

ABSTRACT
The Streptococcus pneumoniae clone Hungary19A-6 expresses unusually high levels of β-lactam resistance, which is in part due to mutations in the MurM gene, encoding a transferase involved in the synthesis of branched peptidoglycan. Moreover, it contains the allele ciaH232, encoding the histidine kinase CiaH (M. Müller, P. Marx, R. Hakenbeck, and R. Brückner, Microbiology 157:3104–3112, 2011, https://doi.org/10.1099/mic.0.053157-0). High-level penicillin resistance primarily requires the presence of low-affinity (mosaic) penicillin binding protein (PBP) genes, as, for example, in strain Hu17, a closely related member of the Hungary19A-6 lineage. Interestingly, strain Hu15 is β-lactam sensitive due to the absence of mosaic PBPs. This unique situation prompted us to investigate the development of cefotaxime resistance in transformation experiments with genes known to play a role in this phenotype, pbp2x, pbp1a, murM, and ciaH, and penicillin-sensitive recipient strains R6 and Hu15. Characterization of phenotypes, peptidoglycan composition, and CiaR-mediated gene expression revealed several novel aspects of penicillin resistance. The murM gene of strain Hu17 (murMHu17), which is highly similar to murM of Streptococcus mitis, induced morphological changes which were partly reversed by ciaH232. murMHu17 conferred cefotaxime resistance only in the presence of the pbp2x of strain Hu17 (pbp2xHu17). The ciaH232 allele contributed to a remarkable increase in cefotaxime resistance in combination with pbp2xHu17 and pbp1a of strain Hu17 (pbp1aHu17), accompanied by higher levels of expression of CiaR-regulated genes, documenting that ciaH232 responds to PBP1aHu17-mediated changes in cell wall synthesis. Most importantly, the proportion of branched peptides relative to the proportion of linear muropeptides increased in cells containing mosaic PBPs, suggesting an altered enzymatic activity of these proteins.
KEYWORDS: Streptococcus pneumoniae, PBP2x, PBP1a, CiaH, MurM, peptidoglycan analysis
INTRODUCTION
Streptococcus pneumoniae is a commensal bacterium that colonizes the human nasopharynx (1, 2). Moreover, it is a leading respiratory human pathogen, causing a variety of diseases mainly in children, elderly people, and immunocompromised patients (3, 4). S. pneumoniae has long been considered a highly β-lactam-susceptible organism. However, high-level penicillin-resistant S. pneumoniae (PRSP) strains frequently express multiple antibiotic resistance genes, and their incidence has increased dramatically since the 1980s worldwide. Only a few clones of serotypes 14, 23F, 19F, 19A, 9V, and 6B that spread globally were mainly responsible for this scenario (5). The introduction of a 7-valent pneumococcal conjugated vaccine in 2000 followed by a 13-valent vaccine in 2010 was associated with a decrease in the incidence of infections due to PRSP but was accompanied by the appearance of antibiotic-resistant clones expressing nonvaccine serotypes (6–8). This development underlines the importance of continuing surveillance for antibiotic resistance and furthering our understanding of resistance mechanisms.
Resistance to β-lactams in S. pneumoniae is primarily driven by alterations in the transpeptidase domain of three penicillin binding proteins (PBPs), PBP2x, PBP2b, and PBP1a (for a review, see reference 9). These altered PBPs display a decrease in affinity to the β-lactam antibiotics, while the alterations apparently leave the enzyme function unaffected. The DNA sequences of PBP genes in penicillin-sensitive S. pneumoniae strains are well conserved. In contrast, the PBP genes of PRSP strains contain mosaic sequence blocks of different lengths that may differ by over 20% at the DNA level or 10% at the amino acid level compared with the sequences of the corresponding regions in the PBP genes of penicillin-sensitive S. pneumoniae strains (see reference 9 and references within). There is evidence that these mosaic blocks were acquired from commensal mitis group streptococci, especially S. mitis, via homologous recombination (10–13). Subsequent intraspecies recombination and mutations lead to the further diversification of PBP genes, resulting in a large number of PBP alleles (14, 15).
PBP2x and PBP2b are essential proteins and represent the primary target of β-lactams (for a review, see reference 16). Mutations in each of these proteins result in low-level β-lactam resistance and can be selected in sensitive strains (17–19). In contrast, mutations in PBP1a do not result in a detectable resistance increase if they are not accompanied by PBP2x and/or PBP2b alterations (20). PBP2b does not interact with third-generation cephalosporins, such as cefotaxime; therefore, it is not a target for this class of compounds (21). Thus, PBP2b mediates resistance primarily to penicillins, such as piperacillin, whereas high-level cefotaxime resistance requires only alterations in PBP2x in combination with alterations in PBP1a (for reviews, see references 9 and 22).
PBPs are crucial enzymes acting in the biosynthesis of peptidoglycan (PG), a major cell wall component that surrounds the cytoplasmic membrane and is required to maintain the shape and osmotic stability of bacteria. Pneumococcal PG forms a multilayered network of glycan chains of alternating N-acetylglucosamine (GlcNAc) and N-acetylmuramic acid (MurNAc) residues connected via short stem peptides consisting of l-Ala–γ-d-iGln–l-Lys–d-Ala–d-Ala (23). Unamidated glutamate is prevalent only in monomers, indicating that the transpeptidases require fully amidated peptide substrates (24). The cross-linking of stem peptides, the crucial reaction that leads to the network structure of PG, is catalyzed by the d,d-transpeptidase activity of PBPs. In S. pneumoniae, the stem peptides can be further modified by replacement of the l-Lys ε-amino group with a dipeptide consisting of l-Ala or l-Ser followed by an invariable l-Ala residue, resulting in branched peptides (24–26). Overall, the pneumococcal PG consists of a complex mixture of mainly monomeric, dimeric, and trimeric peptides with modifications in the glycan and peptide chains (24). Some PRSP clones contain increased levels of branched muropeptides due to the presence of a mosaic MurM gene (25, 27, 28) whose product, MurM, displays increased catalytic activity (29). Hence, it was proposed that low-affinity PBPs from PRSP isolates prefer branched peptides as the substrate (27), but experimental evidence is still lacking. Remarkably, the deletion of murM (also called fibA) in PRSP results in a complete loss of the resistance phenotype (30–32), a phenomenon that is not understood in molecular terms.
In addition to the PBP and MurM genes, other genes have been implicated in the β-lactam resistance of S. pneumoniae (for a review, see reference 9). The first non-PBP gene identified in spontaneous β-lactam-resistant S. pneumoniae laboratory mutants was ciaH (33, 34), encoding the histidine kinase CiaH, part of the two-component regulatory system CiaRH (33, 35). Mutations in CiaH result in higher levels of expression of genes regulated by the cognate response regulator CiaR and confer a pleiotropic phenotype: an increase in the level of β-lactam resistance, prevention of competence development, and protection from lysis-inducing conditions (35–37). A functional CiaRH system is required for proper growth in laboratory mutants containing altered PBP2x (20, 37). Distinct mutations in ciaH were also identified in clinical isolates of S. pneumoniae, including isolate Hungary19A-6 (36, 38). The ciaH alleles from laboratory mutants of the S. pneumoniae R6 strain strongly enhance expression of the CiaR regulon, whereas CiaH variants of clinical strains increase the activity of CiaR-dependent promoters only moderately under the same circumstances (36).
The CiaRH system, part of a complex regulatory network, is ubiquitously present among nonpyogenic streptococci (39, 40). The response regulator CiaR directly controls 15 promoters that drive the transcription of 29 genes; among these are 5 genes specifying small noncoding cia-dependent small RNAs (csRNAs) (35, 41). The csRNAs feed into another regulatory network, including the competence regulon (42). Therefore, it is not surprising that the CiaRH system affects a variety of physiological processes, such as β-lactam resistance (33, 36), genetic competence (33, 37, 43–45), bacteriocin production (46, 47), the maintenance of cell integrity (37, 48), and host colonization (49). The CiaRH system was shown to be highly active and nearly constitutive under a variety of laboratory conditions (50) and in animal models of colonization and virulence (49, 51–53), but the signal detected by the sensor kinase CiaH is still unknown.
High-level penicillin- and multiple-antibiotic-resistant serotype 19A S. pneumoniae strains were prevalent in Hungary during the 1990s (54, 55) and occurred in the Czech Republic and Slovakia as well (56). These type 19A isolates express a PBP3 with an electrophoretic mobility different from that of most other S. pneumoniae isolates and differ in their PBP2x sequences, accompanied by a variable PBP profile (57, 58), and their genomes appear to be surprisingly variable (59). Accordingly, multilocus electrophoretic typing revealed several electrophoretic types (57). Multilocus sequence typing (MLST) identified one major clone, Hungary19A-6 (representative strain HUN663, also named Hungary19A-6), of sequence type (ST) 268 (ST268) (5). In this work, we studied the resistance determinants in two serotype 19A strains from Hungary, strains Hu15 and Hu17. They represent members of the same clone, ST226, a single-locus variant of Hungary19A-6 differing in the ddl allele, encoding the d-Ala–d-Ala ligase (60). Strain Hu15 is penicillin sensitive, whereas strain Hu17 exhibits high-level penicillin resistance; their MIC values for benzylpenicillin are 0.06 μg/ml and 24 μg/ml, respectively (57). This unique situation was used to study the development of cefotaxime resistance and to understand the contribution of genes known to play a role in this phenotype, pbp2x, pbp1a, murM, and ciaH, by analyzing the physiological and biochemical consequences in mutants containing various combinations of these genes.
RESULTS
β-Lactam resistance determinants in S. pneumoniae Hu17 and Hu15.
To identify penicillin resistance determinants, two strains of ST226 were chosen for genome sequencing: Hu17, which is one of the strains with the highest levels of penicillin and cefotaxime resistance held in the Kaiserslautern strain (KL) collection, and penicillin-sensitive strain Hu15 (60). Genes encoding the resistance determinants PBP2x, PBP2b, PBP1a, and MurM were analyzed in detail. All three PBP genes of Hu17 display a mosaic structure very similar to that of the PBP genes of the Hungary19A-6 genome (GenBank accession no. CP000936.1) (Fig. 1; see also Fig. S1 in the supplemental material). They contain one sequence block highly divergent from the sequence of S. pneumoniae R6 that covers the transpeptidase domain (pbp2x of strain Hu17 [pbp2xHu17] and pbp1a of strain Hu17 [pbp1aHu17]) or four smaller divergent sequence blocks in the transpeptidase domain (pbp2b of strain Hu17 [pbp2bHu17]) (Fig. 1 and S1). The mosaic sequence block of pbp2xHu17 contains regions highly similar (<5% divergence at the nucleotide level) to the sequence of pbp2x of S. mitis strains M3 and NCTC10712, which is common among the highly variable pbp2x sequences of serotype 19A isolates from Hungary (58). In the deduced protein sequence, the two mutations T338P and Q552E, close to the active site Ser337 and the K547SG box, respectively, are noteworthy. The combination of both mutations is rare and most likely contributes to the high penicillin resistance level of Hu17. E552 is selectable with cefotaxime (19). Whereas A338 is present in mosaic pbp2x of most PRSP isolates, P338 is uncommon and leads to higher levels of β-lactam resistance compared to those obtained with A338 (20).
