

ABSTRACT
Tuberculosis (TB) continues to be one of the most common bacterial infectious diseases and is the leading cause of death in many parts of the world. A major limitation of TB therapy is slow killing of the infecting organism, increasing the risk for the development of a tolerance phenotype and drug resistance. Studies indicate that Mycobacterium tuberculosis takes several days to be killed upon treatment with lethal concentrations of antibiotics both in vitro and in vivo. To investigate how metabolic remodeling can enable transient bacterial survival during exposure to bactericidal concentrations of compounds, M. tuberculosis strain H37Rv was exposed to twice the MIC of isoniazid, rifampin, moxifloxacin, mefloquine, or bedaquiline for 24 h, 48 h, 4 days, and 6 days, and the bacterial proteomic response was analyzed using quantitative shotgun mass spectrometry. Numerous sets of de novo bacterial proteins were identified over the 6-day treatment. Network analysis and comparisons between the drug treatment groups revealed several shared sets of predominant proteins and enzymes simultaneously belonging to a number of diverse pathways. Overexpression of some of these proteins in the nonpathogenic Mycobacterium smegmatis extended bacterial survival upon exposure to bactericidal concentrations of antimicrobials, and inactivation of some proteins in M. tuberculosis prevented the pathogen from escaping the fast killing in vitro and in macrophages, as well. Our biology-driven approach identified promising bacterial metabolic pathways and enzymes that might be targeted by novel drugs to reduce the length of tuberculosis therapy.
KEYWORDS: M. tuberculosis, synergistic targets, INH, RMP, MXF, TMC207, bedaquiline, RIF, drug targets, proteomics
INTRODUCTION
Tuberculosis (TB) remains one of the most significant diseases, affecting a large portion of the world population. The treatment regimen, which is nearly a half century old, is inadequate to control the epidemic due to socioeconomic factors and the need for prolonged use of combination chemotherapy with a high level of toxicity (1, 2). The breakdown in patient compliance directly contributes to the development and spread of multidrug- and extensively drug-resistant (MDR and XDR) strains of Mycobacterium tuberculosis. In addition, the extended treatment enhances the selection of tolerant (persistent) subpopulations of the drug-susceptible bacteria that manage to survive despite continuous exposure to lethal concentrations of anti-tuberculosis drugs. M. tuberculosis-HIV coinfection further diminishes the practical effectiveness of the current treatment, offering clinicians very limited alternatives. Recently, experimental compounds, such as moxifloxacin (MXF), gatifloxacin, bedaquiline (TMC207), and metronidazole, have emerged as potential new classes of anti-M. tuberculosis drugs, offering an improvement over existing treatments (3–5). Some among the above-cited compounds have novel mechanisms of action and can kill antibiotic-resistant strains (6–9), although clinical trials with moxifloxacin were disappointing (10). A growing pipeline of anti-TB compounds is also under development or undergoing clinical trials, and these experimental compounds are anticipated to improve treatment outcomes (11).
One of the main challenges of anti-TB therapy is the ability of bactericidal concentrations of compounds to rapidly kill M. tuberculosis. The extended time of treatment increases the chances that the pathogen will develop drug tolerance and resistance. Despite discovering new effective compounds against novel targets, the eradication of M. tuberculosis is still challenging. The reason for the delayed killing by currently available compounds remains unclear. The fact is that M. tuberculosis is a metabolically flexible bacterium (12). In an attempt to survive, the pathogen can shift into different metabolic states upon drug treatment. To successfully kill an infecting organism that presumably is in a very different metabolic state becomes a challenge, as the majority of the available antimicrobials target actively growing bacteria in culture. Historically, the introduction of combination therapy to treat tuberculosis was an attempt to prevent the emergence of antibiotic resistance during the course of treatment. In fact, although some of the current anti-TB therapies appear to be effective when administered together; in some instances, the associations have been questioned because of the potential antagonism between drugs (13, 14).
To obtain insights into how the pathogen prolongs survival time following exposure to bactericidal concentrations of antimicrobials, with the goal of developing a rational combination therapy against M. tuberculosis, we investigated the bacterial response to each class of anti-TB drugs. By examining the global proteomic responses of M. tuberculosis to isoniazid (INH), rifampin (RMP), MXF, mefloquine (MQ), and TMC207, we discovered “escape” pathways and enzymes associated with changes in the bacterial metabolic state. The data demonstrate that during exposure to bactericidal compounds, M. tuberculosis upregulates the synthesis of many enzymes, enhancing bacterial survival. Furthermore, gene inactivation of some of these enzymes results in improved drug efficacy against M. tuberculosis. The identified proteins may provide powerful targets for development of synergistic drugs aimed at accelerating bacterial killing and may decrease the chances for development of resistance mechanisms.
RESULTS
Delayed killing of M. tuberculosis by antimicrobials.
Using the broth microdilution method, we determined the MICs at which 90% of the M. tuberculosis H37Rv and M. tuberculosis CDC1551 strains were inhibited to be as follows: isoniazid, 0.2 μg/ml; rifampin, 1 μg/ml; moxifloxacin, 0.5 μg/ml; mefloquine, 16 μg/ml; and TMC270, 60 ng/ml. The killing effects of these drugs were tested for M. tuberculosis H37Rv at twice the MIC over 10 days. As shown in Fig. 1, while significant reduction in bacterial viability was observed at day 8 following INH and RMP treatment, exposure to moxifloxacin, mefloquine, and TMC270 achieved comparable killing effects at day 6 of treatment.
FIG 1.
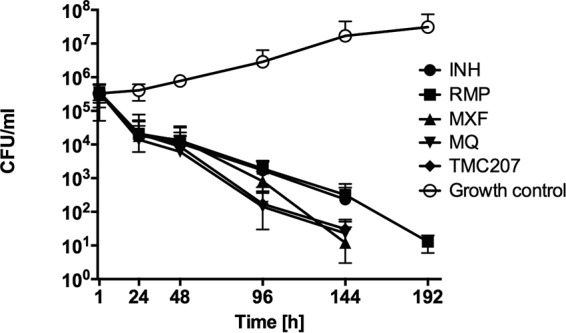
M. tuberculosis delayed killing in vitro. The time-kill curves for M. tuberculosis strain H37Rv against INH, RMP, MXF, MQ, and TMC207 demonstrate bacterial CFU levels over 8 days after incubation with the drugs at 2× MIC. Antimicrobials were added to the broth culture at time zero. Growth control without drug treatment is also shown.
