

ABSTRACT
Viral regulatory complexes perform critical functions during virus replication and are important targets for therapeutic intervention. In HIV, the Tat and Rev proteins form complexes with multiple viral and cellular factors to direct transcription and export of the viral RNA. These complexes are composed of many proteins and are dynamic, making them difficult to fully recapitulate in vitro. Therefore, we developed a cell-based reporter assay to monitor the assembly of viral complexes for inhibitor screening. We screened a small-molecule library and identified multiple hits that inhibit the activity of the viral complexes. A subsequent chemistry effort was focused on a thieno[2,3-b]pyridine scaffold, examples of which inhibited HIV replication and the emergence from viral latency. Notable aspects of the effort to determine the structure-activity relationship (SAR) include migration to the regioisomeric thieno[2,3-c]pyridine ring system and the identification of analogs with single-digit nanomolar activity in both reporter and HIV infectivity assays, an improvement of >100-fold in potency over the original hits. These results validate the screening strategy employed and reveal a promising lead series for the development of a new class of HIV therapeutics.
KEYWORDS: antivirals, HIV, RNA, Rev
INTRODUCTION
RNA-protein complexes are essential to the assembly and activity of many viral regulatory systems and represent an important target class for antiviral drug discovery. In HIV, the Rev-Rev response element (RRE) protein-RNA complex is one such target due to its essential activity in mediating the export of unspliced and partially spliced RNAs from the nucleus to the cytoplasm (1, 2). Disrupting the Rev-RRE interaction prevents the expression of late viral proteins and the packaging of viral RNA, thus inhibiting virus replication.
Rev is a 116-amino-acid RNA-binding protein that is expressed from fully spliced mRNAs early in the virus life cycle (3). Rev binds the RRE, a highly structured ∼350-nucleotide (nt) RNA element encoded within the env gene (reviewed in references 2 and 4). Current models suggest that about six Rev molecules bind the RNA in order to properly position two of the nuclear export sequences on Rev for binding to a dimer of the Crm1-RanGTP export complex (5, 6). This complex is then exported through the nuclear pore, after which it disassembles in the cytoplasm to allow translation of the late HIV proteins and packaging of the viral genome (2, 7).
The formation of the export complex is driven by several critical intermolecular interactions. Rev binds the RNA primarily through its arginine-rich motif (ARM), an α-helical domain that forms several important hydrogen bonds with the RNA (8, 9). The nuclear magnetic resonance (NMR) structures of the Rev peptide complexed to RRE IIB or to an RNA aptamer and a recent cocrystal structure of a Rev-RRE dimer (10) show that the α-helix binds in the widened major groove of the high-affinity IIB site, primarily using arginine and asparagine side chains for base-specific recognition (11–13). The cocrystal structure also shows that a glutamine at position 51 interacts symmetrically across the dimer interface to help stabilize the complex (10). Flanking the ARM of Rev are hydrophobic oligomerization domains that drive the formation of a stable Rev dimer, and Rev dimers in turn form higher-order structures through hydrophobic multimerization domains that are located opposite the oligomerization domain, ultimately assembling a very high affinity (picomolar) complex (14–16). Given the essential nature of this assembly and the requirement for high-affinity binding, small molecules that interfere with any of these interfaces are expected to block formation of competent export complexes and to be of potential therapeutic benefit.
To identify small-molecule inhibitors of the Rev-RRE complex, we developed a high-throughput assay based upon the established Tat hybrid assay platform for characterizing RNA-protein or RNA-peptide interactions in mammalian cells (17, 18). This system takes advantage of the function of the HIV transcription activator Tat, which binds to the TAR RNA hairpin located at the 5′ end of the viral transcripts (19, 20) and enhances the processivity of RNA polymerase II transcription complexes initiated at the HIV long terminal repeat (LTR) (21–23). The Tat activation domain can be used to activate reporter expression via heterologous RNA-protein interactions (24, 25) (Fig. 1A). Reporter activity is proportional to RNA-binding affinity, and thus the assay has been used extensively to characterize RNA-protein interactions, including HIV Tat-TAR, bovine immunodeficiency virus (BIV) Tat-TAR, and HIV Rev-RRE, and to screen for novel proteins that bind the Mason-Pfizer monkey virus (MPMV) constitutive transport element (CTE) (17, 26–32).
FIG 1.
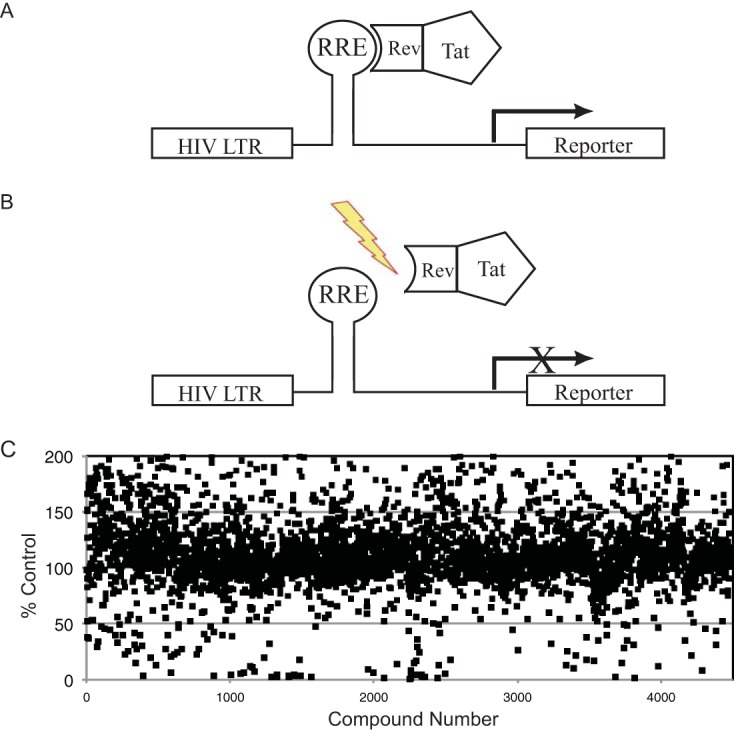
Screening strategy to identify inhibitors of RNA-protein complexes. (A) Schematic diagram of the Tat hybrid system. A plasmid encoding the HIV LTR, RRE RNA target, and a reporter gene is coexpressed with a plasmid encoding a fusion protein of the HIV Tat activation domain and a heterologous RNA-binding domain from the HIV Rev protein. A productive interaction between the RNA and RNA-binding protein confers activation of the reporter gene. (B) Schematic showing that inhibitors of the RNA-protein interaction are identified by a decrease in reporter signal. (C) Screening data from a small-molecule screen for inhibitors of Rev-RRE complexes.
We engineered the Tat hybrid platform with the Rev-RRE interaction and used it to screen a targeted small-molecule compound library. We hypothesized that compounds containing a carboxamide moiety might mimic the essential asparagine or glutamine side chains and compete for Rev-Rev dimerization or Rev binding to the RRE RNA, and we thus constructed a library of ∼4,500 carboxamide-containing small molecules selected from a larger-diversity library. Here, we describe the discovery and structure-activity studies of a thienopyridine inhibitor scaffold in which an unsubstituted carboxamide function was found to be essential for activity. Optimized thienopyridine analogs exhibited low-nanomolar potencies in multiple reporter-based assays as well as in HIV replication assays. Interestingly, related thienopyridine analogs were identified independently as Rev inhibitors using an orthogonal screening approach (33).