FIG 1.
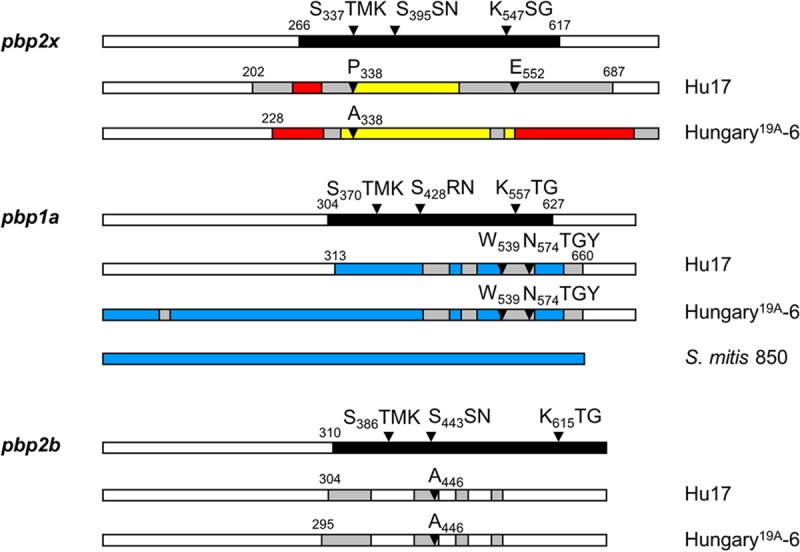
Mosaic PBPs of S. pneumoniae Hu17 and Hungary19A-6. Mosaic gene structures were deduced by comparison of the reference PBP2x sequences of strains S. pneumoniae R6 (white) and S. mitis M3 (red), NCTC10712 (yellow), and 850 (blue). Highly similar sequences (<5% difference) are shown in the same color; gray areas are divergent sequences of unknown origin. The numbers indicate the codons defining the sequence blocks that diverge from the pbp2xR6 nucleotide sequence by >15%. The domain structure and active-site boxes are indicated on top; the black area represents the transpeptidase domain. Mutations potentially relevant for the resistance phenotype are indicated.
The diverse block of pbp1aHu17 contains regions highly similar to the pbp1a sequence of S. mitis 850 (GenBank accession no. JUQO01000231) (61) interspersed with smaller regions of unknown origin (Fig. 1 and S1). PBP1aHu17 contains the mutation L539W and the alteration of four consecutive residues, T574SQF to NTGY, implicated in resistance (62).
The PBP2bHu17 gene of Hu17 has four divergent sequence blocks within the transpeptidase domain, including the mutation T446A, important for penicillin resistance (17). The mosaic blocks of pbp2b are not related to any pbp2b sequence of S. mitis in GenBank, except for a small region between codons 309 and 357 which is similar to the pbp2b sequences of S. mitis strains DD26 (GenBank accession no. LQOD00000000.1) (39) and SK1080 (GenBank accession no. AFQV01000033).
The murM gene of strain Hu17 (murMHu17) is identical to the murM gene of S. pneumoniae Hungary19A-6 and displays a mosaic structure compared to S. pneumoniae R6. Its sequence diverges from that of the murM of strain R6 (murMR6) by 17%, resulting in 74 amino acid (aa) changes. Curiously, the murM allele is almost identical to the murM allele of penicillin-sensitive S. mitis strain SK616 (GenBank accession no. NZ_AICR00000000) (14), which differs by only 17 nucleotides (nt) (or 8 aa) from the murMHu17 sequence (Fig. S2) and by 12 nt (or 5 aa) from the murM sequence of S. pseudopneumoniae 294 (GenBank accession no. JVMO01000033) (61), which is of unknown antibiotic susceptibility. This demonstrates that murM has also been acquired from S. mitis, similar to mosaic blocks in the PBP genes of PRSP isolates.
In addition, Hu17 contains the unique ciaH232 allele, resulting in the mutation N78D within the sensor domain; ciaH232 is slightly less effective than the CiaH wild type in mediating CiaR-dependent transcription in the R6 background (36). The ciaH232 allele does not confer β-lactam resistance in the R6 background, but since other ciaH alleles from laboratory mutants and clinical isolates mediate this phenotype (see reference 36 and references within), it was included in this study.
In penicillin-sensitive strain Hu15, the sequences of the three PBP genes are almost identical to those in S. pneumoniae R6 and do not contain mosaic blocks (Fig. S1). The pbp2x gene of Hu15 differs from that of R6 by 9 nt, resulting in only 1 aa change, I175V, in the N-terminal domain, and pbp1a differs by 9 nt (PBP1a) and 3 aa changes (A124T, A388D, and D533E), which are also present in pbp1aHu17, while pbp2b differs by 3 nt and no amino acid changes. Surprisingly, both murMHu17 and ciaH232 are present in Hu15, suggesting that these genes do not contribute to resistance by themselves, i.e., in the absence of mosaic PBPs. Furthermore, this clearly documents that they were present in this particular clone prior to the introduction of PBP genes, the major β-lactam resistance determinants.
Experimental outline.
In this analysis, we focused on the development of cefotaxime resistance. Since PBP2b is not a target of this particular antibiotic and thus does not contribute to cefotaxime resistance, it was not included in the experiments (21). The genes encoding PBP2x, PBP1a, MurM, and CiaH of resistant strain Hu17 were introduced individually or in various combinations into nonencapsulated laboratory strain R6 (Fig. 2) to see how they affect the resistance level, as described below. In addition, Hu15 was used as the recipient of pbp2xHu17 and pbp1aHu17 to see whether they suffice to confer the cefotaxime resistance level of Hu17. Unlike most clinical isolates, including the resistant members of ST226, Hu15 is readily transformable under standard laboratory conditions and can be used in transformation experiments. Transformants were named according to the transferred genes, as shown in Fig. 2.
FIG 2.
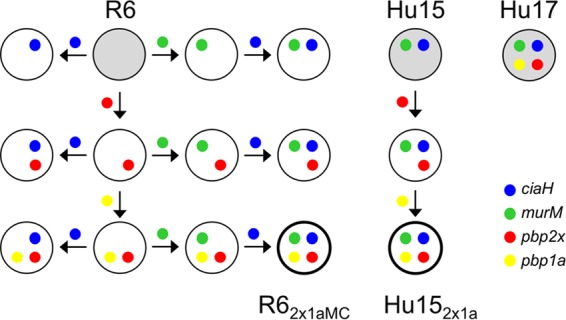
Schematic representation of transformation steps. The genes of PBP2x, PBP1a, MurM, and CiaH present in penicillin-resistant strain Hu17 (top right) were introduced individually or in various combinations into sensitive nonencapsulated laboratory strain S. pneumoniae R6 or into clinical isolate Hu15, as indicated by the colors. The direction of gene transfer is indicated by arrows. Gray circles represent recipient strains R6 and Hu15 and donor strain Hu17. Transformants containing all four genes are identified at the bottom and named according to the order of the transformation steps.
β-Lactam resistance mediated by pbp2xHu17 and pbp1aHu17.
Transformations were carried out with PCR-amplified PBPHu17 genes flanked by R6 or Hu15 sequences, as described in the supplemental material, to ensure that only pbp2xHu17 or pbp1aHu17 and not the flanking genes were introduced into the recipient strain. Cefotaxime was used as the selective antibiotic and was used at concentrations slightly above the MIC values for the recipient strains (for Hu15, 0.019 μg/ml; for R6, 0.018 μg/ml). Between 12 and 20 transformants were analyzed after incubation with BocillinFL, followed by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and fluorography, to identify transformants with low-affinity PBPs, and one such transformant was used in subsequent experiments.
First, the pbp2xHu17 gene was introduced into strain Hu15. The transformant, Hu152x, contained almost the entire transpeptidase domain of pbp2xHu17, including the deduced mutations P338 and E552 (Fig. 3). Subsequent transfer of pbp1aHu17 resulted in Hu152x1a, where pbp1aHu17 was present up to codon 550, a sequence which includes the deduced mutation W539 (Fig. 4). The MIC values of oxacillin and cefotaxime for Hu152x were 1.5 and 0.7 μg/ml, respectively (Fig. 5). The cefotaxime MIC for Hu152x1a was 1.2 μg/ml, which is significantly higher than the MIC for Hu152x and close to the MIC for the parental strain, Hu17 (1.6 μg/ml) (Fig. 5). The slight difference might be attributed to the fact that the entire pbp1aHu17 sequence, including codons 574 to 577, is not present in Hu152x1a. In contrast, no increase in the oxacillin MIC was mediated by pbp1aHu17; rather, a slight but reproducible decrease in the oxacillin MIC was observed (Fig. 5). This suggests that introduction of pbp1aHu17 into this particular genetic background requires cefotaxime selection and that the increased oxacillin resistance is related to other genes not included in the transformation experiments. Nevertheless, these data document that the four genes ciaH232, murMHu17, pbp2xHu17, and pbp1aHu17 suffice to mediate high levels of resistance to cefotaxime in Hu15.
FIG 3.

PBP2x variants obtained in transformation experiments. (A) PBP profiles of transformants containing mosaic PBP2x. PBPs were visualized after incubation with BocillinFL followed by separation by SDS-PAGE and fluorography. Recipient strains S. pneumoniae R6 and Hu15 were included for comparison, as indicated on top. The positions of the PBPs are marked on the left side. Arrowheads, low-affinity PBP2x. (B) Schematic representation of pbp2x in transformants R62x and Hu152x compared to strains R6 (white sequence blocks) and Hu17 (black sequence blocks). (Top row) The transpeptidase domain is shown in black, and the active-site motifs are indicated. (C) Deduced peptide sequences of PBP2x of the transformants compared to the peptide sequences of PBP2x of the penicillin-sensitive strain S. pneumoniae R6 and the donor PBP2x of Hu17. The first three lines indicate the amino acid positions. Only the residues at positions with altered residues are shown; residues identical to those in strain R6 (bold letters) are indicated by dots. The transpeptidase domain is shaded gray.
FIG 4.