Global proteome response of M. tuberculosis against antimicrobials.
The proteomic response of M. tuberculosis exposed to INH, RMP, MXF, MQ, and TMC207 was examined, and quantitative analysis of the protein expression profile between 24 h and 6 days of treatment was performed. Overall, we identified a total of >1,800 proteins and determined the differential expression after drug exposure over time. A list of all the identified proteins, along with their normalized spectral counts and annotations and the fold changes over controls at corresponding time points for each drug group, can be found in Tables S1 and S2 in the supplemental material. The histograms in Fig. 2 demonstrate the distribution of the average fold changes for proteins across all the drugs at each time point.
FIG 2.
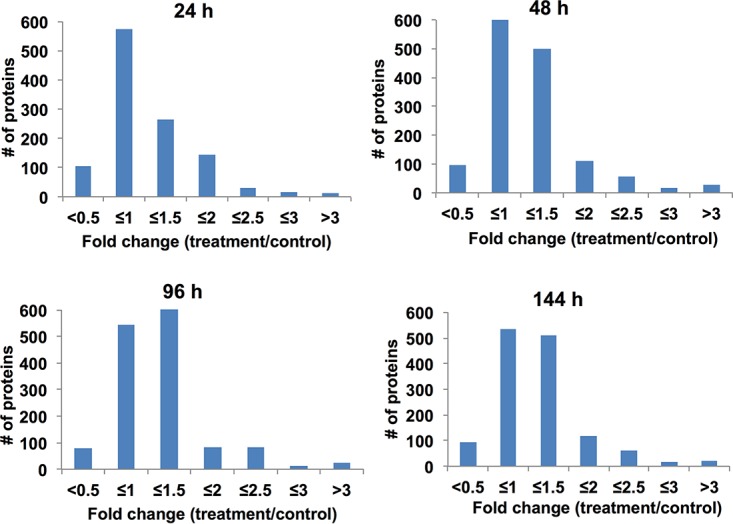
Fold changes of differentially expressed M. tuberculosis proteins. The histograms show the distributions of fold changes of differentially expressed proteins across all the drugs over 6 days of treatment. The fold change was calculated by dividing the expression level in protein overexpression in drug-exposed samples by that in the control sample without any treatment.
For a global overview of proteomic changes in bacterial responses to antibiotics without exclusively focusing on the induced proteins, we compared protein levels in untreated bacteria to each drug treatment using an analysis paired by time points. This analysis revealed proteins with concordant changes across all four time points for each individual drug (P < 0.05). As a result, we obtained 80 proteins for INH, 111 for MQ, 97 for MXF, 116 for RMP, and 110 for TMC207 (Fig. 3A). After intersecting these five lists, we identified 375 proteins that were present in at least one of the lists. Next, we defined which of the proteins were concordantly regulated by all the drugs. Out of the 375 proteins, 173 showed a trend toward the same direction of abundance change across the treatments, with 112 proteins below a P value of 0.05 (false discovery rate [FDR], <7.7%). As shown in Fig. 3B, 62 proteins were concordantly induced while 50 proteins were repressed by all the drugs. To characterize the biological processes in which these proteins were involved, we performed ontology enrichment analysis, which identified translation, amino acid, and carboxylic acid metabolic processes among the ones most overrepresented in the list (Table 1). Analysis of KEGG annotations further supported the Gene Ontology (GO) analysis, pointing to some specific amino acid, carbohydrate, and nucleotide metabolism categories as the most overrepresented pathways (Table 2). We mapped on the general metabolic map 24 proteins that had KEGG annotations (see Fig. S1 in the supplemental material). This further supported the notion that several proteins belonged to nucleotide and amino acid metabolism pathways, with a few others belonging to oxidative phosphorylation, fatty acid degradation, and lipopolysaccharide biosynthesis pathways.
FIG 3.

Proteome response of M. tuberculosis to anti-tuberculosis drug treatments. (A) Volcano plots showing proteins that are induced or repressed by each drug across all time points. Proteins with P values of <0.05 are in blue; each dot is one protein. (B) Heat map showing the levels of proteins that were concordantly regulated across four time points by the five drugs. Raw values were log2 transformed and normalized within each protein. The colors show relative levels as indicated by the scale bar.
TABLE 1.
Ontology enrichment analysis
| GO term | Biological process | Protein count | P value |
|---|---|---|---|
| GO:0006412 | Translation | 7 | 0.002192 |
| GO:0046394 | Carboxylic acid-biosynthetic process | 8 | 0.013694 |
| GO:0016053 | Organic acid-biosynthetic process | 8 | 0.013694 |
| GO:0008652 | Cellular amino acid-biosynthetic process | 7 | 0.017438 |
| GO:0009309 | Amine-biosynthetic process | 7 | 0.019712 |
| GO:0019438 | Aromatic compound-biosynthetic process | 5 | 0.025007 |
TABLE 2.
KEGG annotation analysis
| KEGG annotation | Pathway | Protein count | P value |
|---|---|---|---|
| mtu00281 | Geraniol degradation | 5 | 0.020065 |
| mra00280 | Valine, leucine, and isoleucine degradation | 5 | 0.021223 |
| mtu00640 | Propanoate metabolism | 5 | 0.029036 |
| mtu00650 | Butanoate metabolism | 5 | 0.031976 |
| mra00240 | Pyrimidine metabolism | 4 | 0.03817 |
| mra00400 | Phenylalanine, tyrosine, and tryptophan biosynthesis | 3 | 0.040281 |
| mtu00624 | 1- and 2-methylnaphthalene degradation | 4 | 0.040531 |
To identify key upstream regulators of these 112 altered proteins (62 induced and 50 repressed), we searched the TB Database (http://genome.tbdb.org/tbdb_sysbio/Resources.html) for possible interactions of the proteins. Out of the 112 proteins, 103 could be found in the database, and they connected to a total of 59 upstream regulators. A network to visualize these interactions was constructed (Fig. 4). Ranking of regulators based on their connectivity to the target proteins showed that the top six regulators together were connected to almost 90% of the target proteins (90 out of 103). Interestingly, the top connected regulator, Rv0081, was previously identified among the main hubs coordinating bacterial response to hypoxia (15). This suggests that bacteria might be using universal pathways to respond to different types of stress.
