

ABSTRACT
Within the past few decades, the incidence and complexity of human fungal infections have increased, and therefore, the need for safer and more efficient, broad-spectrum antifungal agents is high. In the study described here, we characterized the new tetrazole-based drug candidate VT-1598 as an inhibitor of sterol 14α-demethylase (CYP51B) from the filamentous fungus Aspergillus fumigatus. VT-1598 displayed a high affinity of binding to the enzyme in solution (dissociation constant, 13 ± 1 nM) and in the reconstituted enzymatic reaction was revealed to have an inhibitory potency stronger than the potencies of all other simultaneously tested antifungal drugs, including fluconazole, voriconazole, ketoconazole, and posaconazole. The X-ray structure of the VT-1598/A. fumigatus CYP51 complex was determined and depicts the distinctive binding mode of the inhibitor in the enzyme active site, suggesting the molecular basis of the improved drug potency and broad-spectrum antifungal activity. These data show the formation of an optimized hydrogen bond between the phenoxymethyl oxygen of VT-1598 and the imidazole ring nitrogen of His374, the CYP51 residue that is highly conserved across fungal pathogens and fungus specific. Comparative structural analysis of A. fumigatus CYP51/voriconazole and Candida albicans CYP51/VT-1161 complexes supports the role of H bonding in fungal CYP51/inhibitor complexes and emphasizes the importance of an optimal distance between this interaction and the inhibitor-heme iron interaction. Cellular experiments using two A. fumigatus strains (strains 32820 and 1022) displayed a direct correlation between the effects of the drugs on CYP51B activity and fungal growth inhibition, indicating the noteworthy anti-A. fumigatus potency of VT-1598 and confirming its promise as a broad-spectrum antifungal agent.
KEYWORDS: fungal infections, Aspergillus fumigatus, VT-1598, sterol 14α-demethylase (CYP51), inhibition, X-ray structure
INTRODUCTION
About 300 fungal species can cause disease in humans (https://www.cdc.gov/fungal/diseases/). The diseases vary from those that are relatively less serious but that at times include tenacious superficial infections of the skin and nails that affect ∼25% of the population worldwide to life-threatening invasive infections that kill 1.5 million to 2 million people each year (1, 2). Despite recent advances in prevention and diagnosis, the incidence of fungal infections continues to rise, particularly due to the increase in the number of immunocompromised patients (3–6). Available clinical antifungal agents include echinocandins, polyenes, and azoles. Echinocandins damage the fungal cell wall, and polyenes remove ergosterol from fungal membranes, disrupting their structure, while azoles (inhibitors of the fungal cytochrome P450 enzyme sterol 14α-demethylase [CYP51, EC 1.14.13.70]) block the biosynthesis of ergosterol de novo, thus depleting the source of ergosterol for the membranes and preventing the formation of regulatory sterols, which are required for cell cycle regulation, cell development, and multiplication (7). Although azoles [1-imidazole and 1-(1,2,4-triazole) derivatives] represent the major class of antifungal drugs (2, 8), each of them still has limitations, such as spectrum of activity, resistance, side effects, drug-drug interactions, or pharmacokinetic profile (3). Consequently, the treatment efficiency remains unacceptably low (the rates of mortality from invasive fungal infections often exceed 50% [1, 2]), and the outcomes strongly depend on how quickly the patient is diagnosed and treated (1, 4). New, safer, and more efficient drugs which would preferably have broad-spectrum antifungal activity are highly needed (9).
Within the past few years, Viamet Pharmaceuticals has successfully developed novel and highly promising 1-tetrazole-based antifungal drug candidates (including VT-1161 for candidiasis [10], VT-1129 for cryptococcosis [11], and VT-1598 for coccidioidomycosis [12]). Because of the use of a tetrazole ring, which represents a metal-binding group less avid than the triazole metal-binding group, these compounds act as selective fungal CYP51-targeting inhibitors with very few off-target effects on human hepatic cytochrome P450 enzymes. This improved selectivity should translate into a lower propensity to cause side effects, such as drug-drug interactions and liver toxicity (13–15; C. M. Yates, E. P. Garvey, S. R. Shaver, R. S. Schotzinger, and W. J. Hoekstra, submitted for publication).