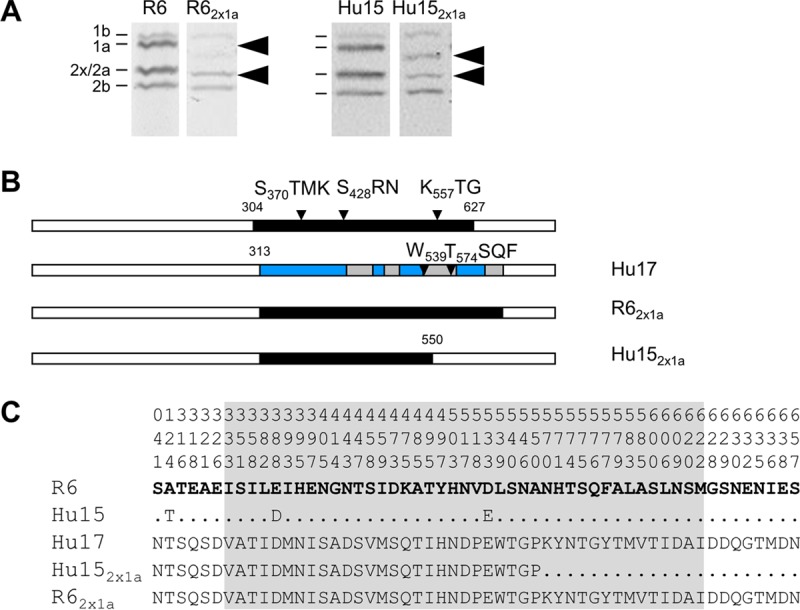
PBP1a variants obtained in transformation experiments. (A) PBP profiles of transformants containing mosaic PBP1a. PBPs were visualized after incubation with BocillinFL followed by separation by SDS-PAGE and fluorography. Recipient strains S. pneumoniae R6 and Hu15 were included for comparison, as indicated on top. The positions of the PBPs are marked on the left side. Arrowheads, low-affinity PBP2x and PBP1a. (B) Schematic representation of pbp1a in transformants compared to strains R6 (white sequence blocks) and Hu17 (black sequence blocks). (Top row) The transpeptidase domain is shown in black, and the active-site motifs are indicated. (C) Deduced peptide sequences of PBP1a of the transformants compared to the peptide sequences of PBP1a of the penicillin-sensitive strain S. pneumoniae R6 and the donor PBP1a of Hu17. The first three lines indicate the amino acid positions. Only the residues at positions with altered residues are shown; residues identical to those in strain R6 (bold letters) are indicated by dots. The transpeptidase domain is shaded gray.
FIG 5.
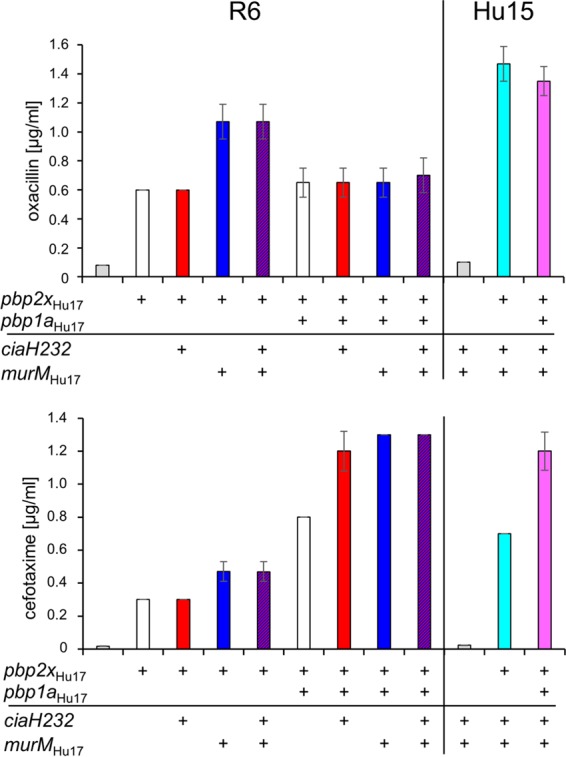
Resistance patterns of various S. pneumoniae transformants. The two β-lactams oxacillin and cefotaxime were used. Mean values from at least three independent experiments are shown. Bars indicate standard deviations. The MIC values of oxacillin and cefotaxime for the donor strain Hu17 were 30 μg/ml and 1.6 μg/ml, respectively.
The transformant R62x contained the pbp2xHu17 sequence up to codon 660, resulting in a considerable increase in cefotaxime and oxacillin MICs (0.3 μg/ml and 0.6 μg/ml, respectively) compared to those for the parental R6 strain (0.018 μg/ml and 0.08 μg/ml, respectively), as expected (Fig. 3 and 5). Successive transformation of the entire pbp1aHu17 sequence increased the resistance levels even more, especially for cefotaxime (0.8 μg/ml); the increase in the oxacillin MIC was less pronounced (0.65 μg/ml) (Fig. 4 and 5). However, these values are below the MICs for Hu152x1a, clearly documenting that at least murMHu17 contributes to β-lactam resistance and ciaH232 potentially contributes to β-lactam resistance.
Effects of murMHu17 and ciaH232 on β-lactam resistance and cell morphology in S. pneumoniae R6.
Although S. pneumoniae strains Hu15 and R6 are penicillin susceptible, they differ slightly in their MIC values for oxacillin and cefotaxime (Fig. 6). To assess the contribution of murMHu17 and ciaH232 to β-lactam resistance, the genes were introduced individually or in combination into S. pneumoniae R6 using the Janus counterselectable procedure (63). This avoids selection with β-lactam antibiotics, and the rpsL41 mutation, which is required for such constructs, has no effect on CiaRH-mediated regulation or β-lactam susceptibility (36).
FIG 6.

Cell morphology and resistance pattern of S. pneumoniae R6 transformants carrying the murM and/or ciaH gene from strain Hu17. (A) The MIC values for the transformants are indicated as colored bars, and those for control strains R6 and Hu15 are indicated as white bars. The two β-lactams cefotaxime and oxacillin were used. Mean values from at least three independent experiments are shown. Bars indicate standard deviations. (B) Representative phase-contrast micrographs are shown. The strains were grown in C+Y medium. Arrows, altered cell morphology. Bar, 5 μm.
The transformant R6 murMHu17 (R6M) grew with a longer generation time of 68 min and a shorter stationary phase (approximately 2 h) compared to those for R6 and R6 ciaH232 (R6C) (51 min and approximately 4 h, respectively) and was only slightly but reproducibly less susceptible to oxacillin and cefotaxime (Fig. 6A). However, the MIC values of cefotaxime and oxacillin for R6M were still below those for Hu15. In addition, the cell morphology of R6M was grossly altered. The cells were round, similar to Hu15 cells, and distinct from the oval-shaped cells of the parental R6 strain, and 50% of the cells grew in chains containing occasionally enlarged or smaller cells (Fig. 6B). In contrast, R6C grew with the same generation time as the parental R6 strain (not shown), and its cell morphology was not affected (Fig. 6B). The visual impression was confirmed by precise determination of the cell dimensions (see Materials and Methods). In fact, the length and width of R6C cells (0.96 μm ± 0.04 and 0.83 μm ± 0.03, respectively; n = 206) were identical to the values obtained for R6 cells (0.95 μm ± 0.04 and 0.80 μm ± 0.03, respectively; n = 220).
Introduction of ciaH232 into R6M (R6MC) had no effect on MIC values but remarkably affected cell morphology. R6MC cells were more oval shaped than R6M cells, and the morphology of R6MC cells was similar to that of cells of the parental R6 strain, whereas chain formation was more pronounced in R6MC cells than parental R6 cells (Fig. 6B). In other words, the morphological defects caused by murMHu17 were almost reversed by ciaH232, while cells still grew in chains, suggesting that the CiaRH system somehow responds to the altered cell wall biochemistry mediated by murMHu17.
Effect of ciaH232 and murMHu17 on β-lactam resistance in S. pneumoniae R62x and R62x1a.
In further experiments, ciaH232 and murMHU17 were transferred into strains R62x and an R6 strain which carried pbp2xHu17 and pbp1aHu17 (R62x1a) individually or in combination to study their impact on resistance in the presence of pbp2xHu17 alone or together with pbp1aHu17. The introduction of murMHu17 into R62x resulted in strain R62xM and conferred increased cefotaxime and oxacillin MICs (Fig. 5). This demonstrates for the first time that pbp2xHu17 alone suffices as a genetic background to reveal that a mosaic murM contributes substantially to β-lactam resistance. In contrast, transfer of ciaH232 into strain R62x (R62xC) or R62xM (R62xMC) had no effect on MIC values (Fig. 5).
When transformed into R62x1a, murMHu17 resulted in a further increase in the level of cefotaxime resistance (1.3 μg/ml compared to 0.8 μg/ml for R62x1a). Surprisingly, transfer of ciaH232 into R62x1a also resulted in a significant increase in the cefotaxime MIC value to 1.2 μg/ml (Fig. 5). Resistance associated with ciaH was first observed in laboratory mutants and led to hyperactivation of the CiaR regulon (64). Since ciaH232 does not mediate resistance by itself in the R6 background (36), this finding suggests that ciaH232 can indeed hyperactivate the CiaR regulon but only in the presence of both pbp1aHu17 and pbp2xHu17 (R62x1aC) and not in the sole presence of pbp2xHu17 (R62xC). This is the first time that a phenotype conferred by a ciaH allele appears to be associated with an altered PBP1a. Moreover, this effect was seen only in the absence of murMHu17; i.e., transfer of ciaH232 into R62x1aM did not result in a further increase in the level of β-lactam resistance (Fig. 5).
Curiously, the oxacillin resistance level of all R6 transformants containing pbp1aHu17 was clearly below the MIC for R62xM or R62xMC mediated by murMHu17, as if the presence of pbp1aHu17 somehow suppresses the murMHu17-mediated resistance (Fig. 5). It should also be noted that the oxacillin resistance level of Hu152x1a was almost 2-fold higher than that of R62x1aMC. This strongly suggests the presence of unknown features that feed into the resistance phenotype of the clone Hungary19A-6.
Activity of CiaR-controlled promoters in the presence of resistance determinants.
Various ciaH alleles identified in laboratory mutants contribute to resistance and result in a hyperactive CiaRH system, whereas these features were less pronounced with three distinct CiaH alleles of clinical isolates or even absent in the case of ciaH232 (36). However, the effect of ciaH232 on cefotaxime resistance when it was introduced into R62x1a as described above suggested that the CiaRH system is also hyperactive under these conditions. Therefore, the expression of two CiaR-dependent genes encoding the serine protease HtrA and a secreted protein of unknown function, spr0931, was tested in different R6 derivatives using lacZ reporter assays (35, 65). The activities of both the PhtrA and Pspr0931 promoters are strongly dependent on CiaR, and there is no evidence that they are controlled by other regulators (35, 50). Each of the promoters was cloned in front of a promoterless Escherichia coli lacZ gene (65) and integrated into the S. pneumoniae R6 strain and its derivatives. We selected for further experiments strains which carried ciaH232 in combination with altered pbp2xHu17 (R62x), strains which carried pbp2xHu17 and pbp1aHu17 (R62x1a), strains which did not carry pbp2xHu17 (R6C), and the parental R6 strain as a control.