RESULTS
We developed a Tat hybrid-based screening assay to identify small molecules that target RNA-protein interactions in general and the Rev-RRE interaction in particular (Fig. 1B). To screen small molecules, we engineered a stable Tat hybrid reporter cell line encoding the RRE IIB RNA target and driving a firefly luciferase (FFL) reporter. The reporter exhibited a good signal-to-noise ratio and was compatible with multiwell plates, robotic liquid handling, and standard luminescence plate readers. The Tat-Rev expression plasmid was then integrated into the reporter cell line, and clonal cell lines were selected that constitutively expressed luciferase to high levels, indicative of delivery of Tat via the heterologous RNA-protein interaction. We validated the assay using small interfering RNA (siRNA) knockdowns, targeting the Tat activation domain, and with 3,6-diaminoacridine, a known small-molecule inhibitor of the Rev-RRE interaction that served as a positive control (see Fig. S1 in the supplemental material) (34).
The RRE IIB-Rev screening cell line was plated in 384-well assay plates, and compounds were added to the cells to a final concentration of 30 μM. Compounds were screened in duplicate, and the luciferase activity of each compound was highly consistent (typically <10% deviation in activity) (Fig. S2). In parallel, MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] cell viability assays were performed to assess the toxicity of compounds and minimize the level of false positives. Screening data for the initial library screen is shown in Fig. 1C.
From the primary screen, we obtained 11 validated hits with confirmed dose-dependent activity and a lack of toxicity in the MTT assay. Of these 11 compounds, two were benzopyrans, one was a tetrasubstituted thiophene, and the remaining eight hits possessed a common thieno[2,3-b]pyridine core (Fig. S3). To further validate the hits, we used an electrophoretic mobility shift assay (EMSA) to evaluate which of the three hit scaffolds were capable of disrupting the Rev-RRE IIB interaction in vitro. Only the thiophene scaffold disrupted the complex, as determined by EMSA (Fig. S4). This result demonstrated that the Tat hybrid screening method could identify bona fide inhibitors of RNA-protein interactions although our experience with EMSA raised questions about this method. We observed that compounds that disrupted the RNA-protein complex by EMSA, namely, 3,6-diaminoacridine, neomycin B, and thiophenes, were toxic to cells. These types of relatively nonspecific intercalating agents (e.g., acridines) and RNA-binding compounds (e.g., aminoglycosides) can be readily picked up as binding inhibitors in EMSAs but may be missed in cell-based assays because of their high toxicity. Conversely, the lack of EMSA inhibition by other types of compounds that scored well in the cell-based assay does not necessarily rule out Rev-RRE as their target since the in vitro EMSA lacks other proteins required for Rev function (e.g., Crm1 and Ran) and potential accessory factors (e.g., hnRNPs and chaperones) that may alter the nature of Rev-RRE complexes for inhibitors to act on. Other experimental evidence supporting Rev-RRE as a plausible target of thienopyridines is provided below. Because of a superior toxicity profile and a nascent structure-activity relationship (SAR) profile, we focused our efforts on the thienopyridine scaffold.
Compounds 1a to c (Fig. 2) are representative of the original thienopyridine hits, which varied in terms of substitution on the pyridine ring but universally possessed amino and carboxamide substitution on the thiophene ring. Commercially available analogs of compound 1a bearing a carboxylic acid (1d) or methyl ketone (1e) in place of the carboxamide were purchased and found not to be significantly active in the Tat hybrid assay. This suggested the importance of the carboxamide function for activity in the Tat hybrid reporter cell line (Fig. 2 and 3; Table 1). Evaluation of additional commercial analogs suggested that modification of the 4 or 5 position of the thienopyridine ring was generally well tolerated, and this afforded some confidence that improvements in potency could be realized with the synthesis of additional analogs (Table S1).
FIG 2.
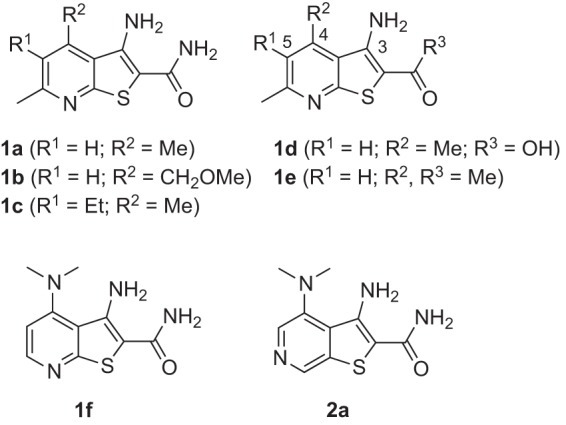
Structures of hits 1a, 1b, 1c, and 1f identified in the primary screen. Commercially available analogs 1d and 1e are congeners of compounds 1a lacking the carboxamide function and were found to be inactive in the reporter assay. Compound 2a is a regioisomer of hit 1f and the heterocyclic scaffold upon which additional synthetic analogs were based.
FIG 3.
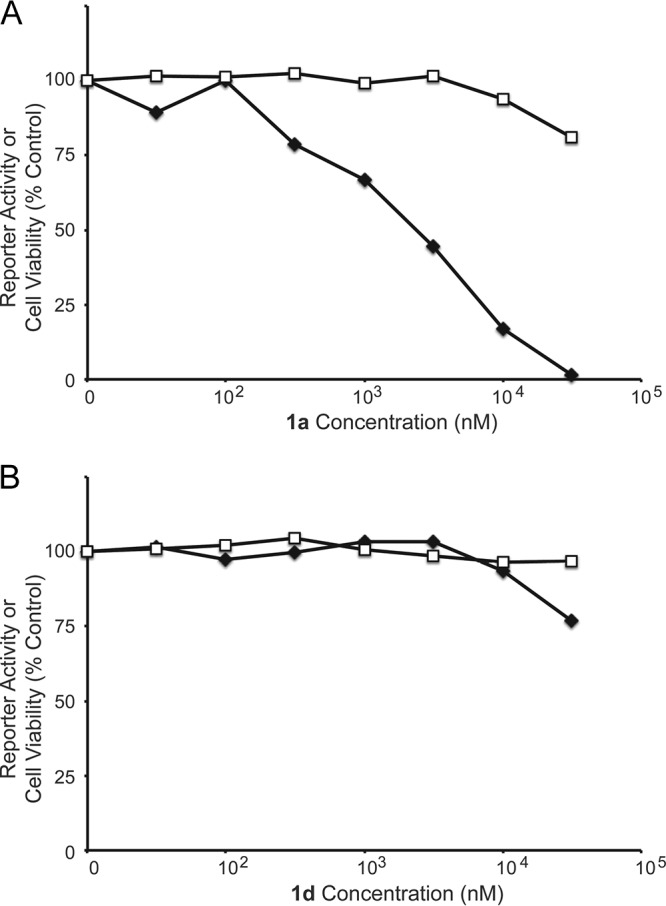
Dose-response curves for the activity and toxicity of thienopyridine analogs 1a (A) and 1d (B). The Tat hybrid reporter cells were plated in 96-well plates, and test compounds were added to the cells at doses ranging from 1 nM to 50,000 nM in duplicate. Cell lysates were collected after 48 h, activity values were determined by luciferase assay, and toxicity was determined by MTT assay. Average activity values (filled symbols) and toxicity values (open symbols) are shown.