In the study described here, we characterized VT-1598 as a ligand and inhibitor of sterol 14α-demethylase from the pathogenic filamentous fungus Aspergillus fumigatus and determined the X-ray costructure of the enzyme/inhibitor complex. The structure shows that, in addition to the coordination bond with the catalytic heme iron and multiple hydrophobic contacts with the protein moiety (the interactions typical for other azole-based CYP51 inhibitors), the VT-1598/CYP51 complex is stabilized by a hydrogen bond between the oxygen of the inhibitor and the nitrogen of His374 of the enzyme, the residue that is highly conserved across the fungal kingdom and fungus specific. The correlation between the inhibitory effects of the drugs on the CYP51B activity and A. fumigatus cellular growth inhibition confirms that VT-1598 is a promising antifungal agent with potent activity against the most prevalent pathogen of human invasive mold infections.
RESULTS
VT-1598 displays a high affinity of binding to A. fumigatus CYP51B.
Upon titration with VT-1598, A. fumigatus CYP51B produces the characteristic type 2 spectral response (16), a red shift in the porphyrin Soret band maximum from 417 to 422 nm (Fig. 1). This shift is indicative of water expulsion and coordination of a moderately basic (17) tetrazole nitrogen of VT-1598 to the distal (axial) face of the cytochrome P450 heme iron. The low nanomolar value of the enzyme-ligand dissociation constant (Kd; 13 ± 1 nM) indicates a strong affinity between VT-1598 and the A. fumigatus CYP51 enzyme. For comparison, the Kd values for complexes of A. fumigatus CYP51B with the first-line clinical antiaspergillosis drug voriconazole and another tetrazole-based CYP51 inhibitor from Viamet (VT-1161), determined under the same experimental conditions, were 56 ± 4 nM (18) and 47 ± 11 nM (19), respectively.
FIG 1.
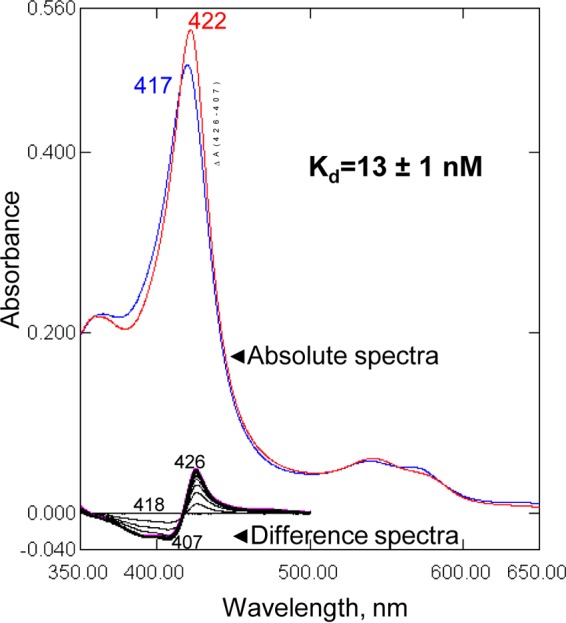
Spectral response of A. fumigatus CYP51B to the addition of VT-1598. Type 2 shifts in the Soret band maximum in the absolute (top, red line) and difference (bottom) absorbance spectra are shown. The P450 concentration was 0.8 μM, the titration step was 0.1 μM, and the optical path length was 5 cm. (Inset) Titration curve. The data for the ligand-induced absorbance changes versus the ligand concentration were fitted to the quadratic equation in GraphPad Prism software, as described in Materials and Methods.
VT-1598 acts as a potent inhibitor of A. fumigatus CYP51B enzymatic activity.