β-Galactosidase activities were measured in cells grown in complex C medium (66) supplemented with 0.1% yeast extract (C+Y medium) at two different time points during exponential growth (when the optical density at 600 nm [OD600] was 0.4 and 0.8) and at the onset of the stationary phase. As shown in Fig. 7A, the levels of transcription from both promoters PhtrA and Pspr1149 were similar in all strains except R62x1aC, where promoter activities increased more than 3-fold and were even higher at the onset of stationary phase. These results confirmed that the CiaRH system is hyperactive in the presence of pbp1aHu17 in combination with pbp2xHu17.
FIG 7.

β-Galactosidase activities expressed from CiaR-regulated promoters and cell morphology of S. pneumoniae R6 derivatives carrying the ciaH232 allele. (A) Strains were grown in C+Y medium. β-Galactosidase activities were determined at three different time points and are given in nanomoles of nitrophenol produced per minute and milligram of protein. Mean values and standard deviations from three independent experiments are presented. The activities of the CiaR-dependent promoters in the R62x1aC strain were significantly different (P < 0.05) from those of the promoters in the other strains. The promoters and relevant genetic markers are indicated. (B) Phase-contrast images taken at the mid-exponential growth phase. The strains were grown in C+Y medium. Arrows, altered cell morphology. Bar, 5 μm.
Effect of CiaH232 and mosaic PBP2xHu17/PBP1aHu17 on morphology and peptidoglycan composition.
The hyperactivation of CiaR observed in strain R62x1aC indicates that the sensor kinase CiaH232 responds to some signal mediated by PBP1aHu17. Since PBP1a acts as a transglycosylase/transpeptidase during PG synthesis, it is possible that R62x1aC produces an altered PG. Therefore, transformants containing pbp2xHu17 or both pbp2xHu17 and pbp1aHu17 with or without ciaH232 were examined microscopically, and the PG composition of the strains was investigated.
The presence of pbp2xHu17 in R62x already resulted in cells that were enlarged and more rounded compared to cells of the parental R6 strain, an effect which was at least partially reverted by ciaH232 in R62xC (Fig. 7B). The additional presence of pbp1aHu17 (R62x1a) resulted in cells with more pointed ends, and chain formation and smaller cells were frequently observed, suggesting some defects related to cell division and separation. Again, the subsequent introduction of ciaH232 resulted in cells that appeared more normal in size and form (Fig. 7B). These results suggest that the function of the products of both pbp2xHu17 and pbp1aHu17 differs from that of the respective R6 proteins and, thus, that these products affect cell morphology, whereas ciaH232 counteracts this defect.
The peptidoglycan was isolated from the transformants, and the compositions of the muropeptides were analyzed as described previously (24). Detailed analysis of the PG of S. pneumoniae by reversed-phase high-performance liquid chromatography (HPLC) revealed 50 different muropeptide structures, including those carrying modifications such as deacetylation of GlcNAc residues and O-acetylation of MurNAc residues, the presence of iGln instead of Glu, and Gly at position 5 of the stem peptide (24). In the present study, 31 peaks which included all major muropeptides, accounting for 68 to 72% of known muropeptides, were used for the final analysis (Table S1).
All mutants retained an overall muropeptide composition similar to that of parental strain R6 (Tables 1 and S1). Small changes (differences of up to 15% from the values for R6) were seen in the total number of monomers, dimers, and trimers between the PG of the strains. However, changes became evident when individual muropeptide classes were analyzed in detail (Table 1). Remarkably, the presence of ciaH232 already resulted in a slight increase in the amount of pentapeptides by 18% in R6C compared to R6 and a relative increase in the ratio of indirectly cross-linked dimers versus directly cross-linked dimers from 1.19 in R6 to 1.61 in R6C (a 35% increase compared to that in R6), in parallel with a higher ratio of branched peptides versus linear peptides (from 0.44 to 0.51, or 16%). Moreover, the proportion of muropeptides with deacetylated GlcNAc increased only slightly from 20.7% (R6) to 22% (Table 1). This suggests that the CiaH232 allele in not neutral for the cell and may alter the cell wall precursor metabolism to be compatible with PBPs that prefer branched substrates.
TABLE 1.
Cell wall muropeptide compositions of S. pneumoniae R6 and mutant strains
| Muropeptide | Relative peak area (%) or ratio in straina: |
|||||
|---|---|---|---|---|---|---|
| R6 | R6C | R62x | R62xC | R62x1a | R62x1aC | |
| Monomers | 56.8 ± 0.8 | 56.9 ± 1.2 | 60.1 ± 0.1 | 61.0 ± 1.7 | 60.5 ± 0.2 | 59.4 ± 1.1 |
| Dimers | 39.0 ± 0.8 | 38.6 ± 1.6 | 35.2 ± 0.1 | 34.4 ± 1.6 | 34.9 ± 0.1 | 35.7 ± 1.2 |
| Trimers | 4.2 ± 0.0 | 4.5 ± 0.3 | 4.7 ± 0.3 | 4.7 ± 0.1 | 4.6 ± 0.2 | 4.9 ± 0.1 |
| Tripeptides | 65.7 ± 0.8 | 65.1 ± 0.3 | 64.4 ± 1.2 | 64.6 ± 0.8 | 64.3 ± 0.6 | 64.4 ± 1.5 |
| Tetrapeptides | 26.1 ± 0.9 | 26.5 ± 1.2 | 25.2 ± 0.6 | 25.1 ± 0.2 | 26.3 ± 1 | 26.1 ± 0.5 |
| Pentapeptides | 6.0 ± 1.4 | 7.1 ± 1.4 | 9.4 ± 1.7 | 9.3 ± 0.6 | 8.7 ± 1.6 | 8.8 ± 1.0 |
| Peptides in cross-links | 43.2 ± 0.8 | 43.1 ± 1.2 | 39.9 ± 0.2 | 39.0 ± 1.7 | 39.5 ± 0.2 | 40.6 ± 1.1 |
| Directly cross-linked dimers | 17.8 ± 1.0 | 14.8 ± 0.8 | 13.2 ± 0.6 | 13.2 ± 0.9 | 11.8 ± 0.5 | 12.5 ± 1.0 |
| Indirectly cross-linked dimers | 21.2 ± 1.8 | 23.8 ± 0.8 | 22.0 ± 0.5 | 21.2 ± 0.7 | 23.2 ± 0.6 | 23.2 ± 0.2 |
| Ratio of indirectly cross-linked dimers/directly cross-linked dimers | 1.19 | 1.61 | 1.7 | 1.61 | 1.97 | 1.86 |
| Linear peptides | 63.9 ± 2.4 | 60.7 ± 1.3 | 59.5 ± 2.1 | 59.5 ± 1.0 | 57.4 ± 0.8 | 57.5 ± 0.6 |
| Branched peptidesb | 27.9 ± 1.9 | 30.9 ± 1.1 | 32.2 ± 2.1 | 31.7 ± 0.7 | 32.6 ± 0.2 | 33.2 ± 0.5 |
| Ratio of branched peptides/linear peptides | 0.44 | 0.51 | 0.54 | 0.53 | 0.57 | 0.58 |
| Deacetylation | 20.7 ± 0.1 | 21.9 ± 0.1 | 23.0 ± 1.9 | 23.3 ± 0.7 | 25.0 ± 3.2 | 24.0 ± 0.2 |
The values are means ± variations for two independent PG preparations. Laura software (Lab Logic Systems Ltd) was used for peak area quantification. The relative peak areas were estimated as the percentage of all known peaks. Underlined values were considered to be significantly different by more than 15% from the value for strain R6; bold underlined values were significantly different by more than 30% from the value for R6.
Muropeptides with an SA or AA branch.
Major differences in muropeptide compositions were observed in constructs containing the mosaic PBPs (Table 1). In all four mutants, the percentage of directly cross-linked dimeric peptides decreased substantially, while the percentage of indirect cross-links slightly increased, resulting in an overall slightly reduced amount of dimers. The percentage of pentapeptides, muropeptides with deacetylated GlcNAc, and branched peptides increased further in all strains containing mosaic PBP genes. The additional presence of ciaH232 (R62xC) had no effect compared to the effect on R62x. In R62x1a, indirectly cross-linked dimers were present almost twice as often as directly cross-linked peptides, and the proportion of muropeptides with deacetylated GlcNAc increased slightly to 25%. In other words, the presence of PBP2xHu17 and PBP1aHu17 as well resulted in a different PG composition, indicating that their alterations apparently affected their substrate specificity, and surprisingly, ciaH232 also had an impact on the muropeptide profile.
Nevertheless, our PG analysis shows the remarkable robustness of PG synthesis in S. pneumoniae that allows the cell to produce an almost normal PG with an altered regulatory CiaRH system and two mosaic synthases, PBP2x and PBP1a, which lead in their original strain background to a different, more branched PG. Yet this specificity of the PBPs does cause structural changes in the PG, including a decrease in direct cross-links and an increase in pentapeptides and deacetylated muropeptides, which may contribute to the morphological defects observed.
DISCUSSION
The results presented here reveal new aspects of resistance development in clinical isolates of S. pneumoniae by analyzing the physiological consequences associated with the transfer of resistance determinants into laboratory strain R6. The high-level penicillin-resistant clone Hungary19A-6 contains four genes involved in cefotaxime resistance: the well-known penicillin target enzymes PBP2x and PBP1a and the non-PBP genes murM and ciaH232. We used the unique situation of penicillin-sensitive strain Hu15, which is of the same sequence type (ST226) as strain Hu17 and which contains both an altered murM and the ciaH232 allele, to show that these two genes were already present before the introduction of altered PBP genes, i.e., most likely long before the extensive use of β-lactam antibiotics triggered the evolution of PRSP. In the following sections we discuss features associated with these four genes.
pbp2x and pbp1a.
Both pbp2xHu17 and pbp1aHu17 represent rare mosaic variants similar to alleles present in other Hungary isolates of ST226 (57, 58) (Fig. 1; see also Fig. S1 in the supplemental material). Both PBPs contain mutations close to active-site motifs. P338 and E552 in PBP2xHu17 have been shown experimentally to play a role in resistance development (19, 20). Also, the change Q599P, a mutation at a site which is affected in high-level cefotaxime-resistant strain S. mitis B6 (W599) (67), might contribute to the high level of cefotaxime resistance of strain Hu17. There are other alterations in PBP2xHu17 frequently found in PBP2x of PRSP strains: A369V, I371T, N444S, and S531K (15). The alterations A279G, I318L, E320K, T401I, Q405E, A410T, V544I, L565T, and S612V have not yet been described, and it remains to be clarified experimentally whether these changes contribute to resistance. All other alterations occur in the parental sequence blocks related to S. mitis strains M3 and NCTC10712.