FIG 4.

Regulatory network of proteins induced or repressed by drugs. Each circle represents a protein; orange, induced by drugs; blue, repressed; yellow, top connected regulators; gray, other regulator proteins. Information about regulator proteins was obtained from the TB Database (http://genome.tbdb.org/tbdb_sysbio/Resources.html). Two proteins in the middle (Rv0324 and Rv2989) were the only regulator proteins that showed trends toward changes by drugs (P = 0.06 and 0.07).
Proteome analysis for M. tuberculosis proteins that were induced in all drug treatment groups by ≥2-fold.
We further analyzed the M. tuberculosis proteome data for proteins that were induced by ≥2-fold in drug treatment groups compared to the nontreatment control. We hypothesized that proteins induced in response to exposure to compounds might likely contain novel drug targets. The heat map in Fig. 5A shows proteins that were induced ≥2-fold by at least one anti-TB drug compared with the control at each time point. There were approximately 300 proteins that fell into this category for each time point. Although there was some variability, overall, the levels of these proteins in control samples were lower than in drug treatment groups. The enrichment of protein functional categories in drug treatment groups is presented in Fig. 5B. The assignment of proteins to KEGG functional categories showed that several metabolic functions were overrepresented. We subsequently focused on proteins that overlapped in 24-h and 48-h drug treatment time point groups and belonged to metabolic pathways (Fig. 5C and Table 3).
FIG 5.

Analysis of M. tuberculosis proteins induced by ≥2-fold by anti-tuberculosis drugs. (A) Heat map of differentially expressed proteins induced by ≥2-fold by at least one drug and their corresponding controls (Cont.). (B) Enrichment of Gene Ontology categories (biological process) for expressed proteins in all drug treatment samples over time. (C) Venn diagrams showing the numbers of overlapping and unique sets of genes modulated >2-fold under multiple drug treatment conditions and at chosen time points.
TABLE 3.
Proteins overlapping in all drug treatment groups at 24 and 48 h

a The target genes that were evaluated in both M. tuberculosis knockout and M. smegmatis overexpressed systems in vitro and in tissue culture are shaded lght gray. The genes tested only in M. smegmatis are shaded medium gray, and the genes tested only in M. tuberculosis are shaded dark gray.
In vitro time-kill curves of Mycobacterium smegmatis overexpressed and M. tuberculosis gene knockout clones.
The bactericidal activities of anti-TB drugs were assessed in two systems using M. smegmatis protein overexpressed clones and M. tuberculosis gene knockout mutants. We hypothesized that proteins that are synthesized following a short exposure to lethal doses of antibiotics and are included in the common response to all drug treatments are likely to belong to escape pathways that could be associated with the extended survival time of the pathogen. To test the concept, we overexpressed seven M. smegmatis genes that had at least 70% homology with M. tuberculosis genes encoding proteins synthesized following exposure to all the drugs for 24 h and 48 h. The Western blot in Fig. 6 confirmed that all the constructed clones had the proteins of interest produced in M. smegmatis. The killing curves for M. smegmatis clones were established over 10 days of exposure to RMP, MXF, and TMC207. In vitro studies revealed no difference between the growth of the M. smegmatis control containing a skeleton plasmid and that of overexpressed clones in liquid culture medium (Fig. 7A). While RMP, MXF, and TMC207 had significant bactericidal activity against wild-type M. smegmatis at 2, 4, and 6 days, overexpression of LpqY, TrxC, and PepB genes delayed killing time by 2 days. Delayed killing was observed when bacteria were exposed to RMP and TMC for GabT and Clip2 clones. This phenomenon was not seen with MXF. M. smegmatis FprA and SdhA clones had outcomes similar to those of the control bacteria.
FIG 6.
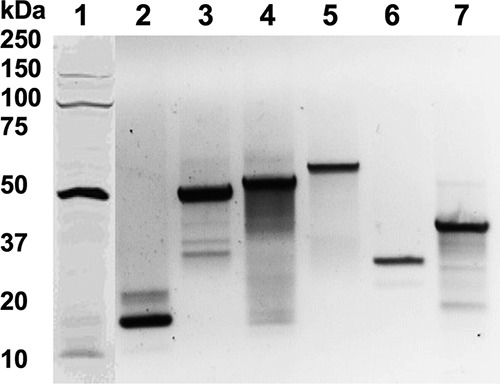
Western blot of target proteins overexpressed in M. smegmatis. M. smegmatis genes highly homologous to M. tuberculosis GabT (46.8 kDa) (lane 1), TrxC (12 kDa) (lane 2), FprA (50.8 kDa) (lane 3), PepB (53.6 kDa) (lane 4), SdhA (64.3 kDa) (lane 5), ClpP2 (23.6 kDa) (lane 6), and LpqY (47.6 kDa) (lane 7) genes were tagged with 6×His and expressed in the pMV261 vector. Positive clones were lysed in HBSS by mechanical disruption using a bead beater. Precleared samples were separated in an SDS-12% PAGE gel (Bio-Rad), transferred to a nitrocellulose membrane (Bio-Rad), and blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) for 1 h. The membrane was probed with 6×His primary antibody (Santa Cruz Biotechnology) at a dilution of 1:300 for 2 h and then with a corresponding IRDye secondary antibody (Li-Cor Biosciences, Inc.) at a dilution of 1:5,000 for 1 h. Proteins were visualized using Odyssey Imager (Li-Cor).
