sodium butyrate (NaBy) (300 μM), three HDAC inhibitors, 12-O-tetradecanoylphorbol-13-acetate (TPA) (20 ng/ml), or hydrocortisone (1 μM) (Sigma-Aldrich) for periods of time varying between 8 h and 96 h and analyzed by RT-qPCR for the expression of viral genes, as described previously (16).
Statistics.
Fisher's exact test was used to compare the effects of treatments on integration frequencies, and chi-square analysis was used to compare clone integration frequency between ddPCR and single-cell cloning procedures. t test was used to compare gene expression levels. A P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
This study was supported by a grant from the Canadian Institutes of Health awarded to L.F. (MOP_123214). S.G.-G. and V.C. are supported by studentships from the Fonds de Recherche Québec-Santé.
A.G. and I.D. performed most experimental studies; S.G.-G., V.C., R.H.-S., J.C., and N.W. contributed by either performing or assisting in certain experiments; A.G., B.B.K., K.R.J., G.B., Y.M., and L.F. made contributions to the design and interpretation of experiments in addition of providing key reagents; L.F. wrote the paper.
REFERENCES
- 1.Luppi M, Marasca R, Barozzi P, Ferrari S, Ceccherini-Nelli L, Batoni G, Merelli E, Torelli G. 1993. Three cases of human herpesvirus-6 latent infection: integration of viral genome in peripheral blood mononuclear cell DNA. J Med Virol 40:44–52. doi: 10.1002/jmv.1890400110. [DOI] [PubMed] [Google Scholar]
- 2.Kaufer BB, Flamand L. 2014. Chromosomally integrated HHV-6: impact on virus, cell and organismal biology. Curr Opin Virol 9C:111–118. doi: 10.1016/j.coviro.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Morissette G, Flamand L. 2010. Herpesviruses and chromosomal integration. J Virol 84:12100–12109. doi: 10.1128/JVI.01169-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daibata M, Taguchi T, Nemoto Y, Taguchi H, Miyoshi I. 1999. Inheritance of chromosomally integrated human herpesvirus 6 DNA. Blood 94:1545–1549. [PubMed] [Google Scholar]
- 5.Daibata M, Taguchi T, Sawada T, Taguchi H, Miyoshi I. 1998. Chromosomal transmission of human herpesvirus 6 DNA in acute lymphoblastic leukaemia. Lancet 352:543–544. doi: 10.1016/S0140-6736(05)79251-5. [DOI] [PubMed] [Google Scholar]
- 6.Gravel A, Dubuc I, Morissette G, Sedlak RH, Jerome KR, Flamand L. 2015. Inherited chromosomally integrated human herpesvirus 6 as a predisposing risk factor for the development of angina pectoris. Proc Natl Acad Sci U S A 112:8058–8063. doi: 10.1073/pnas.1502741112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arbuckle JH, Medveczky MM, Luka J, Hadley SH, Luegmayr A, Ablashi D, Lund TC, Tolar J, De Meirleir K, Montoya JG, Komaroff AL, Ambros PF, Medveczky PG. 2010. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc Natl Acad Sci U S A 107:5563–5568. doi: 10.1073/pnas.0913586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endo A, Watanabe K, Ohye T, Suzuki K, Matsubara T, Shimizu N, Kurahashi H, Yoshikawa T, Katano H, Inoue N, Imai K, Takagi M, Morio T, Mizutani S. 2014. Molecular and virological evidence of viral activation from chromosomally integrated human herpesvirus 6A in a patient with X-linked severe combined immunodeficiency. Clin Infect Dis 59:545–548. doi: 10.1093/cid/ciu323. [DOI] [PubMed] [Google Scholar]
- 9.Gravel A, Hall CB, Flamand L. 2013. Sequence analysis of transplacentally acquired human herpesvirus 6 DNA is consistent with transmission of a chromosomally integrated reactivated virus. J Infect Dis 207:1585–1589. doi: 10.1093/infdis/jit060. [DOI] [PubMed] [Google Scholar]