PBP1aHu17 clearly contributes to cefotaxime resistance but only slightly contributes to oxacillin resistance (58), indicating that its mutations primarily affect the interaction with cefotaxime. Similar results were obtained when mosaic PBP genes of the clone Spain23F-1 were transferred into the R6 strain (20). Curiously, the oxacillin resistance level of all R6 transformants containing pbp1aHu17 was clearly below the MIC of R62xM or R62xMC mediated by murMHu17, as if the presence of pbp1aHu17 somehow suppresses the murMHu17-mediated resistance (Fig. 5). The oxacillin resistance level of Hu152x1a was almost 2-fold higher than that of R62x1aMC, strongly suggesting the presence of unknown features that feed into the resistance phenotype of the ST226 strains.
The A124T and E388D mutations in PBP1a lead to strong suppression of the essentiality of mreCD. PBP1a and MreCD are components proposed to be part of the same protein complex involved in peripheral PG synthesis (68). These changes are not present in strain R6 but are present in strains Hu15 and Hu17. It should also be noted that mraY, located downstream of pbp2x, is highly altered in strains Hu17 and Hu15. MraY is an essential phospho-N-acetylmuramoyl pentapeptide transferase in PG synthesis. These gene products could have an impact on cell morphology and PG composition in the Hungary19A-6 clone and might affect PBP function indirectly.
Although the overall PG composition appeared to be similar in all constructs analyzed, differences which affected the ratio of individual muropeptides, the substrate, and the product of PBPs were revealed. These differences could be due to modifications in the availability of the different substrates or to endopeptidases hydrolyzing the PG cross-links. However, we believe it to be more likely that the PBPs themselves were responsible for these changes. All R6 derivatives containing PBP2xHu17 or both PBP2xHu17 and PBP1aHu17 showed an increased ratio of indirectly over directly cross-linked dimers, ranging from an increase compared to the ratio in R6 of 43% in R62x to 65% in R62x1a (Table 1). This implies that both PBPs have an altered substrate specificity and prefer branched over linear peptides as the substrate, as has been previously suggested (27). We have now demonstrated for the first time experimentally an impact of individual mosaic PBPs on PG composition. Enzymatic analyses of purified PBPs are required to reveal in detail the differences in the kinetic properties of PBPs between resistant strains and sensitive strains.
The R6 strain used in the present study has mostly linear peptides but a higher percentage of branched peptides and indirect cross-links than its progenitor strains, D39 and R36A (24, 26, 28, 69). Therefore, it is possible that mosaic PBPs can affect the PG composition more prominently in different genetic backgrounds.
There were other differences in muropeptide composition that cannot be explained by the sole action of PBP2x and PBP1a. The increase in pentapeptides in strains with the mosaic pbp2x gene suggests either that the trimming of nascent pentapeptides to tetra- and tripeptides by PBP3 (70) and LdcB (DacB) (71) is affected or that the overall synthesis of PG is increased to an extent that pentapeptides are not trimmed fast enough. Notably, the cells of R62x and R62x1a exhibited morphological defects similar to those of cells of a dacB deletion strain (72). Finally, strains with mosaic PBP genes contained more deacetylated muropeptides; whether this was due to an increased activity of the PG deacetylase PgdA remains to be clarified (73).
Effects mediated by murMHu17.
The presence of an altered MurM gene in PRSP isolates of serotype 19A strains from Hungary and its contribution to penicillin resistance in the presence of altered PBP genes have previously been described for strains Hun663 (30, 74) and 3191 (32). In contrast, many other PRSP clones do not contain mosaic MurM genes, including the clone Taiwan19F-14 and serotype 19A switch variants thereof (75) and the clonal complex Spain23F-1. The mosaic murM was present not only in penicillin-resistant strain Hu17, as expected, but also, as shown here for the first time, in penicillin-sensitive strain Hu15, which does not contain mosaic PBP genes. The gene murMHu17 is highly related to murM of the penicillin-sensitive S. mitis strain SK616 (14), differing in only 5 aa, strongly suggesting that it was acquired from S. mitis. This is not surprising, given the fact that Hungary19A-6 had acquired the largest proportion of genes (8.2%) from S. mitis in a comparative genomic analysis of 35 Streptococcus species genomes (76).
Two amino acid changes in MurMHu17 are present in the region between residues 244 and 274 proposed to be important for the contribution to penicillin resistance (77): K266 and P267. When introduced into S. pneumoniae R6, murMHu17 contributed only marginally to β-lactam resistance (Fig. 5); similarly, only a small increase in β-lactam susceptibility has been noted in R6 derivatives with a deleted murM (31). Previous analyses have studied the effect of murM only in the presence of multiple mosaic PBP genes (30, 32). As shown here, MurMHu17 already caused changes in morphology and growth in the R6 background (Fig. 6), and a substantial increase in β-lactam resistance was observed when it was introduced into R62x; i.e., MurMHu17 does not require PBP1a to mediate a clear resistance effect (Fig. 5). Thus, it could well be that the presence of the mosaic murM is not related to a selective advantage during exposure to β-lactams but that it has been circulating in this clone long before the transfer of PBP sequences. In agreement with this, the MurM gene in strains Hu15, Hu17, and Hungary19A-6, including the upstream gene encoding a putative tributyrin esterase, is completely conserved, whereas PBP genes and flanking regions (pbp2x and mraY, clpL; recU and pbp1a; pbp2b and ddl) are diverse, as has been described in other PRSP isolates (78). This scenario suggests that the mosaic PBPs, after all essential enzymes, must cope with MurMHu17 with different enzymatic properties (29), resulting in the abundant presence of branched muropeptides (27), i.e., the substrates of PBPs. In other words, some mutations in the mosaic PBPs might be related to an altered substrate pool and not necessarily to β-lactam resistance in this particular genetic background.
The allele ciaH232.
Almost no effects of ciaH232 on β-lactam resistance or the expression of CiaR-regulated genes were observed when it was introduced into the R6 strain, in contrast to the findings for ciaH alleles from laboratory mutants (36). However, cells containing ciaH232 reacted more strongly to the presence of acetate in the medium (79). In the present study, some new features of ciaH232 that are associated with cell morphology, β-lactam resistance, and cell wall composition were revealed. The first example refers to the comparison of the transformants R6M and R6MC, where R6M cells grew with an aberrant morphology, whereas R6MC cells were indistinguishable from those of the parental R6 strain (Fig. 5). Similarly, the morphological changes induced by PBP2xHu17 and PBP1aHu17 were compensated for by the presence of ciaH232 (Fig. 7B). This suggests that CiaH232 apparently responds to alterations mediated directly or indirectly by the resistance determinants of Hu17 to ensure proper cell growth. Second, cefotaxime resistance was further increased when ciaH232 was transformed into R62x1a but not into R62x (Fig. 6), and the CiaR-regulated genes htrA and spr0931 showed higher expression levels only in R62x1aC (Fig. 7A). The N78D mutation of CiaH232 is located in the sensor domain of the histidine protein kinase, in agreement with an altered signal recognition site outside the cell membrane, mediated by murMHu17 and pbp1aHu17 or pbp2xHu17. Thus, it is possible that the CiaH mutation expressed by ciaH232 is a response to the presence of murMHu17 in the Hungary19A-6 strain.
These data were complemented by cell wall analysis, which revealed changes in the PG composition in R6 derivatives containing pbp1aHu17 and/or pbp2xHu17 (Tables 1 and S1); murM-mediated changes in PG composition have been reported before (30, 74). Curiously, enhanced expression of the CiaR-regulated genes was observed only in R62x1aC and not in R62xC, similar to previous reports that showed that the wild-type ciaH allele did not affect htrA expression in the presence of PBP2x point mutations or another mosaic PBP2x from a clinical isolate (37). However, CiaR activation by CiaH from clinical isolates of S. pneumoniae was far less pronounced than that by CiaH from laboratory mutants (36). This is an indication that mutations in PBP2x and CiaH selected with β-lactams in laboratory mutants affect the function of both proteins differently and with a more detrimental outcome to the cells compared to the outcome resulting from mutated alleles in clinical isolates. Altogether this scenario adds another facet to the interplay between the CiaRH system and PBPs and differences in PBP mutations of laboratory mutants versus clinical isolates.
Most intriguing was the finding that the muropeptide composition was affected by ciaH232 (R6C) in the absence of PBPsHu17: an increase in the amount of pentapeptides by 18%, an increased proportion of indirectly cross-linked dimers (12%), and more muropeptides containing a deacetylated GlcNAc (5.8%). The values are less than those seen for R62x and R62x1a but concern the same muropeptide classes. These data are in agreement with the assumption that the CiaRH system somehow controls the overall composition of the pneumococcal cell wall to ensure its integrity (20, 37).
Concluding remarks.
In the early 1990s, the penicillin resistance of pneumococci was considered to be entirely due to altered PBPs (80). The non-PBP components CiaH and MurM have since been recognized to be relevant players in resistance development in laboratory mutants and clinical isolates, respectively. The present analysis revealed that the CiaH232 allele of the S. pneumoniae clone Hungary19A-6 responds to the presence of MurMHu17. The contribution to β-lactam resistance of these two genes in combination with mosaic PBP genes carrying mutations known to be relevant for resistance development became evident. The interplay between the cytoplasmic MurM involved in the biosynthesis of PG precursors, transpeptidases controlling the PG cross-linkage at the outer surface of the cell wall (PBP2x and PBP1a), and the sensor histidine protein kinase CiaH mediating signals from the outside to the response regulator CiaR is schematically shown in Fig. 8. Taken together, the data reveal a highly complex network that ensures the synthesis of a functional bacterial cell wall under antibiotic stress.
FIG 8.

Schematic view of the interactions between MurM, PBPs, and the CiaRH system. Black, a branched muropeptide; red, l-Ala in the interpeptide bridge added by MurMHu17; gray: cell wall; black line, cell membrane. See the text for details.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
The bacterial strains used in this study are listed in Table 2. S. pneumoniae strains were grown at 37°C without aeration in complex C medium (66) supplemented with 0.1% yeast extract (C+Y medium) or in brain heart infusion (BHI; Roth) medium, and growth was followed by monitoring the optical density at 600 nm (OD600). Strains were grown on D-agar plates (81) supplemented with 3% defibrinated sheep blood.
TABLE 2.