FIG 7.

In vitro time-kill curves for M. smegmatis (MS) overexpressed clones (A) and M. tuberculosis (Mtb) knockout mutants (B). The error bars indicate standard deviations.
To test if the identified genes also influenced M. tuberculosis survival against INH, RMP, MXF, and TMC207, we performed time-kill curves using M. tuberculosis knockout mutants of the LpqY, GabT, FprA, Rv1008, Rv3788, and FabG5 genes. As demonstrated in Fig. 7B, all the drugs had significant bactericidal activity against LpqY and Rv1006 mutants, reducing the killing time by 2 or 4 days depending on the drug and the mutant. The significant reduction in bacterial CFU was observed for GabT and FprA mutants against three of the four tested compounds, and no changes in killing time were detected for the Rv3788 mutant.
Drug activity against mycobacteria in macrophages.
Likewise, we examined the drug activity during macrophage infection with M. smegmatis overexpressed clones and M. tuberculosis knockout mutants. At first, we performed macrophage survival studies and determined that M. smegmatis overexpressed clones had growth and killing kinetics similar to those of the control construct within THP-1 macrophages (Fig. 8A). In macrophage drug treatment studies, while M. smegmatis clones expressing LpqY, FprA, TrxC, and PepB genes survived for an additional 2 days during exposure to RMP, MXF, and TMC207, SdhA and Clip2 clones showed killing effects by drugs similar to those of the M. smegmatis control. The M. tuberculosis survival assay showed an FprA mutant to be attenuated in growth within macrophages, but the rest of the mutants grew in a manner similar that of the wild-type strain (Fig. 8B). The results from INH, RMP, MXF, and TMC207 treatment groups of M. tuberculosis-infected macrophages indicate that, while the FprA mutant had an attenuated phenotype, all the drugs exhibited rapid killing and significantly decreased bacterial CFU numbers by 2 or 4 days (depending on the drug) compared with the wild-type control. Reduced killing time was observed for LpqY and Rv3788 mutants following treatment with INH, RMP, and TMC207, but not MXF. In contrast, GabT and FabG5 mutants had their killing reduced with just one drug, and the CFU numbers for the Rv1006 mutant declined gradually over time, similar to what was seen for the M. tuberculosis wild-type control.
FIG 8.

Killing dynamics of M. smegmatis overexpressed clones (A) and M. tuberculosis knockout mutants (B) in THP-1 cells. The error bars indicate standard deviations.
DISCUSSION
The bacteriological, as well as the socioeconomic, challenges in treating tuberculosis are numerous. A growing pipeline of promising compounds has been developed to offer new and perhaps improved therapies against M. tuberculosis. However, antimicrobials currently being tested in clinical trials do not appear to be more effective than existing therapies. One of the possible explanations is that TB regimens largely consist of randomly discovered compounds that are combined based on clinical experience. A combination therapy has never been developed using a rational approach, except to decrease the chances of emergence of drug resistance. Despite the fact that newly discovered compounds represent a significant advance in the field of killing drug-resistant strains effectively, the reality is that drug resistance will evolve over time and the appearance of resistant strains will soon become a reality.
This study describes a novel approach for development of anti-TB therapies in which the proteomic response of M. tuberculosis to traditional therapy is mapped. It demonstrates how the pathogen protects itself against the actions of antimicrobials, and the mechanisms used in many instances are common among different antimicrobials. The majority of the proteins identified in this study are highly regulated upon therapy and connect several different pathways, allowing the bacterium to shift metabolic strategies as a response to therapy. Although initially identified in in vitro culture media, where one can argue that the availability of nutrients is very different than in the host cell, the results indicate that the observations for several tested genes/proteins are similar to those in macrophages, where the nutrient availability is restricted.
Glutamate plays a central role in a wide range of metabolic processes, in particular, during bacterial stress responses. We identified the 4-aminobutyrate aminotransferase GabT (Rv2589) as a predominant protein modulated more than 2-fold under drug treatment conditions at the 24-h time point. GabT is one of the key enzymes of the glutamate decarboxylase (GAD) system that catalyzes glutamate to the γ-aminobutyrate (GABA). The GABA shunt pathway has been suggested to provide an alternative source of succinate in bacteria and a way to generate energy from glutamate without production of ammonia (16, 17). The in vitro and macrophage assays demonstrated that while delayed killing was observed for an M. smegmatis clone overexpressing the GabT protein during treatment with anti-TB drugs, significant reduction in time was seen in an M. tuberculosis GabT-deficient mutant. The GAD system has been implicated in a variety of stress conditions, such as temperature shock, increasing levels of Ca2+, or hypoxia (18, 19), and increased accumulation of intracellular GABA has been shown to play a significant role in the survival of pathogenic bacteria under acidic conditions (20). In addition, the effect of GABA on bacterial biofilm formation and the lipid composition of the bacterial cell surface has been linked to adhesion potential and cytotoxic activity in Pseudomonas (21).
Our studies have determined that the trehalose ABC transporter sugar-binding lipoprotein LpqY was overexpressed by more than 2-fold at 48 h in all the drug treatment groups. The results indicate that the functional interruption of the LpqY gene accelerates M. tuberculosis killing in macrophages by INH, RMP, and TMC207 by 2 days. LpqY is an essential virulence factor that plays a role in the mycolic acid processing of the mycobacterial cell wall, and it is critical for establishing infection in the host. Trehalose is not found in mammals but can be released by M. tuberculosis from trehalose-containing cell wall glycolipids and reimported inside the cells by the ABC transporter SugABC and the lipoprotein LpqY (22). Trehalose recycling represents one of the strategies of M. tuberculosis for intracellular adaptation in nutrient-restricted environments with poor carbohydrate availability. M. tuberculosis is capable of bypassing glucose phosphorylation and utilizing trehalose as a sole source of carbon and energy (23). Recent work indicates that a functional loss of the LpqY-SugA-SugB-SugC transporter gradually causes carbon starvation in nutrient-limited microenvironments in vivo (24).