VT-1598 completely blocked the initial rate of the A. fumigatus CYP51B reaction at a 1:1 molar ratio of inhibitor/enzyme (data not shown). Because we previously observed such a behavior for many inhibitors of other CYP51 orthologs (20, 21), in the present study we used a long-term reaction (1 h) and a 2-fold molar excess of the inhibitor over the enzyme, as these conditions allow screening out of the inhibitors that can be replaced in the CYP51 active site by the substrate over time and thus reveal the most potent compounds (18, 22). Figure 2 shows that VT-1598 had the strongest inhibitory effect on the activity of A. fumigatus CYP51B: 100% inhibition of substrate conversion at a P450/inhibitor/eburicol molar ratio of 1:2:50. It was followed by voriconazole (93% inhibition), posaconazole (89% inhibition), and VT-1161 (84% inhibition). Consistent with their known activity against A. fumigatus, ketoconazole and fluconazole were significantly less effective inhibitors. Thus, the potency of VT-1598 was found to be higher than the potencies of all tested clinical antifungal azoles.
FIG 2.

Comparative inhibitory effects of VT-1598, VT-1161, and clinical antifungal drugs on the activity of A. fumigatus CYP51B. The molar ratio of enzyme/inhibitor/substrate was 1:2:50, and the P450 concentration was 0.5 μM (60-min reaction). The experiments were performed in triplicate, and the results are presented as means ± SDs.
Structural basis for the potent inhibitory effect of VT-1598.
In order to understand the molecular basis of the VT-1598 inhibitory potency, the crystal structure of its complex with A. fumigatus CYP51B was determined. For crystallization purposes, the 49-amino-acid membrane anchor sequence at the N terminus of A. fumigatus CYP51B was replaced with the 5-amino-acid sequence fragment MAKKT- and the 6 amino acid residues at the C terminus (-ESATKA) were replaced with a 6His tag (18). To obtain the enzyme/inhibitor complex, VT-1598 was added to the buffer solution upon the last step of the purification procedure, as described in Materials and Methods. The complex was crystallized in the hexagonal P3121 space group and diffracted to a maximum resolution of 2.99 Å (Table 1). The asymmetric unit consisted of one monomer (Fig. 3a), and the protein chain was seen from Lys50 (KT- in the N-terminal MAKKT- sequence) to His519 (the C-terminal His tag). The electron density for VT-1598 was well defined, showing full occupancy and a single orientation of the inhibitor molecule within the CYP51 binding cavity (Fig. 3b).
TABLE 1.
Data collection and refinement statistics
| Parametera | Values(s) for A. fumigatus CYP51B- VT-1598 |
|---|---|
| Data collection statistics | |
| Wavelength (Å) | 0.97856 |
| Space group | P3121 |
| Unit cell dimensions | |
| a, b, c (Å) | 109.86, 109.86, 84.838 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| No. of molecules per asymmetric unit | 1 |
| No. of reflections | 12,238 |
| Resolution (outer shell) (Å) | 95.15–2.99 (3.07–2.99)b |
| Rmerge (outer shell) | 0.033 (0.648)b |
| I/σ (outer shell) | 26.1 (1.9)b |
| Completeness (outer shell) (%) | 99.4 (100.0)b |
| Redundancy (outer shell) | 5.4 (5.6)b |
| Refinement statistics | |
| Rwork | 0.258 |
| Rfree | 0.280 |
| RMSD from ideal geometry | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.754 |
| Ramachandran plot (%) | |
| Residues in favorable/allowed regions | 95.7/99.8 |
| Outliers | 0.2 |
| No. of atoms (mean B-factor [Å2]) | 3,838 (153.7) |
| No. of residues per molecule (mean B-factor [Å2]) | |
| Protein | 470 (155.6) |
| Heme | 1 (139.6) |
| Ligand | VT2 1 (140.8) |
| PDB accession no. | 5FRB |
I, intensity of the a reflection; RMSD, root mean square deviation.
The values in parentheses represent the highest-resolution shell.
FIG 3.

Complex of A. fumigatus CYP51 with VT-1598. (a) Overall view in a ribbon representation (top) and surface representation (bottom). (b) 2Fo − Fc electron density map (gray mesh) of VT-1598 contoured at 1.2σ. (c) Enlarged view of the binding site. Eighteen VT-1598-contacting residues (<4.5 Å from the inhibitor) and the corresponding secondary structural elements are marked. The H bond is depicted as a pink dashed line. The heme iron coordination bonds are depicted as orange wires. VT-1598 and amino acid side chains are presented as stick models, and the heme (except for panel c) is shown as spheres. The carbon atoms of the inhibitor are green; the carbon atoms of the enzyme are gray. The color code and orientation are the same in all panels.