S. pneumoniae strains used in this study
| Strain | Relevant characteristics | Reference(s) or source |
|---|---|---|
| R6a | Unencapsulated laboratory strain, β-lactam susceptible | 85, 86 |
| Hu15 | Serotype 19A isolate from Hungary, β-lactam susceptible | 57 |
| Hu17 | Serotype 19A isolate from Hungary, β-lactam resistant | 57 |
| CCCOmurM::Janus | CCCO murM::Kanr rpsL+ rpsL41 Kanr Strr | 12 |
| CCCOmurMHu17 | CCCO murMHu17 rpsL41 Strr | 12 |
| RKL243 | R6 ciaH232 rpsL41 Strr | 36 |
| RKL248 | RKL243 bgaA::tet(M)-PhtrA Strr Tetr | 36 |
| RKL249 | RKL243 bgaA::tet(M)-Pspr0931 Strr Tetr | 36 |
| RKL161 | R6 ciaH::Kanr rpsL+ rpsL41 Kanr Strs | 36 |
| R6strR | R6 rpsL41 Strr | This study |
| R6M | R6 murMHu17 rpsL41 Strr | This study |
| R6MC | R6M ciaH232 Strr | This study |
| R62xa | R6 pbp2xHu17 | This study |
| R62xCa | R62x ciaH232 rpsL41 Strr | This study |
| R62xM | R62x murMHu17 rpsL41 Strr | This study |
| R62xMC | R62xM ciaH232 Strr | This study |
| R62x1aa | R62x pbp1aHu17 | This study |
| R62x1aM | R62x1a murMHu17 rpsL41 Strr | This study |
| R62x1aCa | R62x1a ciaH232 Strr | This study |
| R62x1aMC | R62x1aM ciaH232 Strr | This study |
| Hu152x | Hu15 pbp2xHu17 | This study |
| Hu152x1a | Hu152x pbp1aHu17 | This study |
The promoter-probe plasmids carrying htrA or spr0931 promoter fragments (35) were integrated into the bgaA locus of R6 derivatives; therefore, the strains are deficient in endogenous β-galactosidase activity due to disruption of bgaA.
Promoter-probe plasmid pPP2 containing the htrA and spr0931 promoters and plasmid pPP2 have been described previously (35, 65). For cloning and propagation of plasmids, Escherichia coli strain DH5α was used as a host. E. coli strains were grown aerobically at 37°C either in LB medium or on LB agar plates (82). The growth of E. coli was followed by measuring the OD600 using a spectrophotometer.
Microscopy and growth curves.
For physiological and morphological analysis, cells were inoculated in prewarmed C+Y or BHI medium and grown at 37°C without aeration. Cell growth was monitored by spectroscopy at OD600. At an OD600 of 0.7, cultures were diluted 1:20 in the respective prewarmed medium, and growth was followed throughout the growth cycle. For microscopic analysis, 5 μl of exponential growing cells at an OD600 of 0.7 was transferred to poly-l-lysine-coated slides and analyzed by phase-contrast microscopy using an Eclipse E600 (Nikon) microscope equipped with a 100× oil immersion objective (numerical aperture, 1.4). Photographs were taken with a DXM1200C camera (Nikon). Image analysis and determination of the cell size were carried out using Nikon Nis-Elements BR (version 3.2) imaging software. For physiological and morphological analyses, the cells from three separate cultures were analyzed, and at least two photographs of each culture were taken.
Transformation procedure.
Transformation of the S. pneumoniae strains was carried out as described previously (37). When required, the growth media for S. pneumoniae were supplemented with the following antibiotics: 200 μg/ml kanamycin (Kan), 200 μg/ml streptomycin (Str), 3 μg/ml tetracycline (Tet), or 20 μg/ml spectinomycin (Spc). The β-lactam concentrations used to select mosaic pbp2x and pbp1a are specified in the supplemental material.
E. coli DH5α was transformed by using chemically competent cells (82), and transformants were selected in the presence of 100 μg/ml ampicillin or 50 μg/ml spectinomycin.
Antibiotic susceptibility.
The MICs of the β-lactam antibiotics were determined by the agar dilution method on D-agar plates supplemented with 3% sheep blood under a natural atmosphere. Cells were grown in C+Y medium to an OD600 of 0.3, and after 1,000-fold dilution, 30-μl aliquots were spotted onto D-agar plates containing the appropriate antibiotic. In order to detect minor differences in β-lactam susceptibilities, a narrow range of antibiotic concentrations was used. In detail, for strains R6, R6C, R6M, R6MC, and Hu15, concentration steps of 0.01 μg/ml for oxacillin and 0.012 μg/ml for cefotaxime were used, and for the remaining strains, concentration steps of 0.1 μg/ml for oxacillin and cefotaxime were used. The MICs for S. pneumoniae Hu17 were determined with Etest strips (Oxoid GmbH). The MIC values were obtained after incubation at 37°C for 48 h. Mean values from at least three experiments were used.
Determination of β-galactosidase activity.
Determination of β-galactosidase activity in strains carrying CiaR-controlled promoters in front of a promoterless E. coli lacZ gene was performed as described previously (65). The cultures were grown in C+Y or BHI medium, and β-galactosidase activity was measured at three time points: when the OD600 was 0.4 and 0.8 and at the beginning of the stationary phase. The volume of the culture harvested for one measurement was adjusted to contain the equivalent of 2 ml cells at an OD600 of 0.8. Specific β-galactosidase activities are expressed in nanomoles of nitrophenol released per minute and milligram of protein. Protein concentrations were determined by the method of Bradford (83). Student's t test was applied to determine the significance of the results.
Detection of penicillin binding proteins.
Preparation of samples, PBP labeling with BocillinFL, and separation of proteins by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) were carried out as described previously (84). BocillinFL-PBP complexes were visualized by fluorography detection with a FluorImager 595 fluorescence scanner (Molecular Dynamics) at 488 nm.
Preparation of pneumococcal cell wall.
Pneumococcal cell wall preparation and analysis of muropeptides were carried out as described previously (24). Briefly, pneumococci were cultured in 2 liters of C+Y medium to the mid-exponential growth phase (OD600, 0.6 to 0.7) and then harvested and resuspended in 40 ml of ice-cold 50 mM Tris-HCl, pH 7.0. The cell suspension was added dropwise into a flask with 150 ml of boiling 5% SDS, and the sample was boiled for an additional 30 min. The samples were centrifuged at 130,000 × g at 25°C, and the pellet was washed with deionized water until it was free of SDS. The lysed cells were disrupted with glass beads, and the sample was treated with DNase I (10 μg/ml) and RNase I (50 μg/ml) for 2 h at 37°C with stirring in 100 mM Tris-HCl, pH 7.5, containing 20 mM MgSO4. Trypsin (100 μg/ml) and CaCl2 (10 mM) were added, and the sample was incubated overnight at 37°C with stirring. SDS was added to yield a final concentration of 1%, and the samples were boiled for 15 min. The resulting purified cell wall was recovered by ultracentrifugation, washed so that it was free of SDS (see above), and lyophilized.
Preparation of muropeptides and analysis of peptidoglycan composition.
Experiments for preparation of muropeptides and analysis of the peptidoglycan composition were performed as described previously (24). In brief, secondary cell wall polymers were removed by incubation with 48% hydrofluoric acid for 48 h at 4°C. The resulting PG was recovered by centrifugation, washed, and digested with the muramidase Cellosyl for 48 h at 37°C with stirring. The samples were boiled for 10 min at 100°C, and the muropeptides were reduced with sodium borohydride. Reduced muropeptides were separated by HPLC on a 250- by 4.6-mm 3-μm-particle-size Prontosil 120-3-6 C18 AQ reversed-phase column (Bischoff, Leonberg, Germany). The eluted muropeptides were detected by their absorbance at 205 nm and assigned by their retention times.
DNA manipulations and construction of mutants.
All DNA techniques and the construction of strains are described in the supplemental material.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the contribution of Achim Herrmann, Tobias Helfert, and especially Julia Pipo for the construction of strains. We thank Ulrike Klein for technical assistance.
This work was supported by grants HA1011/11-2 and HA1011/11-3 from the Deutsche Forschungsgemeinschaft, a fellowship from the Stiftung Alfried-Krupp Kolleg Greifswald to R.H., and a Wellcome Trust award (101824/Z/13/Z) to W.V.
We have no conflict of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00414-17.