Among the proteins identified, thioredoxin (TrxC), NADPH-adrenodoxin oxidoreductase (FprA), and the hypothetical Rv3788 are strong synergistic candidates that have potential to improve the effectiveness of frontline drugs and to reduce the duration of treatment. M. tuberculosis has the ability to survive intracellular redox stress, in part by expressing TrxC under a variety of oxidative conditions (25). Thioredoxin (Trx) genes are highly expressed during M. tuberculosis infection and contribute to natural resistance to oxidative killing that the pathogen encounters in the phagosomal environment. In vivo studies have demonstrated that, by targeting thioredoxin reductase TrxB2, M. tuberculosis can be cleared during acute and chronic mouse infections (26). Interestingly, it was also found that the partial inactivation of TrxB2 increases M. tuberculosis susceptibility to rifampin and synergizes bacterial killing with this frontline antituberculosis drug (26).
The M. tuberculosis FprA enzyme is a homologue of eukaryotic adrenodoxin reductases (AdR proteins) (27). Although the physiological role of FprA is still unclear, data suggest a relevant role of FprA in Fe metabolism, turnover of multiple cytochrome P450-dependent reactions, and oxidative-stress response (28–30). Recently, the FprA protein has been classified as a novel ferredoxin reductase (29). FprA provides electrons to several pivotal pathways for M. tuberculosis viability in the host via interactions with different iron-sulfur proteins, either adrenodoxin or ferredoxins, and represents a potential target for antimycobacterial drugs. In addition, a mycobacterial anaerobic-type ferredoxin reductase enzyme has been demonstrated to drive the unconventional tricarboxylic acid (TCA) cycle, indicating the presence of an alternate pathway that can operate in the absence of oxidation (31).
An important concept for anti-tuberculosis therapy is the early use of a high-potency bactericidal drug, such as INH. INH typically causes 1-log10-unit reduction in colony counts during the initial few days of therapy, killing actively replicating bacteria (32, 33). Six months of therapy, however, is necessary to eradicate the nonreplicating persisters (34). M. tuberculosis exploits a diverse group of specific changes in the metabolic pathway as a weapon to survive for a prolonged period of time, which may also facilitate the acquisition of resistance (35, 36). By using M. tuberculosis knockout strains and targeting alternative phenotypes essential for the pathogen's survival during exposure to primary antimicrobials, we can demonstrate in vitro and in macrophages that our strategy to shorten the duration of the therapy can be successful.
The roles of particular gene-encoded proteins in the regulatory process are still unknown. However, the fact that the overexpression of target proteins in M. smegmatis resulted in increases in the period necessary for an antibiotic to kill bacteria is very suggestive that those proteins play an important role in the process of escape. The connection of more significant proteins associated with the increase in resistance was addressed by constructing a network and ranking the regulators based on their connectivity to target proteins. Rv0081 and Rv0023 were identified, and Rv0081 has been recently demonstrated to play a role in the regulation of the M. tuberculosis response to hypoxia (15).
The identification of the “rational targets” may offer an opportunity to develop compounds that interfere with the bacterial response when exposed to therapeutic antimicrobials. It may have a significant impact on reducing the time needed for therapy and, consequently, the emergence of resistance and compliance. The development of inhibitors for novel targets is a subject for future study.
MATERIALS AND METHODS
Strains and culture conditions.
M. tuberculosis strain H37Rv was purchased from the American Type Culture Collection (ATCC 27294). M. smegmatis strain mc2155 was provided by William Jacobs, Jr., Albert Einstein College of Medicine, Bronx, NY. M. tuberculosis strain CDC1551 and transposon mutants of the Rv1006, Rv1235, Rv2589, Rv2766c, Rv3106, and Rv3788 knockout genes were obtained from the Biodefense and Emerging Infections Research Resources Repository (BEI Resources), NIAID, NIH. The bacteria were homogenized and passed through a syringe with a 24-gauge needle and then cultured to mid-exponential phase in Middlebrook 7H9 broth supplemented with 10% OADC (oleic acid, albumin, dextrose, catalase) (Hardy Diagnostics), 0.1% glycerol, and 0.025% Tween 80. The viable counts of bacteria were determined on 7H11 agar supplemented with OADC and glycerol by plating of serial dilutions. Kanamycin (Sigma-Aldrich) at a 50-μg/ml concentration was added to M. smegmatis gene overexpression clone cultures, where appropriate.
Antibiotic killing kinetics in vitro.
An M. tuberculosis inoculum of 106 bacteria was cultured in liquid medium containing 2× MIC of the following drugs: INH, 0.2 μg/ml; RMP, 1 μg/ml; MXF, 0.5 μg/ml; MQ, 16 μg/ml; and TMC207, 60 ng/ml. Samples were taken after 24 h, 48 h, and 4 and 6 days of drug exposure and plated onto 7H11 agar plates for CFU determination. The drug concentrations tested for M. smegmatis were 125 μg/ml RMP, 0.8 μg/ml MXF, and 8 μg/ml TMC207, and INH was excluded from the experiments. The experiments were carried out in duplicate and repeated three times. All the drugs used in this study were purchased from the Sigma-Aldrich Company.
Sample preparation and proteome analysis.
Approximately 108 cells of M. tuberculosis H37Rv were inoculated in 10 ml of 7H9 medium with or without drugs. Twice the MIC of INH, RMP, MXF, MQ, or TMC270 was added to the medium, and the bacteria were exposed to the drugs for 24 h, 48 h, and 4 and 6 days. At the indicated time points, samples were washed with Hanks' balanced salt solution (HBSS) (Corning) and pelleted. To prepare the lysis solution, 1 mg of ProteaseMax surfactant (Promega) was added to 1 ml of the mixture of ammonium bicarbonate (50 mM) with acetonitrile at a ratio of 9:1 (vol/vol). The bacterial pellets were resuspended in 1 ml lysis solution and mechanically disturbed in a bead beater. Samples were cleared by centrifugation and filtration through 0.45-μm filters. The total concentration of proteins was measured prior to in-solution digestion. Proteins were reduced and alkylated via incubation with equal amounts of dithiothreitol (DTT) and iodoacetamide at a final concentration of 10 mM and then digested with trypsin at a ratio of 1:40 (wt/wt) at 37°C for 9 h. The trypsinization was terminated with 5% acetic acid (vol/vol). All the samples were heated and centrifuged at 500 rpm for 30 min at 45°C in order to precipitate the detergent, and peptide mixtures were purified using spin filtration (Pall Nanosep, 10 kDa, with an Omega membrane). Samples were dried using a SpeedVac and resuspended in water with 0.1% formic acid (Sigma-Aldrich). The mass spectrometric analysis was performed using a Thermo Fisher LTQ Velos mass spectrometer at the Oregon Health and Science University (OHSU) proteomics facility.