TABLE 1.
Activity of thienopyridine group 1 and 2 analogs in reporter and antiviral assays
| Compound | Tat hybrid assay EC50 (nM) | U1 replication assay IC50 (nM) | Rev reporter assay EC50 (nM) | HIV-1 replication assay IC50 (nM) | LipEa |
|---|---|---|---|---|---|
| 1a | 2,170 | 1,473 | 4.7 | ||
| 1b | 1,160 | 528 | 5.2 | ||
| 1c | 700 | 750 | 4.5 | ||
| 1d | >100,000 | ||||
| 1e | 31,200 | 2.4 | |||
| 1f | 210 | 659 | 530 | 6.2 | |
| 2a | 710 | 450 | 5.8 | ||
| 2b | 65 | 28 | 680 | 77 | 5.9 |
| 2c | 330 | 391 | 6.5 | ||
| 2d | 930 | 6.2 | |||
| 2e | 7,620 | 5.3 | |||
| 2f | 140 | 219 | 634 | 5.2 | |
| 2g | 370 | 173 | 5.75 | ||
| 2h | 350 | 219 | 5.0 | ||
| 2i | 70 | 13 | 5.3 | ||
| 2j | 100 | 34 | 4.6 | ||
| 2k | 730 | 1,087 | 4.8 |
LipE = pKi − clogP. pKi was estimated from the Tat hybrid assay EC50; clogP values were determined in Vortex (Dotmatics) using the calculated property XlogP.
Because of the reasonable activity of thienopyridine compounds in the Tat hybrid reporter assay, low toxicity in the MTT assay, and a nascent SAR profile, we embarked on structure-activity studies of the thienopyridine series. One of the first analogs synthesized was compound 2a derived from a thieno[2,3-c]pyridine ring system that is regioisomeric with the original hit compounds (Fig. 2). Gratifyingly, 2a exhibited submicromolar potency in both the Tat hybrid assay and in a viral replication assay in U1 cells (Table 1). The U1 cells contain an integrated and inducible HIV-1 provirus, meaning that inhibition of viral replication in theses cells is indicative of compound action subsequent to retroviral genome integration, as expected for a Rev-RRE inhibitor. As the potency of analog 2a was similar to that of its directly analogous thieno[2,3-b]thionopyridine congener 1f in both assays (Fig. 2 and Table 1), we focused our SAR studies on the comparatively unexplored thieno[2,3-c]thionopyridine scaffold represented by compound 2a.
All thieno[2,3-c]pyridine analogs described herein were prepared by one of the three synthetic approaches described below (Fig. 4) (35). Thus, the preparation of the group 2 amine-bearing analogs involved SNAr reaction of secondary amines with 3,5-dichloro-4-pyridinecarbonitrile, followed by reaction with 2-mercaptoacetamide in the presence of sodium methoxide. Aryl and heteroaryl-substituted group 3 analogs were prepared similarly, but beginning with Suzuki coupling reactions. Finally, des-amino group 4 variants were prepared from 3,5-dihalopyridinecarboxaldehyde via initial formation of the thieno[2,3-c]pyridine ring as before, followed by Suzuki coupling (36).
FIG 4.
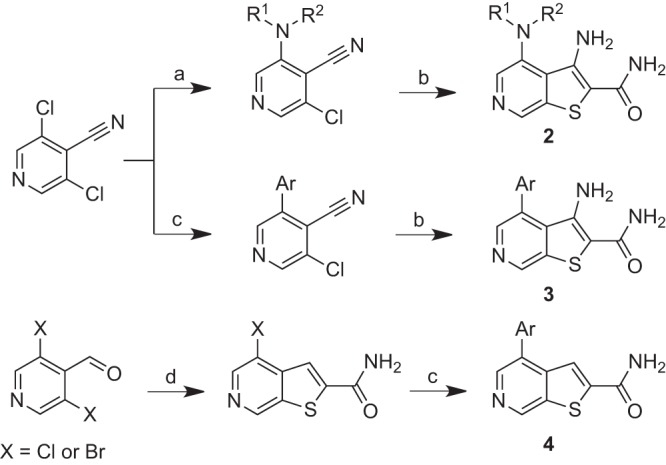
Synthesis of thieno[2,3-c]pyridine analogs 2 to 4. Conditions: a, 1 to 2 equivalents R1R2NH, Et3N, dimethylformamide, 25 to 40°C, 12 to 24 h; b, 2 equivalents 2-mercaptoacetamide, 10 wt% in MeOH/NH3, 2 equivalents NaOMe, dimethylformamide, μW, 80°C, 10 to 60 min; c, 1 to 2 equivalents Ar-B(OH)2, 0.025 equivalent PCy3, 0.01 equivalent Pd2(dba)3, 1.7 equivalents K2CO3, dioxane, μW, 150°C, 30 min; d; 0.75 equivalent 2-mercaptoacetamide, 10 wt% in MeOH/NH3, 1.2 equivalents NaOMe, dimethylformamide, μW, 80°C, 10 to 60 min.
New analogs were evaluated in both the Tat hybrid assay, which reports on the Rev-RRE interaction, and in the viral activation assay in U1 cells (Table 1). Representative dose-response curves in the U1 assay are shown for analogs 1a, 2b, and 4e (Fig. 5 and Table S2). We found that the activities of ∼50 synthetic thienopyridine analogs were well correlated in the Tat hybrid and U1 assays (Fig. 6), suggesting that compound action remained on target during optimization and consistent with the postintegration pharmacology expected of a Rev-RRE inhibitor.
FIG 5.
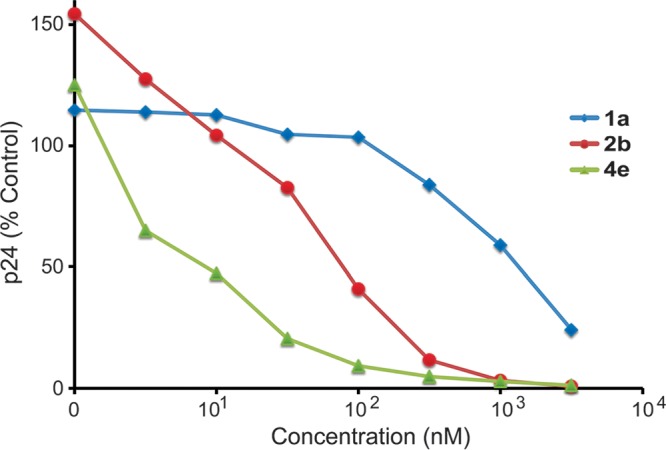
Activity of thienopyridine analogs 1a, 2b, and 4e in the U1 activation assay. U1 cells were plated in 96-well plates and activated by the addition of phytohemagglutinin. The test compounds were added to the cells at doses ranging from 1 nM to 3,160 nM in triplicate, and supernatants were collected 72 h later. p24 values were determined by ELISA, and average values are shown.
FIG 6.