REFERENCES
- 1.Högberg L, Geli P, Ringberg H, Melander E, Lipsitch M, Ekdahl K. 2007. Age- and serogroup-related differences in observed durations of nasopharyngeal carriage of penicillin-resistant pneumococci. J Clin Microbiol 45:948–952. doi: 10.1128/JCM.01913-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Short KR, Diavatopoulos DA. 2015. Nasopharyngeal colonization with Streptococcus pneumoniae, p 279–291. In Brown J, Hammerschmidt S, Orihuela CJ (ed), Streptococcus pneumoniae molecular mechanisms of host-pathogen interactions. Academic Press, London, United Kingdom. [Google Scholar]
- 3.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T, Hib and Pneumococcal Global Burden of Disease Study Team. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 4.Henriques-Normark B, Tuomanen E. 2013. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3:a010215. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGee L, McDougal L, Zhou J, Spratt BG, Tenover FC, George R, Hakenbeck R, Hryniewicz W, Lefévre JC, Tomasz A, Klugman KP. 2001. Nomenclature of major antimicrobial-resistant clones of Streptococcus pneumoniae defined by the pneumococcal molecular epidemiology network. J Clin Microbiol 39:2565–2571. doi: 10.1128/JCM.39.7.2565-2571.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyaw MH, Lynfield R, Schaffner W, Craig AS, Hadler J, Reingold A, Thomas AR, Harrison LH, Bennett NM, Farley MM, Facklam RR, Jorgensen JH, Besser J, Zell ER, Schuchat A, Whitney CG, Active Bacterial Core Surveillance of the Emerging Infections Program Network. 2006. Effect of introduction of the pneumococcal conjugate vaccine on drug-resistant Streptococcus pneumoniae. N Engl J Med 354:1455–1463. doi: 10.1056/NEJMoa051642. [DOI] [PubMed] [Google Scholar]
- 7.Hampton LM, Farley MM, Schaffner W, Thomas A, Reingold A, Harrison LH, Lynfield R, Bennett NM, Petit S, Gershman K, Baumbach J, Beall B, Jorgensen J, Glennen A, Zell ER, Moore M. 2012. Prevention of antibiotic-nonsusceptible Streptococcus pneumoniae with conjugate vaccines. J Infect Dis 205:401–411. doi: 10.1093/infdis/jir755. [DOI] [PubMed] [Google Scholar]
- 8.Torres A, Bonanni P, Hryniewicz W, Moutschen M, Reinert RR, Welte T. 2015. Pneumococcal vaccination: what have we learnt so far and what can we expect in the future? Eur J Clin Microbiol Infect Dis 34:19–31. doi: 10.1007/s10096-014-2208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hakenbeck R, Bruckner R, Denapaite D, Maurer P. 2012. Molecular mechanisms of beta-lactam resistance in Streptococcus pneumoniae. Future Microbiol 7:395–410. doi: 10.2217/fmb.12.2. [DOI] [PubMed] [Google Scholar]
- 10.Dowson CG, Coffey TJ, Kell CM, Whiley RA. 1993. Evolution of penicillin resistance in Streptococcus pneumoniae; the role of Streptococcus mitis in the formation of a low affinity PBP2B in S. pneumoniae. Mol Microbiol 9:635–643. [DOI] [PubMed] [Google Scholar]
- 11.Sibold C, Henrichsen J, Konig A, Martin C, Chalkley L, Hakenbeck R. 1994. Mosaic pbpX genes of major clones of penicillin-resistant Streptococcus pneumoniae have evolved from pbpX genes of a penicillin-sensitive Streptococcus oralis. Mol Microbiol 12:1013–1023. doi: 10.1111/j.1365-2958.1994.tb01089.x. [DOI] [PubMed] [Google Scholar]
- 12.Sauerbier J, Maurer P, Rieger M, Hakenbeck R. 2012. Streptococcus pneumoniae R6 interspecies transformation: genetic analysis of penicillin resistance determinants and genome-wide recombination events. Mol Microbiol 86:692–706. doi: 10.1111/mmi.12009. [DOI] [PubMed] [Google Scholar]
- 13.Todorova K, Maurer P, Rieger M, Becker T, Bui NK, Gray J, Vollmer W, Hakenbeck R. 2015. Transfer of penicillin resistance from Streptococcus oralis to Streptococcus pneumoniae identifies murE as resistance determinant. Mol Microbiol 97:866–880. doi: 10.1111/mmi.13070. [DOI] [PubMed] [Google Scholar]
- 14.Jensen A, Valdórsson O, Frimodt-Møller N, Hollingshead S, Kilian M. 2015. Commensal streptococci serve as a reservoir for β-lactam resistance genes in Streptococcus pneumoniae. Antimicrob Agents Chemother 59:3529–3540. doi: 10.1128/AAC.00429-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Linden M, Otten J, Bergmann C, Latorre C, Liñares J, Hakenbeck R. 2017. PBP2x in Streptococcus pseudopneumoniae—an insight into the diversity of PBP2x alleles 2 and mutations in viridans streptococci. Antimicrob Agents Chemother 61:e02646-16. doi: 10.1128/AAC.02646-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denapaite D, Chi F, Maurer P, Nolte O, Hakenbeck R. 2007. Mechanism of penicillin resistance in Streptococcus pneumoniae: targets, gene transfer, and mutations, p 290–303. In Hakenbeck R, Chhatwal GS (ed), Molecular biology of streptococci. Horizon Bioscience, Wymondham, Norfolk, United Kingdom. [Google Scholar]
- 17.Grebe T, Hakenbeck R. 1996. Penicillin-binding proteins 2b and 2x of Streptococcus pneumoniae are primary resistance determinants for different classes of β-lactam antibiotics. Antimicrob Agents Chemother 40:829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laible G, Spratt BG, Hakenbeck R. 1991. Interspecies recombinational events during the evolution of altered PBP2x genes in penicillin-resistant clinical isolates of Streptococcus pneumoniae. Mol Microbiol 5:1993–2002. doi: 10.1111/j.1365-2958.1991.tb00821.x. [DOI] [PubMed] [Google Scholar]
- 19.Sifaoui F, Kitzis M-D, Gutmann L. 1996. In vitro selection of one-step mutants of Streptococcus pneumoniae resistant to different oral β-lactam antibiotics is associated with alterations of PBP2x. Antimicrob Agents Chemother 40:152–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zerfass I, Hakenbeck R, Denapaite D. 2009. An important site in PBP2x of penicillin-resistant clinical isolates of Streptococcus pneumoniae: mutational analysis of Thr338. Antimicrob Agents Chemother 53:1107–1115. doi: 10.1128/AAC.01107-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hakenbeck R, Tornette S, Adkinson NF. 1987. Interaction of non-lytic β-lactams with penicillin-binding proteins in Streptococcus pneumoniae. J Gen Microbiol 133:755–760. [DOI] [PubMed] [Google Scholar]
- 22.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 23.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 24.Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, Simorre JP, Vollmer W. 2012. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Anal Biochem 421:657–666. doi: 10.1016/j.ab.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 25.Garcia-Bustos JF, Chait BT, Tomasz A. 1987. Structure of the peptide network of pneumococcal peptidoglycan. J Biol Chem 262:15400–15405. [PubMed] [Google Scholar]
- 26.Severin A, Tomasz A. 1996. Naturally occurring peptidoglycan variants of Streptococcus pneumoniae. J Bacteriol 178:168–174. doi: 10.1128/jb.178.1.168-174.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garcia-Bustos J, Tomasz A. 1990. A biological price of antibiotic resistance: major changes in the peptidoglycan structure of penicillin-resistant pneumococci. Proc Natl Acad Sci U S A 87:5415–5419. doi: 10.1073/pnas.87.14.5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Severin A, Figueiredo AMS, Tomasz A. 1996. Separation of abnormal cell wall composition from penicillin resistance through genetic transformation of Streptococcus pneumoniae. J Bacteriol 178:1788–1792. doi: 10.1128/jb.178.7.1788-1792.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lloyd AJ, Gilbey AM, Blewett AM, De Pascale G, El Zoeiby A, Levesque RC, Catherwood AC, Tomasz A, Bugg TD, Roper DI, Dowson CG. 2008. Characterization of tRNA dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J Biol Chem 283:6402–6417. doi: 10.1074/jbc.M708105200. [DOI] [PubMed] [Google Scholar]
- 30.Filipe SR, Tomasz A. 2000. Inhibition of the expression of penicillin resistance in Streptococcus pneumoniae by inactivation of cell wall muropeptide branching genes. Proc Natl Acad Sci U S A 97:4891–4896. doi: 10.1073/pnas.080067697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weber B, Ehlert K, Diehl A, Reichmann P, Labischinski H, Hakenbeck R. 2000. The fib locus in Streptococcus pneumoniae is required for peptidoglycan crosslinking and PBP-mediated beta-lactam resistance. FEMS Microbiol Lett 188:81–85. [DOI] [PubMed] [Google Scholar]
- 32.Smith AM, Klugman KP. 2001. Alterations in MurM, a cell wall muropeptide branching enzyme, increase high-level penicillin and cephalosporin resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 45:2393–2396. doi: 10.1128/AAC.45.8.2393-2396.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guenzi E, Gasc AM, Sicard MA, Hakenbeck R. 1994. A two-component signal-transducing system is involved in competence and penicillin susceptibility in laboratory mutants of Streptococcus pneumoniae. Mol Microbiol 12:505–515. doi: 10.1111/j.1365-2958.1994.tb01038.x. [DOI] [PubMed] [Google Scholar]
- 34.Zähner D, Kaminski K, van der Linden M, Mascher T, Merai M, Hakenbeck R. 2002. The ciaR/ciaH regulatory network of Streptococcus pneumoniae. J Mol Microbiol Biotechnol 4:211–216. [PubMed] [Google Scholar]
- 35.Halfmann A, Kovacs M, Hakenbeck R, Brückner R. 2007. Identification of the genes directly controlled by the response regulator CiaR in Streptococcus pneumoniae: five out of 15 promoters drive expression of small non-coding RNAs. Mol Microbiol 66:110–126. doi: 10.1111/j.1365-2958.2007.05900.x. [DOI] [PubMed] [Google Scholar]
- 36.Müller M, Marx P, Hakenbeck R, Brückner R. 2011. Effect of new alleles of the histidine kinase gene ciaH on the activity of the response regulator CiaR in Streptococcus pneumoniae R6. Microbiology 157:3104–3112. doi: 10.1099/mic.0.053157-0. [DOI] [PubMed] [Google Scholar]
- 37.Mascher T, Heintz M, Zähner D, Merai M, Hakenbeck R. 2006. The CiaRH system of Streptococcus pneumoniae prevents lysis during stress induced by treatment with cell wall inhibitors and by mutations in pbp2x involved in β-lactam resistance. J Bacteriol 188:1959–1968. doi: 10.1128/JB.188.5.1959-1968.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moscoso M, Domenech M, García E. 2010. Vancomycin tolerance in clinical and laboratory Streptococcus pneumoniae isolates depends on reduced enzyme activity of the major LytA autolysin or cooperation between CiaH histidine kinase and capsular polysaccharide. Mol Microbiol 77:1052–1064. doi: 10.1111/j.1365-2958.2010.07271.x. [DOI] [PubMed] [Google Scholar]
- 39.Denapaite D, Rieger M, Köndgen S, Brückner R, Ochigava I, Kappeler P, Mätz-Rensing K, Leendertz F, Hakenbeck R. 2016. Highly variable Streptococcus oralis strains are common among viridans streptococci isolated from primates. mSphere 1(2):e00041-15. doi: 10.1128/mSphere.00041-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marx P, Nuhn M, Kovács M, Hakenbeck R, Brückner R. 2010. Identification of genes for small non-coding RNAs that belong to the regulon of the two-component regulatory system CiaRH in Streptococcus. BMC Genomics 11:661. doi: 10.1186/1471-2164-11-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Denapaite D, Brückner R, Hakenbeck R, Vollmer W. 2012. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb Drug Resist 18:344–358. doi: 10.1089/mdr.2012.0026. [DOI] [PubMed] [Google Scholar]
- 42.Schnorpfeil A, Kranz M, Kovács M, Kirsch C, Gartmann J, Brunner I, Bittmann S, Brückner R. 2013. Target evaluation of the non-coding csRNAs reveals a link of the two-component regulatory system CiaRH to competence control in Streptococcus pneumoniae R6. Mol Microbiol 89:334–349. doi: 10.1111/mmi.12277. [DOI] [PubMed] [Google Scholar]