Construction of M. smegmatis overexpression clones.
M. smegmatis genes with more than 70% protein sequence homology to M. tuberculosis proteins were selected for construction of protein overexpression clones in M. smegmatis. MSMEG_2959 (Rv2589/GabT), MSMEG_6934 (Rv3914/TrxC), MSMEG2086 (Rv3106/FprA), MSMEG_1670 (Rv3318/SdhA), MSMEG_4281 (Rv2213/PepB), MSMEG_4672 (Rv2460c/ClpP2), and MSMEG_5061 (Rv1235/LpqY) genes were amplified from M. smegmatis genomic DNA and ligated at the HindIII and EcoRI restriction sites of the pMV261 vector with 6×His. The recombinant plasmids were transformed to M. smegmatis, and the resulting clones were tested for sensitivity to antibiotics.
Infection of macrophages.
The THP-1 human monocytic cell line (ATCC TIB-202) was maintained in RPMI 1640 (Corning) supplemented with 10% fetal bovine serum (Gemini) and 2 mM l-glutamine. THP-1 cells were treated with 10 ng/ml of phorbol 12-myristate 13-acetate (Sigma-Aldrich) and seeded at 80 to 100% confluence into 24-well plates. After 24 h, the monolayers were replenished with new medium for an additional 48 h to allow monocytic cells to differentiate into macrophages. Adherent monolayers were infected with M. tuberculosis and wild-type M. smegmatis or gene knockout or overexpressed clones at a multiplicity of infection of 10 bacteria to 1 cell and incubated at 37°C and 5% CO2 for 1 h. Extracellular bacteria were subsequently removed by washing the monolayers three times with HBSS and treating the cells with amikacin (50 μg/ml for M. smegmatis or 200 μg/ml for M. tuberculosis) for 1 h. Infected cells were treated with anti-TB drugs at the concentrations listed above, lysed at several time points, and plated on 7H11 agar plates to determine the intracellular bacterial CFU. Infected monolayers without drug treatment served as a control.
Proteome data analysis.
An in-house script was used to create DTA format files from the RAW files using extract_msn.exe (version 5.0; ThermoFisher) with a molecular weight range of 550 to 4,000, an absolute threshold of 500, a group scan setting of 1, and a minimum of 25 ions. A human species subset of the Sprot (version 2011.06) protein database (20,235 proteins) was prepared with concatenated reversed entries (and common contaminants) using scripts available from the Proteomic Analysis Workbench website and searched with SEQUEST (version 28, revision 12; ThermoFisher). Parent ion and fragment ion tolerances of 2.5 and 1.0 Da were used with calculated average and monoisotopic masses, respectively. Cysteine had a static modification mass of +57 Da, and trypsin specificity was used. An in-house suite of programs was used to provide Peptide Prophet-like discriminant function scoring to identify “correct” peptides and discard “incorrect” peptides using sequence-reversed matches to estimate the FDR. Protein identification lists were prepared using standard parsimony principles. Proteins were required to have two or more fully tryptic peptides with distinct sequences. Different charge states of the same peptide sequence were not considered unique peptides. An in-house algorithm was used to compare identified peptide sets between proteins that had peptides in common and to group highly homologous identifications into families. Shared and unique peptide status was recomputed across all proteins after family grouping. Shared peptide counts were fractionally split based on the relative total unique counts of the proteins that had those peptides in common. These corrected total spectral counts per protein were used in the quantitative comparisons.
Construction of Venn diagrams.
Venn diagrams were generated using the tool at http://bioinformatics.psb.ugent.be/cgi-bin/liste/Venn/calculate_venn.htpl. This tool calculates the intersection(s) of the list of proteins that change in response to the drugs and generates a textual output, indicating which proteins are in each intersection or are unique to a certain drug treatment group. Inclusion criteria include observation of ≥2.5 normalized spectral counts and ≥2-fold increase over the control.
Protein selection.
Using BRB-ArrayTools, the experimental data were compared between the control and five drug treatment groups individually for the same time points, with a t test paired at a univariate P value of <0.05. Proteins that had concordant directions of change across five treatments were selected (173 proteins). Then, using Fisher's combined probability test, we calculated the combined P value using the following formula: , where k is 5 (drugs), pi is the P value of a protein being regulated by drug i, and is a random variable following the chi-square distribution with 2 k degrees of freedom. Out of 173 proteins, 112 were under the cutoff threshold (P < 0.05; FDR < 7.7%) and were selected for further analysis.
Gene Ontology and KEGG pathway enrichment analyses.
We used DAVID Bioinformatics Resources (https://david.ncifcrf.gov/) to perform enrichment analyses of GO biological process terms and KEGG pathways for the list of 112 proteins with P values of <0.05. In addition, the KEGG pathways of M. tuberculosis (KEGG entry, mtu) were downloaded using the R package KEGGREST in Bioconductor. As a result, a total of 4,008 M. tuberculosis proteins were found, 1,081 of which had KEGG pathway annotations. The metabolic pathway map was performed in http://www.kegg.jp/kegg/tool/map_pathway2.html against mtu by selecting mtu01100 metabolic pathways for visualization.
Network reconstruction.