- 10.Greco A, Fester N, Engel AT, Kaufer BB. 2014. Role of the short telomeric repeat region in Marek's disease virus replication, genomic integration, and lymphomagenesis. J Virol 88:14138–14147. doi: 10.1128/JVI.02437-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaufer BB, Jarosinski KW, Osterrieder N. 2011. Herpesvirus telomeric repeats facilitate genomic integration into host telomeres and mobilization of viral DNA during reactivation. J Exp Med 208:605–615. doi: 10.1084/jem.20101402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallaschek N, Sanyal A, Pirzer F, Gravel A, Mori Y, Flamand L, Kaufer BB. 2016. The telomeric repeats of human herpesvirus 6A (HHV-6A) are required for efficient virus integration. PLoS Pathog 12:e1005666. doi: 10.1371/journal.ppat.1005666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trempe F, Gravel A, Dubuc I, Wallaschek N, Collin V, Gilbert-Girard S, Morissette G, Kaufer BB, Flamand L. 2015. Characterization of human herpesvirus 6A/B U94 as ATPase, helicase, exonuclease and DNA-binding proteins. Nucleic Acids Res 43:6084–6098. doi: 10.1093/nar/gkv503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallaschek N, Gravel A, Flamand L, Kaufer BB. 2016. The putative U94 integrase is dispensable for human herpesvirus 6 (HHV-6) chromosomal integration. J Gen Virol 97:1899–1903. doi: 10.1099/jgv.0.000502. [DOI] [PubMed] [Google Scholar]
- 15.Sedlak RH, Hill JA, Nguyen T, Cho M, Levin G, Cook L, Huang ML, Flamand L, Zerr DM, Boeckh M, Jerome KR. 2016. Detection of human herpesvirus 6B (HHV-6B) reactivation in hematopoietic cell transplant recipients with inherited chromosomally integrated HHV-6A by droplet digital PCR. J Clin Microbiol 54:1223–1227. doi: 10.1128/JCM.03275-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaworska J, Gravel A, Flamand L. 2010. Divergent susceptibilities of human herpesvirus 6 variants to type I interferons. Proc Natl Acad Sci U S A 107:8369–8374. doi: 10.1073/pnas.0909951107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Y, Hidalgo-Bravo A, Zhang E, Cotton VE, Mendez-Bermudez A, Wig G, Medina-Calzada Z, Neumann R, Jeffreys AJ, Winney B, Wilson JF, Clark DA, Dyer MJ, Royle NJ. 2014. Human telomeres that carry an integrated copy of human herpesvirus 6 are often short and unstable, facilitating release of the viral genome from the chromosome. Nucleic Acids Res 42:315–327. doi: 10.1093/nar/gkt840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynaud JM, Jegou JF, Welsch JC, Horvat B. 2014. Human herpesvirus 6A infection in CD46 transgenic mice: viral persistence in the brain and increased production of proinflammatory chemokines via Toll-like receptor 9. J Virol 88:5421–5436. doi: 10.1128/JVI.03763-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang E, Cotton VE, Hidalgo-Bravo A, Huang Y, Bell AJ, Jarrett RF, Wilkie GS, Davison AJ, Nacheva EP, Siebert R, Majid A, Kelpanides I, Jayne S, Dyer MJ, Royle NJ. 2016. HHV-8-unrelated primary effusion-like lymphoma associated with clonal loss of inherited chromosomally-integrated human herpesvirus-6A from the telomere of chromosome 19q. Sci Rep 6:22730. doi: 10.1038/srep22730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. 2012. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 21.Wang RC, Smogorzewska A, de Lange T. 2004. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 119:355–368. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 22.Neumann AA, Watson CM, Noble JR, Pickett HA, Tam PP, Reddel RR. 2013. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev 27:18–23. doi: 10.1101/gad.205062.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsao EH, Kellam P, Sin CS, Rasaiyaah J, Griffiths PD, Clark DA. 2009. Microarray-based determination of the lytic cascade of human herpesvirus 6B. J Gen Virol 90:2581–2591. [DOI] [PubMed] [Google Scholar]