The N-4 atom of the tetrazole ring of VT-1598 forms the sixth axial coordination bond with the heme iron on the distal surface of the porphyrin plane (Fig. 3c), with the fifth axial coordination bond (on the proximal surface of the heme plane) being formed with the thiolate ion of Cys463. Interestingly, considering that the relatively lower electronegativity of the tetrazole ring N-4 atom would predict a greater distance between atoms, the length of the Fe–VT-1598 coordination bond is 2.1 Å, which is more typical for coordination bonds between the CYP51 iron and 1,2,4-triazoles (13, 17, 22). The difluorinated β-phenyl ring of VT-1598 protrudes deeper into the CYP51 substrate binding cavity, while the long arm lies within the substrate access channel, with its benzonitrile portion being seen above the surface of the channel entry (Fig. 3a, bottom). VT-1598 forms van der Waals contacts with 19 amino acid residues of A. fumigatus CYP51B (Fig. 3c): Y68 and G69 from helix A′; Y122, T126, and F130 from helix B′; V135 and Y136 from the αB″ turn (P450 substrate recognition site 1 [SRS1] [23]); F234 from helix F″ (SRS2); A303, A307, and S311 from helix I (SRS4); H374 and S375 from the αK/β1-4 loop; M360 from β1-4 (SRS5); and F504, Y500, S501, S502, and L503 from the β4 hairpin (SRS6), with the αA′, αF″, and β4 hairpins forming the access channel entry. In addition, the CYP51/VT-1598 interaction is strengthened by the H bond between the imidazole ring of H374 (proton donor) and the phenoxymethyl oxygen of VT-1598 (proton acceptor) (Fig. 3c). Thus, the tight binding of VT-1598 to A. fumigatus CYP51B and its profound inhibitory effect on the activity of the enzyme are due to the combination of its (i) coordination bond with the iron of the heme moiety, (ii) van der Waals interactions with the 19 amino acid residues of the protein moiety, and (iii) the hydrogen bond with H374 (2.96 Å, >4 kcal/mol), which is at least 1 order of magnitude stronger than the average strength of a van der Waals contact.
A. fumigatus cellular growth inhibition.
The potencies of VT-1598, voriconazole, and VT-1161 against A. fumigatus cells were tested using two strains of the fungus (strains 1022 and 32820) (Fig. 4), and with both strains, cell growth inhibition was found to correlate with the inhibitory effects of the compounds on CYP51B activity. VT-1598 inhibition was essentially the same as that observed with the first-line antiaspergillosis drug voriconazole, and the levels of inhibition provided by both VT-1598 and voriconazole were superior to the level of inhibition provided by VT-1161.
FIG 4.

Inhibitory effects of VT-1598, VT-1161, and voriconazole at a 50 μM concentration on A. fumigatus cellular growth. A. fumigatus strains 1022 and 32820 were exposed to the drugs in 96-well plate cultures for 24 h, and then alamarBlue was added to the cultures for 6 h to evaluate fungal growth. Experiments were performed in triplicate, and the bars represent the means ± SEs from triplicate experiments. The P values were determined using two-way analysis of variance.
DISCUSSION
Unlike the vast majority of nonphotosynthetic species (24), A. fumigatus has two CYP51 genes, each of which is present as a single copy (chromosomes 4 and 7) and which are named CYP51A and CYP51B, respectively (25). The CYP51B gene is expressed constitutively, is found in all sequenced fungi, and has been reported to encode the enzyme primarily responsible for sterol 14α-demethylation, while the CYP51A gene, which appears in some filamentous ascomycetes, was suggested to be expressed only under evolutionary pressure (26, 27). The presence of two CYP51 genes implies a possibility for the faster biosynthesis of ergosterol in A. fumigatus and might be one of the reasons for the high level of resistance of this pathogen to treatment (18). The fungal growth data (i) support the importance of the A. fumigatus CYP51B enzyme, (ii) suggest that VT-1598 must inhibit CYP51A as well, and (iii) characterize VT-1598 as a promising drug candidate for the treatment of aspergillosis and a noteworthy alternative to voriconazole, which, in addition to the high frequency of A. fumigatus resistance to voriconazole (28), often produces adverse side effects (visual disturbances, skin rashes, hepatotoxicity, vomiting, abdominal pain, etc.) which result in the need for permanent therapeutic drug monitoring (29).