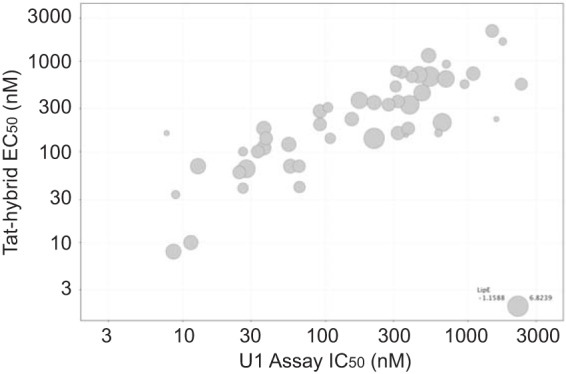
Comparison of activities for thienopyridine analogs in the Tat hybrid and U1 viral activation assays. Each data point is for one compound, and the relative symbol size indicates the LipE value for that compound.
To further explore amine substitution as in analog 2a, additional analogs such as the piperidine 2b, morpholine 2d, and N-Me piperazine analog 2e were prepared and evaluated (Fig. 7 and Table 1). Piperidine analog 2b was nearly 10-fold more potent than the dimethylamino analog 2a, an effect that appears to derive from the aliphatic nature of the piperidine ring, substitution of which (2c) or replacement with a morpholine (2d) or piperazine (2e) ring resulted in less potent analogs. Spirocyclic (2g) and geminal-diflouro (2h) substitutions at the 4 position proved inferior to results with the parent piperidine 2b. Tetrahydroquinoline (2i) and phenyl ether (2j) analogs were equipotent to 2b but with their additional mass and lipophilicity produced inferior lipophilic efficiency (LipE) values (LipE = pKi − clogP) (37). Finally, introduction of a heteroaryl ring at the 3 position of the piperidine ring as in compound 2k was poorly tolerated. Although N-acylpiperazine analog 2f was notably more potent than the basic N-methylpiperazine analog 2e, the exploration of additional amide variants did not yield useful gains in potency.
FIG 7.
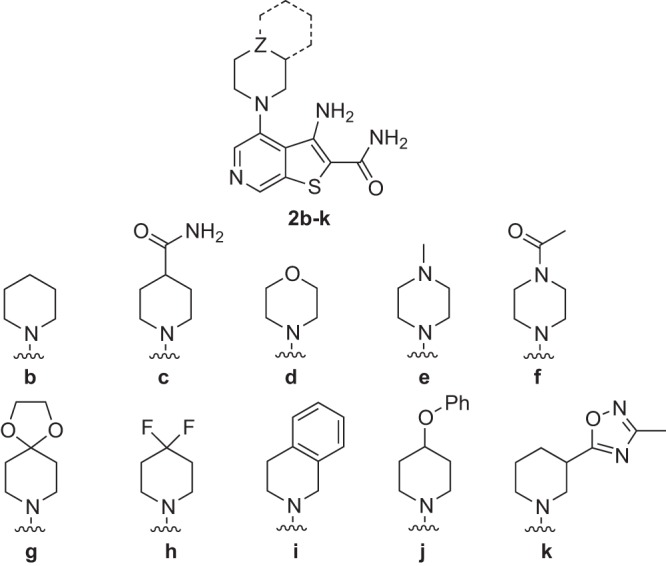
Structures of thieno[2,3-c]pyridine analogs 2b to k.
Having identified analogs like 2b and 2i that were up to 10-fold more potent than 2a, we next explored 4-aryl-substituted thienopyridine analogs either with (3a to j) or without (4a to j) a 3-amino group (Fig. 8 and Table 2). In the case of substituted phenyl rings, we found substitution at the para and/or meta position(s) was favored over ortho substitution (compare compounds 4c and 4d). Moderately electron-rich analogs like 3a/4a and 3e/4e were more potent than either electron-deficient congeners (4c) or more electron-rich analogs like 3f/4f. Consistent with substitution effects in the piperazine series (2k), bulky substitution at the 3 position (meta) as in 3j/4j was poorly tolerated. The most consistent effect in this series was the favorable effect of removing the 3-amino group (as in group 4 analogs, X represents H). Particularly potent were des-amino analogs bearing a moderately electron-rich aryl ring, as in the analogs 4a and 4e, which exhibited low-nanomolar potency in both the Tat hybrid and U1 assays and were the most potent analogs evaluated (Table 2).
FIG 8.
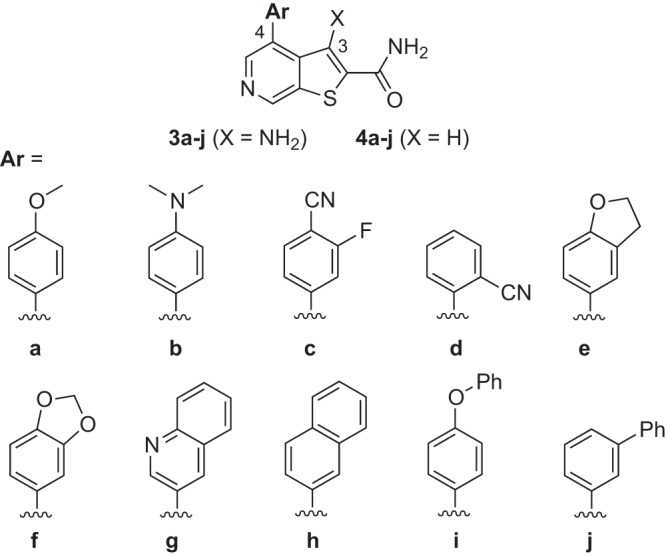
Structures of thieno[2,3-c]pyridine analogs of groups 3 and 4.
TABLE 2.
Activity of thienopyridine group 3 and 4 analogs in reporter and antiviral assays
| Compound | Tat hybrid assay EC50 (nM) | U1 replication assay IC50 (nM) | Rev reporter assay EC50 (nM) | HIV-1 replication assay IC50 (nM) | LipEa |
|---|---|---|---|---|---|
| 3a | 70 | 57 | 4.9 | ||
| 3b | 140 | 38 | 4.6 | ||
| 3e | 200 | 91 | 4.6 | ||
| 3f | 330 | 277 | 4.5 | ||
| 3j | 4,700 | 1.15 | |||
| 4a | 10 | 11 | 115 | 4.5 | 5.0 |
| 4c | 140 | 108 | 4.0 | ||
| 4d | 560 | 946 | 3.5 | ||
| 4e | 8 | 9 | 150 | 3.9 | 5.3 |
| 4f | 70 | 65 | 4.5 | ||
| 4g | 41 | 66 | 6.4 | 4.2 | |
| 4h | 34 | 8.9 | 3.4 | 3.2 | |
| 4i | 160 | 7.7 | 2.2 | ||
| 4j | 150 | 368 | 1.9 |
LipE = pKi − clogP. pKi was estimated from the Tat hybrid EC50; clogP values were determined in Vortex using the calculated property XlogP.