- 24.Mirandola P, Menegazzi P, Merighi S, Ravaioli T, Cassai E, Di Luca D. 1998. Temporal mapping of transcripts in herpesvirus 6 variants. J Virol 72:3837–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohyashiki JH, Takaku T, Ojima T, Abe K, Yamamoto K, Zhang Y, Ohyashiki K. 2005. Transcriptional profiling of human herpesvirus type B (HHV-6B) in an adult T cell leukemia cell line as in vitro model for persistent infection. Biochem Biophys Res Commun 329:11–17. doi: 10.1016/j.bbrc.2005.01.090. [DOI] [PubMed] [Google Scholar]
- 26.Baumann P, Benson FE, West SC. 1996. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87:757–766. doi: 10.1016/S0092-8674(00)81394-X. [DOI] [PubMed] [Google Scholar]
- 27.Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. 2012. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res 40:7347–7357. doi: 10.1093/nar/gks353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kofod-Olsen E, Pettersson S, Wallace M, Abduljabar AB, Oster B, Hupp T, Hollsberg P. 2014. Human herpesvirus-6B protein U19 contains a p53 BOX I homology motif for HDM2 binding and p53 stabilization. Virology 448:33–42. doi: 10.1016/j.virol.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 29.Takemoto M, Koike M, Mori Y, Yonemoto S, Sasamoto Y, Kondo K, Uchiyama Y, Yamanishi K. 2005. Human herpesvirus 6 open reading frame U14 protein and cellular p53 interact with each other and are contained in the virion. J Virol 79:13037–13046. doi: 10.1128/JVI.79.20.13037-13046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clark DA, Nacheva EP, Leong HN, Brazma D, Li YT, Tsao EH, Buyck HC, Atkinson CE, Lawson HM, Potter MN, Griffiths PD. 2006. Transmission of integrated human herpesvirus 6 through stem cell transplantation: implications for laboratory diagnosis. J Infect Dis 193:912–916. doi: 10.1086/500838. [DOI] [PubMed] [Google Scholar]
- 31.Kuhl U, Lassner D, Wallaschek N, Gross UM, Krueger GR, Seeberg B, Kaufer BB, Escher F, Poller W, Schultheiss HP. 2015. Chromosomally integrated human herpesvirus 6 in heart failure: prevalence and treatment. Eur J Heart Fail 17:9–19. doi: 10.1002/ejhf.194. [DOI] [PubMed] [Google Scholar]
- 32.Strenger V, Caselli E, Lautenschlager I, Schwinger W, Aberle SW, Loginov R, Gentili V, Nacheva E, DiLuca D, Urban C. 2014. Detection of HHV-6-specific mRNA and antigens in PBMCs of individuals with chromosomally integrated HHV-6 (ciHHV-6). Clin Microbiol Infect 20:1027–1032. doi: 10.1111/1469-0691.12639. [DOI] [PubMed] [Google Scholar]
- 33.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 34.Tang H, Kawabata A, Yoshida M, Oyaizu H, Maeki T, Yamanishi K, Mori Y. 2010. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 407:360–367. doi: 10.1016/j.virol.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 35.Gravel A, Sinnett D, Flamand L. 2013. Frequency of chromosomally-integrated human herpesvirus 6 in children with acute lymphoblastic leukemia. PLoS One 8:e84322. doi: 10.1371/journal.pone.0084322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sedlak RH, Cook L, Huang ML, Magaret A, Zerr DM, Boeckh M, Jerome KR. 2014. Identification of chromosomally integrated human herpesvirus 6 by droplet digital PCR. Clin Chem 60:765–772. doi: 10.1373/clinchem.2013.217240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufer BB. 2013. Detection of integrated herpesvirus genomes by fluorescence in situ hybridization (FISH). Methods Mol Biol 1064:141–152. doi: 10.1007/978-1-62703-601-6_10. [DOI] [PubMed] [Google Scholar]