We extracted interactions for the 103 proteins from the regulatory network map, which can be downloaded from TBDB (http://tuberculosis.bu.edu/share/ChIPseq_4dec13/interactions.txt). An M. tuberculosis comprehensive regulatory network map was generated by using chromatin immunoprecipitation sequencing (ChIP-seq) data combined with transcriptomic data following transcription factor-induced expression (15). Gene information (e.g., GeneID, gene symbol, and locus tag) for the proteins of interest was extracted from the NCBI gene database (ftp://ftp.ncbi.nih.gov/gene/), and then network visualization was carried out in Cytoscape.
Supplementary Material
ACKNOWLEDGMENT
The following reagents were obtained through BEI Resources, NIAID, NIH: Mycobacterium tuberculosis strain CDC1551, transposon mutant 1040 (MT1035 and Rv1006) NR-14700, transposon mutant 3247 (MT1273 and Rv1235) NR-18886, transposon mutant 2902 (MT2666 and Rv2589) NR-18723, transposon mutant 1206 (MT2836 and Rv2766c) NR-18033, transposon mutant 1319 (MT3189 and Rv3106) NR-15077, and transposon mutant 1921 (MT3896 and Rv3788) NR-18279.
This work was supported by the grant R21AI 110078 from the National Institutes of Health.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00430-17.
REFERENCES
- 1.Saukkonen JJ, Cohn DL, Jasmer RM, Schenker S, Jereb JA, Nolan CM, Peloquin CA, Gordin FM, Nunes D, Strader DB, Bernardo J, Venkataramanan R, Sterling TR, ATS (American Thoracic Society) Hepatotoxicity of Antituberculosis Therapy Subcommittee. 2006. An official ATS statement: hepatotoxicity of antituberculosis therapy. Am J Respir Crit Care Med 174:935–952. doi: 10.1164/rccm.200510-1666ST. [DOI] [PubMed] [Google Scholar]
- 2.Joshi JM. 2011. Tuberculosis chemotherapy in the 21 century: back to the basics. Lung India 28:193–200. doi: 10.4103/0970-2113.83977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merle CS, Fielding K, Sow OB, Gninafon M, Lo MB, Mthiyane T, Odhiambo J, Amukoye E, Bah B, Kassa F, N′Diaye A, Rustomjee R, de Jong BC, Horton J, Perronne C, Sismanidis C, Lapujade O, Olliaro PL, Lienhardt C, OFLOTUB/Gatifloxacin for Tuberculosis Project. 2014. A four-month gatifloxacin-containing regimen for treating tuberculosis. N Engl J Med 371:1588–1598. doi: 10.1056/NEJMoa1315817. [DOI] [PubMed] [Google Scholar]
- 4.Ginsberg AM. 2008. Emerging drugs for active tuberculosis. Semin Respir Crit Care Med 29:552–559. doi: 10.1055/s-0028-1085706. [DOI] [PubMed] [Google Scholar]
- 5.Ruan Q, Liu Q, Sun F, Shao L, Jin J, Yu S, Ai J, Zhang B, Zhang W. 2016. Moxifloxacin and gatifloxacin for initial therapy of tuberculosis: a meta-analysis of randomized clinical trials. Emerg Microbes Infect 5:e12. doi: 10.1038/emi.2016.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palomino JC, Martin A. 2013. TMC207 becomes bedaquiline, a new anti-TB drug. Future Microbiol 8:1071–1080. doi: 10.2217/fmb.13.85. [DOI] [PubMed] [Google Scholar]
- 7.Diacon AH, Pym A, Grobusch M, Patientia R, Rustomjee R, Page-Shipp L, Pistorius C, Krause R, Bogoshi M, Churchyard G, Venter A, Allen J, Palomino JC, De Marez T, van Heeswijk RP, Lounis N, Meyvisch P, Verbeeck J, Parys W, de Beule K, Andries K, McNeeley DF. 2009. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med 360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 8.Manjunatha U, Boshoff HI, Barry CE. 2009. The mechanism of action of PA-824: novel insights from transcriptional profiling. Commun Integr Biol 2:215–218. doi: 10.4161/cib.2.3.7926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Somasundaram S, Anand RS, Venkatesan P, Paramasivan CN. 2013. Bactericidal activity of PA-824 against Mycobacterium tuberculosis under anaerobic conditions and computational analysis of its novel analogues against mutant Ddn receptor. BMC Microbiol 13:218. doi: 10.1186/1471-2180-13-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillespie SH. 2016. The role of moxifloxacin in tuberculosis therapy. Eur Respir Rev 25:19–28. doi: 10.1183/16000617.0085-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swindells S. 2012. New drugs to treat tuberculosis. F1000 Med Rep 4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cook GM, Berney M, Gebhard S, Heinemann M, Cox RA, Danilchanka O, Niederweis M. 2009. Physiology of mycobacteria. Adv Microb Physiol 55:81–182, 318–389. doi: 10.1016/S0065-2911(09)05502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mitchison DA. 2004. Fluoroquinolones in the treatment of tuberculosis: a study in mice. Am J Respir Crit Care Med 169:334–335. doi: 10.1164/rccm.2312005. [DOI] [PubMed] [Google Scholar]
- 14.Grosset J, Truffot-Pernot C, Lacroix C, Ji B. 1992. Antagonism between isoniazid and the combination pyrazinamide-rifampin against tuberculosis infection in mice. Antimicrob Agents Chemother 36:548–551. doi: 10.1128/AAC.36.3.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, Gomes A, Rustad T, Dolganov G, Glotova I, Abeel T, Mahwinney C, Kennedy AD, Allard R, Brabant W, Krueger A, Jaini S, Honda B, Yu WH, Hickey MJ, Zucker J, Garay C, Weiner B, Sisk P, Stolte C, Winkler JK, Van de Peer Y, Iazzetti P, Camacho D, Dreyfuss J, Liu Y, Dorhoi A, Mollenkopf HJ, Drogaris P, Lamontagne J, Zhou Y, Piquenot J, Park ST, Raman S, Kaufmann SH, Mohney RP, Chelsky D, Moody DB, Sherman DR, Schoolnik GK. 2013. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature 499:178–183. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feehily C, O'Byrne CP, Karatzas KA. 2013. Functional gamma-aminobutyrate shunt in Listeria monocytogenes: role in acid tolerance and succinate biosynthesis. Appl Environ Microbiol 79:74–80. doi: 10.1128/AEM.02184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waagepetersen HS, Sonnewald U, Schousboe A. 1999. The GABA paradox: multiple roles as metabolite, neurotransmitter, and neurodifferentiative agent. J Neurochem 73:1335–1342. doi: 10.1046/j.1471-4159.1999.0731335.x. [DOI] [PubMed] [Google Scholar]