Analogs 4a and 4e combine exceptional potencies with reasonable calculated lipophilicities, thus affording LipE values of 5.0 and 5.3, respectively. These analogs, along with the potent but more lipophilic analogs 4g and 4h, were evaluated in an HIV-1 replication assay. Jurkat cells were infected with HIV-1 laboratory isolate NL4-3 in the presence of the test compounds, and viral production was monitored by collecting viral supernatants and measuring p24 levels by enzyme-linked immunosorbent assay (ELISA) (Fig. 9 and Table S3). All four analogs displayed low-nanomolar efficacy in this assay, and moreover, their rank order potencies in the infectivity assay were correlated with 50% inhibitory concentrations (IC50s) in the U1 activation assay (Table 2). Replication curves for the des-amino analogs 4e and 4h are shown in Fig. 9B and C. These compounds exhibited IC50s of 3.9 nM and 3.4 nM, respectively, in the HIV-1 replication assay compared to 50% toxic concentration (TC50) values of 19,200 and 20,500 nM in the MTT cell viability assay. For comparison, AZT exhibited an IC50 of 3 nM and TC50 of >100,000 nM in these same assays. The piperazine analog 2b was also evaluated in the HIV-1 replication assay and exhibited activities in the mid- to high-nanomolar range (77 nM) and modest toxicity (41,400 nM) (Fig. 9A).
FIG 9.

Replication assays with HIV-1 isolate NL4-3 in Jurkat cells. Replication spreading assays were performed by infecting Jurkat cells with NL4-3 in the presence of compound at doses ranging from 3.16 nM to 1,000 nM in triplicate. Supernatants were collected every 48 to 72 h, and p24 values were determined by ELISA. Average replication curves are shown for the indicated compounds.
We next tested compound 4e in replication assays using primary cells and clinical isolates of HIV-1. Peripheral blood mononuclear cells (PBMCs) were stimulated with phytohemagglutinin (PHA) and infected with several clinical isolates of HIV-1 in the presence of the compounds. IC50s for the compound were determined by collecting viral supernatants and measuring p24 levels by ELISA (Table 3). Results of this experiment are shown in Fig. 10 where the dose-response and toxicity curves for isolate 93BR021 (CCR5-tropic, group M, subtype B) are shown. Here, we observed an IC50 of 55 nM and TC50 of 32,300 nM. AZT was again used as a positive control and exhibited an IC50 of 18.5 nM, and the TC50 was determined to be >50,000 nM by MTT assay.
TABLE 3.
Activity of thienopyridine analog 4e in replication assays in PBMCs
| Viral isolate | Coreceptor(s) | Compound 4e IC50 (nM) |
|---|---|---|
| Ba-L | CCR5 | 48.8 |
| 93BR021 | CCR5 | 54.8 |
| 93BR028 | CCR5 | 9.2 |
| 92TH014 | CCR5 | 19.6 |
| 92UG005 | CXCR4 | 70.8 |
| 92HT599 | CXCR4 | 25.9 |
| LAI | CXCR4 | 291 |
| 92HT593 | CXCR4, CCR5 | 13.4 |
FIG 10.
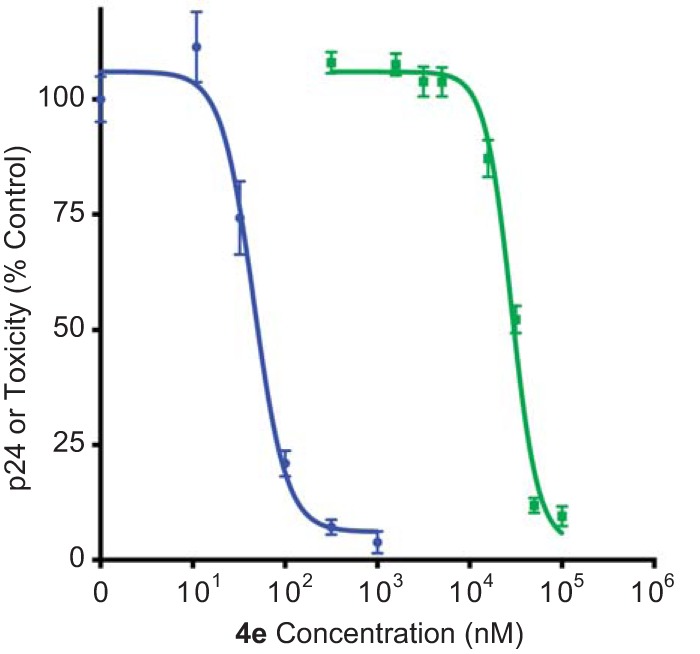
Replication assay with HIV-1 primary isolate 93BR021 in PBMCs. Replication assays were performed in PBMCs by stimulating cells with phytohemagglutinin and then infecting them with clinical isolates of HIV. Compound 4e was added to PBMCs at doses ranging from 1 nM to 1,000 nM in triplicate or more, and supernatants were collected every 48 to 72 h. p24 values were determined by ELISA. Average replication curves are shown in blue, and toxicity is shown in green.
We next evaluated analogs 2b, 2f, 4a, and 4e in a panel of standard in vitro absorption, distribution, metabolism, and excretion (ADME) assays to assess drug-like properties and the potential for efficacy in animals. All four analogs are rule-of-five compliant and exhibited moderate to high permeability across Caco-2 monolayers, a model of intestinal absorption (Table 4). Analogs 4a and 4e, with only two hydrogen bond donors, were more permeable but also notably less soluble than analogs 2b and 2f. The more lipophilic analogs 4a and 4e also appear more prone to oxidative metabolism in the presence of human liver microsomes. This is perhaps unsurprising given the presence of methoxyphenyl (4a) and benzylic methylene (4e) functions, both potential sites of oxidative metabolism.
TABLE 4.
In vitro ADME properties of selected thienopyridine analogs
| Compound | Tat hybrid assay EC50 (nM) | Aqueous solubility at pH 7.4 (nM) | Papp of Caco-2 cells (nm/s)a | HLM t1/2 (min)b |
|---|---|---|---|---|
| 2b | 65 | 34,000 | 505 | 28 |
| 2f | 140 | 258,000 | 97 | >60 |
| 4a | 10 | 440 | 594 | 8 |
| 4e | 8 | 280 | 666 | 18 |
Papp, apparent permeability.
HLM t1/2, half-life in human liver microsomes.
DISCUSSION
Here, we used a screening approach based on the HIV LTR Tat hybrid system to identify small molecules putatively targeting a protein-RNA interaction. A focused library of carboxamide-containing compounds was screened based on the expectation that such molecules can interact with RNA bases and could potentially disrupt or alter Rev-RNA binding interactions. Of the various hits identified in the screen, a series of thienopyridine carboxamides emerged as the most promising hit scaffold. We then used an HIV reporter cell line and the U1 replication assay to validate the screening hits and to evaluate an additional ∼100 commercial analogs. These analyses revealed that an unsubstituted carboxamide group at the 2 position of the thienopyridine ring was essential for activity. Furthermore, substitution on the thienopyridine ring was well tolerated, particularly at positions 4 and 5. This initial survey of commercially available analogs provided a preliminary SAR profile that encouraged further synthetic efforts on the scaffold.