Interestingly, similar to VT-1598, the binding of voriconazole to A. fumigatus CYP51B also involves H-bond formation (between the nitrogen of the 5-fluoropyrimidine ring of voriconazole and the side chain hydroxyl oxygen of Tyr122 [18]). The crucial role of H bonding in strengthening the CYP51/inhibitor complexes was first discovered for the VNI scaffold inhibitors of protozoan CYP51 (21, 30). A strong H bond (2.8 Å) between the trifluoroethoxyphenyl oxygen of VT-1161 and His377 was found in its complex with Candida albicans CYP51 (22). Since His377 in C. albicans corresponds to His374 in A. fumigatus CYP51B (Fig. 5a and b) and VT-1161 is one of the strongest C. albicans CYP51 inhibitors identified (22), it is, at first glance, unclear why VT-1161 produces a weaker effect on the A. fumigatus ortholog (Fig. 2). Comparative structural analysis of these two complexes (A. fumigatus CYP51B/VT-1598 and C. albicans CYP51/VT-1161) suggests that this might be associated with the relatively shorter length of the VT-1161 fragment connecting the heme coordinating nitrogen and the H-bond-forming oxygen (13.1 Å with VT-1161 versus 14.4 Å with VT-1598 [Fig. 5a and b]). Indeed, modeling of VT-1161 into the A. fumigatus CYP51B active site shows that the distance between the VT-1161 oxygen and the nitrogen of A. fumigatus CYP51B His374 is likely much greater than 3 Å, so the H bond would be either weak at best or not formed at all (Fig. 5c). Thus, the longer fragment of VT-1598 apparently provides a better opportunity for optimized H-bond formation, which is also supported by the high affinity of VT-1598 binding to C. albicans CYP51 (Kd = 25 nM). This feature is likely highly advantageous for broad-spectrum antifungal activity because the histidine residue is conserved among fungal sterol 14α-demethylases (including CYP51A sequences) and is fungi specific (Fig. 6, see also Fig. 5d). This finding is consistent with the broadly acting and highly selective antifungal activity of VT-1598 (C. M. Yates, E. P. Garvey, S. R. Shaver, R. S. Schotzinger, and W. J. Hoekstra, unpublished data). Given this profile, VT-1598 has the potential to become a life-saving drug that could be applied in a timely fashion for the treatment of various invasive fungal infections.
FIG 5.

Structural basis for the broad-spectrum antifungal activity of VT-1598. (a) VT-1598 bound in the active site of A. fumigatus CYP51B; (b) VT-1161 bound in the active site of C. albicans CYP51 (PDB accession number 5TZ1); (c) VT-1161 (cyan) modeled into the active site of A. fumigatus CYP51B (tan); (d) VT-1598 in the model of A. fumigatus CYP51A (aquamarine). The oxygen atom of each inhibitor that forms an H bond with the histidine residue of the protein and the heme iron coordinating nitrogens (in a to c) are circled (in green); the distances are shown (orange); the color code and orientation are the same in all panels.
FIG 6.