- 18.Shelp BJ, Bown AW, McLean MD. 1999. Metabolism and functions of gamma-aminobutyric acid. Trends Plant Sci 4:446–452. doi: 10.1016/S1360-1385(99)01486-7. [DOI] [PubMed] [Google Scholar]
- 19.Maras B, Sweeney G, Barra D, Bossa F, John RA. 1992. The amino acid sequence of glutamate decarboxylase from Escherichia coli. Evolutionary relationship between mammalian and bacterial enzymes. Eur J Biochem 204:93–98. [DOI] [PubMed] [Google Scholar]
- 20.Feehily C, Karatzas KA. 2013. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J Appl Microbiol 114:11–24. doi: 10.1111/j.1365-2672.2012.05434.x. [DOI] [PubMed] [Google Scholar]
- 21.Dagorn A, Chapalain A, Mijouin L, Hillion M, Duclairoir-Poc C, Chevalier S, Taupin L, Orange N, Feuilloley MG. 2013. Effect of GABA, a bacterial metabolite, on Pseudomonas fluorescens surface properties and cytotoxicity. Int J Mol Sci 14:12186–12204. doi: 10.3390/ijms140612186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grzegorzewicz AE, Pham H, Gundi VA, Scherman MS, North EJ, Hess T, Jones V, Gruppo V, Born SE, Kordulakova J, Chavadi SS, Morisseau C, Lenaerts AJ, Lee RE, McNeil MR, Jackson M. 2012. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat Chem Biol 8:334–341. doi: 10.1038/nchembio.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marrero J, Trujillo C, Rhee KY, Ehrt S. 2013. Glucose phosphorylation is required for Mycobacterium tuberculosis persistence in mice. PLoS Pathog 9:e1003116. doi: 10.1371/journal.ppat.1003116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR Jr. 2010. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 107:21761–21766. doi: 10.1073/pnas.1014642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akif M, Khare G, Tyagi AK, Mande SC, Sardesai AA. 2008. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J Bacteriol 190:7087–7095. doi: 10.1128/JB.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin K, O'Brien KM, Trujillo C, Wang R, Wallach JB, Schnappinger D, Ehrt S. 2016. Mycobacterium tuberculosis thioredoxin reductase is essential for thiol redox homeostasis but plays a minor role in antioxidant defense. PLoS Pathog 12:e1005675. doi: 10.1371/journal.ppat.1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ziegler GA, Vonrhein C, Hanukoglu I, Schulz GE. 1999. The structure of adrenodoxin reductase of mitochondrial P450 systems: electron transfer for steroid biosynthesis. J Mol Biol 289:981–990. doi: 10.1006/jmbi.1999.2807. [DOI] [PubMed] [Google Scholar]
- 28.McLean KJ, Warman AJ, Seward HE, Marshall KR, Girvan HM, Cheesman MR, Waterman MR, Munro AW. 2006. Biophysical characterization of the sterol demethylase P450 from Mycobacterium tuberculosis, its cognate ferredoxin, and their interactions. Biochemistry 45:8427–8443. doi: 10.1021/bi0601609. [DOI] [PubMed] [Google Scholar]
- 29.Fischer F, Raimondi D, Aliverti A, Zanetti G. 2002. Mycobacterium tuberculosis FprA, a novel bacterial NADPH-ferredoxin reductase. Eur J Biochem 269:3005–3013. doi: 10.1046/j.1432-1033.2002.02989.x. [DOI] [PubMed] [Google Scholar]
- 30.Bossi RT, Aliverti A, Raimondi D, Fischer F, Zanetti G, Ferrari D, Tahallah N, Maier CS, Heck AJ, Rizzi M, Mattevi A. 2002. A covalent modification of NADP+ revealed by the atomic resolution structure of FprA, a Mycobacterium tuberculosis oxidoreductase. Biochemistry 41:8807–8818. doi: 10.1021/bi025858a. [DOI] [PubMed] [Google Scholar]
- 31.Baughn AD, Garforth SJ, Vilcheze C, Jacobs WR Jr. 2009. An anaerobic-type alpha-ketoglutarate ferredoxin oxidoreductase completes the oxidative tricarboxylic acid cycle of Mycobacterium tuberculosis. PLoS Pathog 5:e1000662. doi: 10.1371/journal.ppat.1000662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jindani A, Dore CJ, Mitchison DA. 2003. Bactericidal and sterilizing activities of antituberculosis drugs during the first 14 days. Am J Respir Crit Care Med 167:1348–1354. doi: 10.1164/rccm.200210-1125OC. [DOI] [PubMed] [Google Scholar]
- 33.Donald PR, Diacon AH. 2008. The early bactericidal activity of anti-tuberculosis drugs: a literature review. Tuberculosis 88(Suppl 1):S75–S83. doi: 10.1016/S1472-9792(08)70038-6. [DOI] [PubMed] [Google Scholar]
- 34.Connolly LE, Edelstein PH, Ramakrishnan L. 2007. Why is long-term therapy required to cure tuberculosis? PLoS Med 4:e120. doi: 10.1371/journal.pmed.0040120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honer zu Bentrup K, Russell DG. 2001. Mycobacterial persistence: adaptation to a changing environment. Trends Microbiol 9:597–605. doi: 10.1016/S0966-842X(01)02238-7. [DOI] [PubMed] [Google Scholar]
- 36.Baek SH, Li AH, Sassetti CM. 2011. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol 9:e1001065. doi: 10.1371/journal.pbio.1001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.