A variety of analogs was prepared based on a regioisomeric thieno[2,3-c]pyridine ring system. Substitution of the 4 position with N-linked heteroaliphatic rings (group 2 compounds) or C-linked aryl and heteroaryl rings (groups 3 and 4) was well tolerated, with the C-linked aryl analogs in general possessing superior potencies. Most significantly, we found that the 3-amino group is not essential for activity and that, in fact, des-amino analogs possess notably superior potencies in both the Tat hybrid reporter assay and the U1 assay. The elimination of two hydrogen bond donors in such analogs likely improves intrinsic cell permeability, which may wholly or partly explain the enhanced potency of group 4 analogs in cell-based assays. Of the des-amino analogs, compound 4e showed the best potencies and therapeutic indices in the reporter and U1 assays and in the HIV replication assays, with a 50% effective concentration (EC50) of 8 nM in the reporter assay and IC50s of 9 nM and 3.9 nM in the U1 and HIV replication assays, respectively (Table 2). By comparison, the most potent of the N-linked analogs, piperazine 2b, displayed an EC50 of 65 nM in the reporter assay and IC50s of 28 nM and 77 nM in the U1 and HIV replication assays, respectively (Table 1). Overall, our structure activity studies improved potencies from the low-micromolar to the low-nanomolar regime, likely by improved target-level binding affinity and improved cellular permeability. While further optimization of in vivo drug-like properties will be required, leads like analog 4e appear promising as a starting point for such efforts.
Thienopyridine analogs that are closely related to our initial screening hits were described as inhibitors of Rev-RRE by another group of investigators (33) using a reporter assay that is dependent upon the activity of Rev (38). We therefore tested selected compounds from our SAR studies (1f, 2b, 4a and 4e) in a similar Rev-dependent reporter assay (Tables 1 and 2). Several additional experiments were described that strongly suggest that Rev is the target of the thienopyridine analogs, including additional reporter experiments that showed inhibition of a Rev-dependent, but not a CTE-dependent, reporter as well as experiments that showed that the protein composition of inhibitor-treated samples was consistent with an inhibitor of Rev function (33).
Biochemical assays including EMSAs did not show inhibition of the Rev-RRE interaction by the thienopyridine class in vitro. These assays generally capture a snapshot of Rev activity and lack critical interactions with cellular cofactors such as Crm1 and Ran. We have found that compounds that inhibit formation of RNA-protein complexes are often toxic, suggesting that EMSAs or other in vitro assays may lack the sensitivity to reveal subtle effects of small-molecule inhibitors. In some cases, such as with 3,6-diaminoacridine and neomycin B, inhibition of Rev-RRE formation is observed by EMSA, but both are toxic at therapeutic concentrations against HIV. Similarly, the thiophene class identified by the small-molecule screen also inhibits the Rev-RRE interaction, as determined by EMSA (see Fig. S4 in the supplemental material), but several of the commercially available thiophene analogs showed substantial toxicity and thus were not pursued further. Interestingly, SAR analysis of the thiophenes and thienopyridines identified the thienopyridine as a better scaffold that likely maintained similar binding characteristics (39).
Although our studies and the previous work implicate Rev as the likely target of thienopyridine carboxamides, other targets cannot be ruled out. For example, our screen is dependent upon the activity of the HIV LTR, and thus an inhibitor of HIV transcription would also produce a positive response in the assay. However, this would appear unlikely because we showed, using the Tat hybrid assay (Fig. S5), that the thienopyridine compound 4a specifically targets the Rev-RRE reporter but not the HIV Tat-TAR reporter. The previous screen did not use the HIV LTR, and these investigators further uncovered a thienopyridine resistance mutation in the RRE, consistent with the Rev-RRE interaction as the target of thienopyridines (40). We have also generated this RRE-defective virus and shown that it is resistant to compound 4e (Fig. S6). Though strongly supportive that Rev is the target of the compounds, these data do not explicitly rule out the possibility of alternative targets; for example, thienopyridines may directly target structured RNA, or multiple regions of the HIV genome may be targeted by the compounds in addition to the RRE. In summary, we identified and optimized a class of thienopyridine carboxamides that potently inhibits HIV replication in cells. This work represents an early but important step toward the development of a new class of HIV therapeutics that could eventually find a place in future HIV combination therapies. While the weight of evidence suggests that these compounds act by inhibition of the Rev-RRE interaction, further studies will be required to definitively establish their molecular pharmacology.
MATERIALS AND METHODS
Small-molecule screen.
RNA reporter plasmids were constructed using the pHIV LTR vector (18). To express HIV Tat fusion proteins, we constructed a mammalian codon-optimized pcDNA-Tat vector containing the first 48 amino acids of HIV Tat with a fusion to HIV Rev (residues 3 to 70). HIV Tat fusion proteins have been previously described (17, 41). A HeLa cell line expressing the pcDNA3 HIV-1 LTR-RREIIB-FFL reporter was generated to obtain a consistent background for library screens. The plasmid was transfected into HeLa cells, and stable integrants were selected using neomycin (G418) (800 μg/ml) for 10 days. Resistant cells were transfected with the pcDNA3 Tat-Rev3-70 plasmid harboring a hygromycin resistance marker. Individual clonal cell lines were assayed for luciferase signal, and 3,6-diaminoacridine, a nonspecific small-molecule inhibitor of the reporter, was used to identify cell lines with high signal-to-noise ratios (34). A Z′ value of 0.75 was obtained for the cell line used for the small-molecule screen (see Fig. S1 in the supplemental material).
A library of approximately 4,500 carboxamide-containing compounds was identified and purchased from ChemDiv. Approximately 5,000 cells were plated in a white 384-well plate, and compounds were added to the cells to a final concentration of 30 μM using a Biomek FX robotic liquid handler (Beckman Coulter). The cells were incubated with test compounds for 48 h and assayed for luciferase activity (Bright Glo; Promega). The cytotoxicity of each compound was determined in parallel via 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT)-based cell viability assays (42). Additional compounds structurally related to screening hits were purchased from Hit2Lead/Chembridge, Asinex, and Specs.
The test compounds that displayed activity were reconfirmed in duplicate or triplicate at multiple concentrations using the Tat hybrid reporter cell lines or by transient transfection. Previously characterized thienopyridines and 3,6-diaminoacridine (34) were used as controls. Cells were counted and plated into white 384-well or 96-well cell culture-treated assay plates, and compounds were added. After 48 h, the cells were prepared for luminescence assays that were performed on a plate reader (Tecan Evolution or MD Analyst). The cytotoxicity of each compound was determined in parallel via MTT-based cell viability assays.
Rev reporter assay.
293T cells were plated in 96-well plates and cotransfected with an HIV RRE (Rev-dependent) reporter plasmid (43) and a pSV2 Rev expression plasmid. Compounds were incubated with the cells for 48 h, and reporter activity was assessed by p24 ELISA. Leptomycin B, an inhibitor of the Crm1 pathway that inhibits Rev-dependent export, was included as a positive control. The cytotoxicity of each compound was determined in parallel via MTT-based cell viability assays.
U1 activation assay.
U1 cells obtained from the AIDS Research and Reference Reagent Program were maintained under standard culture conditions in RPMI 1640 medium supplemented with 10% fetal bovine serum (heat inactivated), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. U1 cells contain an integrated copy of a phorbol ester-inducible HIV-1 provirus, and addition of phytohemagglutinin (PHA) to the cell culture was used to induce virus production. On the day of the assay, the U1 cells were activated with PHA, and 2.5 × 105 cells were plated in 96-well plates. Test compounds diluted in medium were immediately applied to the cells at final concentrations of from 1 to 10,000 nM. Cultures were incubated for 3 days, and supernatants were harvested. Cell-free virus production was measured by p24 ELISA in culture supernatants. Compound toxicity was determined by MTT cell viability assay in parallel assays.
Virus spreading assay.