- 43.Sebert ME, Patel KP, Plotnick M, Weiser JN. 2005. Pneumococcal HtrA protease mediates inhibition of competence by the CiaRH two-component signaling system. J Bacteriol 187:3969–3979. doi: 10.1128/JB.187.12.3969-3979.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cassone M, Gagne AL, Spruce LA, Seeholzer SH, Sebert ME. 2012. The HtrA protease from Streptococcus pneumoniae digests both denatured proteins and the competence-stimulating peptide. J Biol Chem 287:38449–38459. doi: 10.1074/jbc.M112.391482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laux A, Sexauer A, Sivaselvarajah D, Kaysen A, Brückner R. 2015. Control of competence by related non-coding csRNAs in Streptococcus pneumoniae R6. Front Genet 6:246. doi: 10.3389/fgene.2015.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dawid S, Sebert ME, Weiser JN. 2009. Bacteriocin activity of Streptococcus pneumoniae is controlled by the serine protease HtrA via posttranscriptional regulation. J Bacteriol 191:1509–1518. doi: 10.1128/JB.01213-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kochan TJ, Dawid S. 2013. The HtrA protease of Streptococcus pneumoniae controls density-dependent stimulation of the bacteriocin blp locus via disruption of pheromone secretion. J Bacteriol 195:1561–1572. doi: 10.1128/JB.01964-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dagkessamanskaia A, Moscoso M, Hénard V, Guiral S, Overweg K, Reuter M, Martin B, Wells J, Claverys JP. 2004. Interconnection of competence, stress and CiaR regulons in Streptococcus pneumoniae: competence triggers stationary phase autolysis of ciaR mutant cells. Mol Microbiol 51:1071–1086. doi: 10.1111/j.1365-2958.2003.03892.x. [DOI] [PubMed] [Google Scholar]
- 49.Sebert ME, Palmer LM, Rosenberg M, Weiser JN. 2002. Microarray-based identification of htrA, a Streptococcus pneumoniae gene that is regulated by the CiaRH two-component system and contributes to nasopharyngeal colonization. Infect Immun 70:4059–4067. doi: 10.1128/IAI.70.8.4059-4067.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Halfmann A, Schnorpfeil A, Müller M, Marx P, Günzler U, Hakenbeck R, Brückner R. 2011. Activity of the two-component regulatory system CiaRH in Streptococcus pneumoniae R6. J Mol Microbiol Biotechnol 20:96–104. doi: 10.1159/000324893. [DOI] [PubMed] [Google Scholar]
- 51.Throup JP, Koretke KK, Bryant AP, Ingraham KA, Chalker AF, Ge Y, Marra A, Wallis NG, Brown JR, Holmes DJ, Rosenberg M, Burnham MK. 2000. A genomic analysis of two-component signal transduction in Streptococcus pneumoniae. Mol Microbiol 35:566–576. [DOI] [PubMed] [Google Scholar]
- 52.Marra A, Asundi J, Bartilson M, Lawson S, Fang F, Christine J, Wiesner C, Brigham D, Schneider WP, Hromockyj AE. 2002. Differential fluorescence induction analysis of Streptococcus pneumoniae identifies genes involved in pathogenesis. Infect Immun 70:1422–1433. doi: 10.1128/IAI.70.3.1422-1433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ibrahim YM, Kerr AR, McCluskey J, Mitchell TJ. 2004. Control of virulence by the two-component system CiaR/H is mediated via HtrA, a major virulence factor of Streptococcus pneumoniae. J Bacteriol 186:5258–5266. doi: 10.1128/JB.186.16.5258-5266.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marton A, Gulyas M, Munoz R, Tomasz A. 1991. Extremely high incidence of antibiotic resistance in clinical isolates of Streptococcus pneumoniae in Hungary. J Infect Dis 163:542–548. doi: 10.1093/infdis/163.3.542. [DOI] [PubMed] [Google Scholar]
- 55.Marton A, Meszner Z. 1999. Epidemiological studies on drug resistance in Streptococcus pneumoniae in Hungary: an update for the 1990s. Microb Drug Resist 5:201–205. [DOI] [PubMed] [Google Scholar]
- 56.Figueiredo AM, Austrian R, Urbaskova P, Teixeira LA, Tomasz A. 1995. Novel penicillin-resistant clones of Streptococcus pneumoniae in the Czech Republic and Slovakia. Microb Drug Resist 1:71–78. doi: 10.1089/mdr.1995.1.71. [DOI] [PubMed] [Google Scholar]
- 57.Reichmann P, Varon E, Günther E, Reinert RR, Lüttiken R, Marton A, Geslin P, Wagner J, Hakenbeck R. 1995. Penicillin-resistant Streptococcus pneumoniae in Germany: genetic relationship to clones from other European countries. J Med Microbiol 43:377–385. doi: 10.1099/00222615-43-5-377. [DOI] [PubMed] [Google Scholar]
- 58.Reichmann P, König A, Marton A, Hakenbeck R. 1996. Penicillin-binding proteins as resistance determinants in clinical isolates of Streptococcus pneumoniae. Microb Drug Resist 2:177–181. doi: 10.1089/mdr.1996.2.177. [DOI] [PubMed] [Google Scholar]
- 59.Hakenbeck R, Balmelle N, Weber B, Gardès C, Keck W, de Saizieu A. 2001. Mosaic genes and mosaic chromosomes: intra- and interspecies genomic variation of Streptococcus pneumoniae. Infect Immun 69:2477–2486. doi: 10.1128/IAI.69.4.2477-2486.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rieger M, Denapaite D, Brückner R, Maurer P, Hakenbeck R. 2017. Draft genome sequences of two Streptococcus pneumoniae serotype 19A sequence type 226 clinical isolates from Hungary, Hu17 with high-level beta-lactam resistance and Hu15 of a penicillin-sensitive phenotype. Genome Announc 5:e00401-17. doi: 10.1128/genomeA.00401-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, Shendure J, Salipante SJ. 2015. A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet 11:e1005413. doi: 10.1371/journal.pgen.1005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smith AM, Klugman KP. 2003. Site-specific mutagenesis analysis of PBP 1A from a penicillin-cephalosporin-resistant pneumococcal isolate. Antimicrob Agents Chemother 48:387–389. doi: 10.1128/AAC.47.1.387-389.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sung CK, Li H, Claverys JP, Morrison DA. 2001. An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl Environ Microbiol 67:5190–5196. doi: 10.1128/AEM.67.11.5190-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mascher T, Zähner D, Merai M, Balmelle N, de Saizieu AB, Hakenbeck R. 2003. The Streptococcus pneumoniae cia regulon: CiaR target sites and transcription profile analysis. J Bacteriol 185:60–70. doi: 10.1128/JB.185.1.60-70.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halfmann A, Hakenbeck R, Brückner R. 2007. A new integrative reporter plasmid for Streptococcus pneumoniae. FEMS Microbiol Lett 268:217–224. doi: 10.1111/j.1574-6968.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- 66.Lacks S, Hotchkiss RD. 1960. A study of the genetic material determining an enzyme activity in pneumococcus. Biochim Biophys Acta 39:508–518. doi: 10.1016/0006-3002(60)90205-5. [DOI] [PubMed] [Google Scholar]
- 67.Hakenbeck R, König A, Kern I, van der Linden M, Keck W, Billot-Klein D, Legrand R, Schoot B, Gutmann L. 1998. Acquisition of five high-Mr penicillin-binding protein variants during transfer of high-level β-lactam resistance from Streptococcus mitis to Streptococcus pneumoniae. J Bacteriol 180:1831–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Land AD, Winkler ME. 2011. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol 193:4166–4179. doi: 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barendt SM, Land AD, Sham LT, Ng WL, Tsui HC, Arnold RJ, Winkler ME. 2009. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J Bacteriol 191:3024–3040. doi: 10.1128/JB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Severin A, Schuster C, Hakenbeck R, Tomasz A. 1992. Altered murein composition in a dd-carboxypeptidase mutant of Streptococcus pneumoniae. J Bacteriol 174:5152–5155. doi: 10.1128/jb.174.15.5152-5155.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoyland CN, Aldridge C, Cleverley RM, Duchêne MC, Minasov G, Onopriyenko O, Sidiq K, Stogios PJ, Anderson WF, Daniel RA, Savchenko A, Vollmer W, Lewis RJ. 2014. Structure of the LdcB ld-carboxypeptidase reveals the molecular basis of peptidoglycan recognition. Structure 22:949–960. doi: 10.1016/j.str.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barendt SM, Sham LT, Winkler ME. 2011. Characterization of mutants deficient in the l,d-carboxypeptidase (DacB) and WalRK (VicRK) regulon, involved in peptidoglycan maturation of Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol 193:2290–2300. doi: 10.1128/JB.01555-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vollmer W, Tomasz A. 2000. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J Biol Chem 275:20496–20501. doi: 10.1074/jbc.M910189199. [DOI] [PubMed] [Google Scholar]
- 74.Filipe SR, Severina E, Tomasz A. 2000. Distribution of the mosaic structured murM genes among natural populations of Streptococcus pneumoniae. J Bacteriol 182:6798–6805. doi: 10.1128/JB.182.23.6798-6805.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Croucher NJ, Chewapreecha C, Hanage WP, Harris SR, McGee L, van der Linden M, Song JH, Ko KS, de Lencastre H, Turner C, Yang F, Sá-Leão R, Beall B, Klugman KP, Parkhill J, Turner P, Bentley SD. 2014. Evidence for soft selective sweeps in the evolution of pneumococcal multidrug resistance and vaccine escape. Genome Biol Evol 6:1589–1602. doi: 10.1093/gbe/evu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kilian M, Riley DR, Jensen A, Brüggemann H, Tettelin H. 2014. Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. mBio 5(4):e01490-14. doi: 10.1128/mBio.01490-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Filipe SR, Severina E, Tomasz A. 2001. Functional analysis of Streptococcus pneumoniae MurM reveals the region responsible for its specificity in the synthesis of branched cell wall peptides. J Biol Chem 276:39618–39628. doi: 10.1074/jbc.M106425200. [DOI] [PubMed] [Google Scholar]
- 78.Chewapreecha C, Marttinen P, Croucher NJ, Salter SJ, Harris SR, Mather AE, Hanage WP, Goldblatt D, Nosten FH, Turner C, Turner P, Bentley SD, Parkhill J. 2014. Comprehensive identification of single nucleotide polymorphisms associated with beta-lactam resistance within pneumococcal mosaic genes. PLoS Genet 10:e1004547. doi: 10.1371/journal.pgen.1004547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Marx P, Meiers M, Brückner R. 2015. Activity of the response regulator CiaR in mutants of Streptococcus pneumoniae R6 altered in acetyl phosphate production. Front Microbiol 5:772. doi: 10.3389/fmicb.2014.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coffey TJ, Dowson CG, Daniels M, Spratt BG. 1995. Genetics and molecular biology of beta-lactam-resistant pneumococci. Microb Drug Resist 1:29–34. doi: 10.1089/mdr.1995.1.29. [DOI] [PubMed] [Google Scholar]
- 81.Alloing G, Granadel C, Morrison DA, Claverys JP. 1996. Competence pheromone, oligopeptide permease, and induction of competence in Streptococcus pneumoniae. Mol Microbiol 21:471–478. doi: 10.1111/j.1365-2958.1996.tb02556.x. [DOI] [PubMed] [Google Scholar]
- 82.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 83.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 84.Peters K, Schweizer I, Beilharz K, Stahlmann C, Veening JW, Hakenbeck R, Denapaite D. 2014. Streptococcus pneumoniae PBP2x mid-cell localization requires the C-terminal PASTA domains and is essential for cell shape maintenance. Mol Microbiol 92:733–755. doi: 10.1111/mmi.12588. [DOI] [PubMed] [Google Scholar]
- 85.Ottolenghi E, Hotchkiss RD. 1962. Release of genetic transforming agent from pneumococcal cultures during growth and disintegration. J Exp Med 116:491–519. doi: 10.1084/jem.116.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.