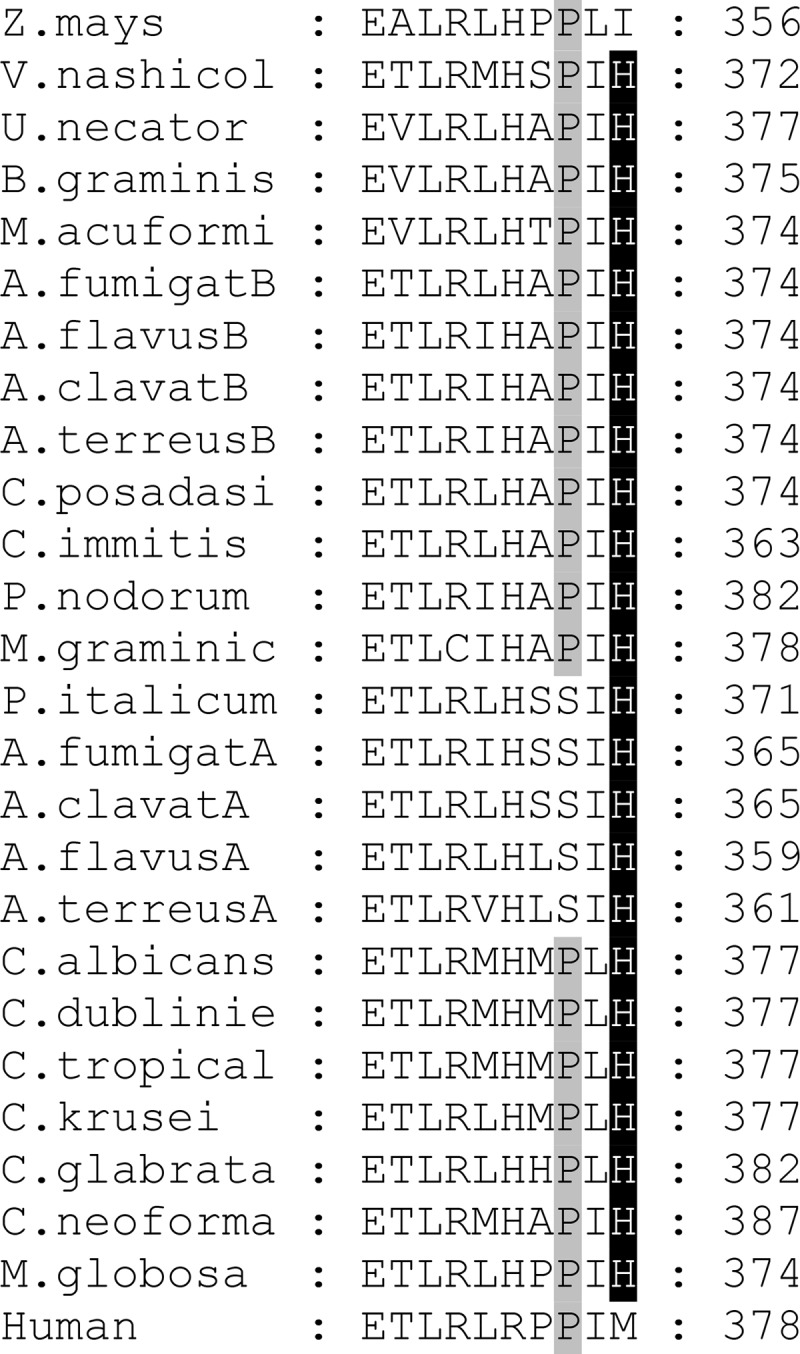
A fragment of multiple-sequence alignment showing the conservation of His374 (A. fumigatus CYP51B numbering) in fungal CYP51 enzymes. Human and Zea mays CYP51 sequences are included for the comparison with fungal CYP51 enzyme sequences. The type B (versus type A) CYP51-defining proline is shadowed in gray. The sequences were aligned using the Clustal Omega program, and the figure was prepared in the GeneDoc program. Z. mays, Zea mays; V.nashicol, Venturia nashicola; U.necator, Uncinula necator; B.graminis, Blumeria graminis; M.acuformi, Melanella acuformis; A.fumigatB, Aspergillus fumigatus B; A.flavusB, Aspergillus flavus B; A.clavatB, Aspergillus clavatus B; A.terreusB, Aspergillus terreus B; C.posadasi, Coccidioides posadasii; C.immitis, Coccidioides immitis; F.nodorum, Fusarium nodorum; M.graminic, Mycosphaerella graminicola; P.italicum, Penicillium italicum; A.fumigatA, Aspergillus fumigatus A; A.clavatA, Aspergillus clavatus A; A.flavusA, Aspergillus flavus A; A.terreusA, Aspergillus terreus A; C.albicans, Candida albicans; C.dublinie, Candida dubliniensis; C.tropical, Candida tropicalis; C.krusei, Candida krusei; C.glabrata, Candida glabrata; C.neoforma, Cryptococcus neoformans; M.globosa, Malassezia globosa.