Jurkat (E6-1) cells (NIH AIDS Reagent Program) were cultured in RPMI medium supplemented with 25 mM HEPES, pH 7.4, 10% heat-inactivated fetal calf serum (HyClone, Waltham MA), and 1% penicillin-streptomycin. A total of 2.5 × 105 Jurkat cells were inoculated with 250 pg of p24 in 250 μl of medium in a 96-well flat-bottom polystyrene cell culture microplate. Input virus was removed after 18 h via washing cells in phosphate-buffered saline (PBS). Cells were resuspended in 250 μl of medium containing compounds at the concentrations described in Fig. 9. Cells were incubated at 37°C in 5% CO2, supernatant samples were removed every 48 h, and medium was replaced with fresh medium containing the compounds. Viral replication was determined using an ELISA to quantify p24 viral capsid protein in the culture supernatant. Mouse monoclonal and rabbit polyclonal anti-p24 antibodies used in the ELISA were obtained from the NIH AIDS Reagent Program. Toxicity was monitored using the MTT cell viability assay.
HIV-1 replication assay in PBMCs.
Virus isolates were obtained from the NIH AIDS Reagent Program. Human PBMCs, seronegative for HIV and hepatitis B virus (Astarte Biologics, LLC), were stimulated in R-3 medium (RPMI 1640 medium with 25 mM HEPES and l-glutamine, 20% heat-inactivated fetal bovine serum, 1% penicillin-streptomycin, 20 IU/ml interleukin-2 [IL-2]) with 5 μg/ml phytohemagglutinin (PHA) for 48 to 72 h. Stimulated PBMCs were resuspended and diluted in fresh R-3 medium and added to 96-well plates at 5 × 104 cells/well. The 50% tissue culture infective dose (TCID50) of each virus stock was measured by endpoint dilution assay as described by Reed and Muench (44). Spinoculation (1,200 × g, 2 h) was applied to improve the efficiency of infection.
Cells were infected with 40 TCID50s of virus stock under spinoculation in the presence of different concentrations of test compounds (biological quintuplicate wells/concentration) and incubated at 37°C in 5% CO2 for 7 days. On day 4, half of the supernatant was removed and replaced with fresh R-3 medium containing the appropriate concentration of test compounds. On day 7, cell-free supernatant samples were collected for analysis of p24 antigen expression measured by ELISA as described above. Antiviral activity was assessed by the inhibition of p24 expression.
In vitro ADME assays.
Aqueous solubility studies at pH 7.4 in phosphate buffer, Caco-2 permeability studies, and human liver microsome (HLM) stability studies were performed by Absorption Systems (Exton, PA) using their standard methods.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Rekosh and Marie-Louise Hammarskjold of the University of Virginia for providing the Rev reporter construct and control small molecules, Kip Guy for critical help in designing the library screen, and members of the Frankel Lab for providing critical feedback and comments.
This work was supported by National Institutes of Health grants CA103407 to A.D.F. and R.L.N., AI076143 to R.L.N. and A.D.F., AI076087 to A.R.R. and R.L.N., and P50GM082250 to A.D.F., by a U.S. Treasury Department Qualified Therapeutic Discovery Grant to Advanced Genetics Systems, and by China Scholarship Council financial support to S.Y.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02366-16.
REFERENCES
- 1.Cullen BR. 2003. Nuclear mRNA export: insights from virology. Trends Biochem Sci 28:419–424. doi: 10.1016/S0968-0004(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 2.Pollard VW, Malim MH. 1998. The HIV-1 Rev protein. Annu Rev Microbiol 52:491–532. doi: 10.1146/annurev.micro.52.1.491. [DOI] [PubMed] [Google Scholar]
- 3.Feinberg MB, Jarrett RF, Aldovini A, Gallo RC, Wong-Staal F. 1986. HTLV-III expression and production involve complex regulation at the levels of splicing and translation of viral RNA. Cell 46:807–817. doi: 10.1016/0092-8674(86)90062-0. [DOI] [PubMed] [Google Scholar]
- 4.Fernandes J, Jayaraman B, Frankel A. 2012. The HIV-1 Rev response element: an RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol 9:6–11. doi: 10.4161/rna.9.1.18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daugherty MD, Liu B, Frankel AD. 2010. Structural basis for cooperative RNA binding and export complex assembly by HIV Rev. Nat Struct Mol Biol 17:1337–1342. doi: 10.1038/nsmb.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booth DS, Cheng Y, Frankel AD. 2014. The export receptor Crm1 forms a dimer to promote nuclear export of HIV RNA. eLife 3:e04121. doi: 10.7554/eLife.04121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fornerod M, Ohno M, Yoshida M, Mattaj IW. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060. doi: 10.1016/S0092-8674(00)80371-2. [DOI] [PubMed] [Google Scholar]
- 8.Kjems J, Brown M, Chang DD, Sharp PA. 1991. Structural analysis of the interaction between the human immunodeficiency virus Rev protein and the Rev response element. Proc Natl Acad Sci U S A 88:683–687. doi: 10.1073/pnas.88.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malim MH, Tiley LS, McCarn DF, Rusche JR, Hauber J, Cullen BR. 1990. HIV-1 structural gene expression requires binding of the Rev trans-activator to its RNA target sequence. Cell 60:675–683. doi: 10.1016/0092-8674(90)90670-A. [DOI] [PubMed] [Google Scholar]
- 10.Jayaraman B, Crosby DC, Homer C, Ribeiro I, Mavor D, Frankel AD. 2014. RNA-directed remodeling of the HIV-1 protein Rev orchestrates assembly of the Rev-Rev response element complex. eLife 3:e04120. doi: 10.7554/eLife.04120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baba M, Pauwels R, Balzarini J, Arnout J, Desmyter J, De Clercq E. 1988. Mechanism of inhibitory effect of dextran sulfate and heparin on replication of human immunodeficiency virus in vitro. Proc Natl Acad Sci U S A 85:6132–6136. doi: 10.1073/pnas.85.16.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Battiste JL, Mao H, Rao NS, Tan R, Muhandiram DR, Kay LE, Frankel AD, Williamson JR. 1996. Alpha helix-RNA major groove recognition in an HIV-1 rev peptide-RRE RNA complex. Science 273:1547–1551. doi: 10.1126/science.273.5281.1547. [DOI] [PubMed] [Google Scholar]
- 13.Ye X, Gorin A, Ellington AD, Patel DJ. 1996. Deep penetration of an alpha-helix into a widened RNA major groove in the HIV-1 rev peptide-RNA aptamer complex. Nat Struct Biol 3:1026–1033. doi: 10.1038/nsb1296-1026. [DOI] [PubMed] [Google Scholar]
- 14.Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. 2010. HIV Rev response element (RRE) directs assembly of the Rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci U S A 107:12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daugherty MD, D'Orso I, Frankel AD. 2008. A solution to limited genomic capacity: using adaptable binding surfaces to assemble the functional HIV Rev oligomer on RNA. Mol Cell 31:824–834. doi: 10.1016/j.molcel.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jain C, Belasco JG. 2001. Structural model for the cooperative assembly of HIV-1 Rev multimers on the RRE as deduced from analysis of assembly-defective mutants. Mol Cell 7:603–614. doi: 10.1016/S1097-2765(01)00207-6. [DOI] [PubMed] [Google Scholar]
- 17.Tan R, Frankel AD. 1998. A novel glutamine-RNA interaction identified by screening libraries in mammalian cells. Proc Natl Acad Sci U S A 95:4247–4252. doi: 10.1073/pnas.95.8.4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landt SG, Tan R, Frankel AD. 2000. Screening RNA-binding libraries using a Tat-fusion system in mammalian cells. Methods Enzymol 318:350–363. doi: 10.1016/S0076-6879(00)18062-0. [DOI] [PubMed] [Google Scholar]
- 19.Rosen CA, Sodroski JG, Haseltine WA. 1985. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 41:813–823. doi: 10.1016/S0092-8674(85)80062-3. [DOI] [PubMed] [Google Scholar]
- 20.Roy S, Delling U, Chen CH, Rosen CA, Sonenberg N. 1990. A bulge structure in HIV-1 TAR RNA is required for Tat binding and Tat-mediated trans-activation. Genes Dev 4:1365–1373. doi: 10.1101/gad.4.8.1365. [DOI] [PubMed] [Google Scholar]
- 21.Kao SY, Calman AF, Luciw PA, Peterlin BM. 1987. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 22.Feinberg MB, Baltimore D, Frankel AD. 1991. The role of Tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc Natl Acad Sci U S A 88:4045–4049. doi: 10.1073/pnas.88.9.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marciniak RA, Sharp PA. 1991. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J 10:4189–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Southgate C, Zapp ML, Green MR. 1990. Activation of transcription by HIV-1 Tat protein tethered to nascent RNA through another protein. Nature 345:640–642. doi: 10.1038/345640a0. [DOI] [PubMed] [Google Scholar]
- 25.Selby MJ, Peterlin BM. 1990. Trans-activation by HIV-1 tat via a heterologous RNA binding protein. Cell 62:769–776. doi: 10.1016/0092-8674(90)90121-T. [DOI] [PubMed] [Google Scholar]
- 26.Calnan BJ, Tidor B, Biancalana S, Hudson D, Frankel AD. 1991. Arginine-mediated RNA recognition: the arginine fork. Science 252:1167–1171. doi: 10.1126/science.252.5009.1167. [DOI] [PubMed] [Google Scholar]
- 27.Chen L, Frankel AD. 1994. An RNA-binding peptide from bovine immunodeficiency virus Tat protein recognizes an unusual RNA structure. Biochemistry 33:2708–2715. doi: 10.1021/bi00175a046. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Frankel AD. 1995. A peptide interaction in the major groove of RNA resembles protein interactions in the minor groove of DNA. Proc Natl Acad Sci U S A 92:5077–5081. doi: 10.1073/pnas.92.11.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan R, Frankel AD. 1994. Costabilization of peptide and RNA structure in an HIV Rev peptide-RRE complex. Biochemistry 33:14579–14585. doi: 10.1021/bi00252a025. [DOI] [PubMed] [Google Scholar]
- 30.Smith CA, Calabro V, Frankel AD. 2000. An RNA-binding chameleon. Mol Cell 6:1067–1076. doi: 10.1016/S1097-2765(00)00105-2. [DOI] [PubMed] [Google Scholar]
- 31.Peled-Zehavi H, Smith CA, Harada K, Frankel AD. 2000. Screening RNA-binding libraries by transcriptional antitermination in bacteria. Methods Enzymol 318:297–308. doi: 10.1016/S0076-6879(00)18059-0. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura RL, Landt SG, Mai E, Nejim J, Chen L, Frankel AD. 2012. A cell-based method for screening RNA-protein interactions: identification of constitutive transport element-interacting proteins. PLoS One 7:e48194. doi: 10.1371/journal.pone.0048194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shuck-Lee D, Chen FF, Willard R, Raman S, Ptak R, Hammarskjold ML, Rekosh D. 2008. Heterocyclic compounds that inhibit Rev-RRE function and human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother 52:3169–3179. doi: 10.1128/AAC.00274-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeJong ES, Chang CE, Gilson MK, Marino JP. 2003. Proflavine acts as a Rev inhibitor by targeting the high-affinity Rev binding site of the Rev responsive element of HIV-1. Biochemistry 42:8035–8046. doi: 10.1021/bi034252z. [DOI] [PubMed] [Google Scholar]
- 35.Zheng GZ, Bhatia P, Daanen J, Kolasa T, Patel M, Latshaw S, El Kouhen OF, Chang R, Uchic ME, Miller L, Nakane M, Lehto SG, Honore MP, Moreland RB, Brioni JD, Stewart AO. 2005. Structure-activity relationship of triazafluorenone derivatives as potent and selective mGluR1 antagonists. J Med Chem 48:7374–7388. doi: 10.1021/jm0504407. [DOI] [PubMed] [Google Scholar]
- 36.Stewart AO, Bhatia PA, McCarty CM, Patel MV, Staeger MA, Arendsen DL, Gunawardana IW, Melcher LM, Zhu GD, Boyd SA, Fry DG, Cool BL, Kifle L, Lartey K, Marsh KC, Kempf-Grote AJ, Kilgannon P, Wisdom W, Meyer J, Gallatin WM, Okasinski GF. 2001. Discovery of inhibitors of cell adhesion molecule expression in human endothelial cells. 1. Selective inhibition of ICAM-1 and E-selectin expression. J Med Chem 44:988–1002. [DOI] [PubMed] [Google Scholar]
- 37.Ryckmans T, Edwards MP, Horne VA, Correia AM, Owen DR, Thompson LR, Tran I, Tutt MF, Young T. 2009. Rapid assessment of a novel series of selective CB(2) agonists using parallel synthesis protocols: a lipophilic efficiency (LipE) analysis. Bioorg Med Chem Lett 19:4406–4409. doi: 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
- 38.Hope TJ, Huang XJ, McDonald D, Parslow TG. 1990. Steroid-receptor fusion of the human immunodeficiency virus type 1 Rev transactivator: mapping cryptic functions of the arginine-rich motif. Proc Natl Acad Sci U S A 87:7787–7791. doi: 10.1073/pnas.87.19.7787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morwick T, Berry A, Brickwood J, Cardozo M, Catron K, DeTuri M, Emeigh J, Homon C, Hrapchak M, Jacober S, Jakes S, Kaplita P, Kelly TA, Ksiazek J, Liuzzi M, Magolda R, Mao C, Marshall D, McNeil D, Prokopowicz A III, Sarko C, Scouten E, Sledziona C, Sun S, Watrous J, Wu JP, Cywin CL. 2006. Evolution of the thienopyridine class of inhibitors of IκB kinase-β: part I: hit-to-lead strategies. J Med Chem 49:2898–2908. doi: 10.1021/jm0510979. [DOI] [PubMed] [Google Scholar]
- 40.Shuck-Lee D, Chang H, Sloan EA, Hammarskjold ML, Rekosh D. 2011. Single-nucleotide changes in the HIV Rev-response element mediate resistance to compounds that inhibit Rev function. J Virol 85:3940–3949. doi: 10.1128/JVI.02683-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frankel AD, Pabo CO. 1988. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 42.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 43.Hope TJ, Bond BL, McDonald D, Klein NP, Parslow TG. 1991. Effector domains of human immunodeficiency virus type 1 Rev and human T-cell leukemia virus type I Rex are functionally interchangeable and share an essential peptide motif. J Virol 65:6001–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Hyg 27:493–497. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.