MATERIALS AND METHODS
Reagents.
VT-1598 and VT-1161 were synthesized by Viamet Pharmaceuticals. Voriconazole, ketoconazole, and posaconazole were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); fluconazole was from ICN Biomedicals. Hydroxypropyl-β-cyclodextrin (HPCD) was purchased from Cyclodextrin Technology Development (Gainesville, FL). A. fumigatus CYP51B (the full-length and truncated constructs) and rat NADPH-cytochrome P450 reductase (CPR) were expressed in Escherichia coli and purified as described previously (18). The full-length protein was used for functional studies, including ligand binding, reconstitution of enzymatic activity, and inhibition. The truncated protein was used for crystallization purposes.
Spectroscopic measurements and ligand binding assay.
UV-visible absorption spectra were recorded using a dual-beam Shimadzu UV-2401PC spectrophotometer in 50 mM potassium phosphate buffer (pH 7.2) containing 10% (vol/vol) glycerol and 0.1% (vol/vol) Triton X-100. P450 concentrations were estimated from the Soret band intensity using a ε417 value of 117 mM−1 cm−1 for the low-spin ferric form of the protein or a Δε450–490 value of 91 mM−1 cm−1 for the reduced carbon monoxide difference spectra (18, 31). Titration with VT-1598 was carried out at an ∼0.8 μM P450 concentration in 5-cm-optical-path-length cuvettes. Difference spectra were generated by recording the absorbance of P450 in a sample cuvette versus the absorbance in a reference cuvette; both cuvettes contained the same amount of the protein. Aliquots of VT-1598 (dissolved in dimethyl sulfoxide [DMSO]) were added to the sample cuvette over a concentration range 0.1 to 1.3 μM, with each titration step being 0.1 μM. At each step, the corresponding volume of DMSO was added to the reference cuvette. The apparent dissociation constants (Kds) of the enzyme-ligand complex were calculated in GraphPad Prism (version 6) software by fitting the data for the ligand-induced changes in the absorbance of the difference spectra [Δ(Amax − Amin)], which is the difference in the maximum absorbance (Amax) and the minimum absorbance (Amin), versus the ligand concentration to the quadratic equation in equation 1 (tightly binding ligands):
| (1) |
where ΔA and ΔAmax are the change in the absorbance and the change in the maximum absorbance, respectively, and L and E are the total concentrations of ligand and enzyme used for the titration, respectively.
Reconstitution of catalytic activity and inhibition assay.
The standard reaction mixture contained 0.5 μM A. fumigatus CYP51B, 1.0 μM rat CPR, 100 μM l-α-1,2-dilauroyl-sn-glycerophosphocholine, 0.4 mg/ml isocitrate dehydrogenase, and 25 mM sodium isocitrate in 50 mM potassium phosphate buffer (pH 7.2) containing 10% (vol/vol) glycerol and 0.1% (vol/vol) Triton X-100 (18). After addition of the radiolabeled ([3-3H]) sterol substrate (eburicol at ∼4,000 dpm/nmol dissolved in 45% [wt/vol] HPCD; final concentration, 25 μM) (32) and an inhibitor (from a 0.2 mM stock solution in DMSO; final concentration, 1 μM), the mixture was preincubated for 30 s at 37°C in a shaking water bath, and the reaction was initiated by the addition of 100 μM NADPH and stopped by the extraction of the sterols with 5 ml of ethyl acetate. The extracted sterols were dried, dissolved in methanol, and analyzed by a reversed-phase high-performance liquid chromatography system (Waters) equipped with a β-RAM detector (IN/US Systems, Inc.) using a NovaPak octyldecyl silane (C18) column (particle size, 4 μm; 3.9 mm by 150 mm) and a linear gradient of water-acetonitrile-methanol (1.0:4.5:4.5, vol/vol/vol) (solvent A) to methanol (solvent B), increasing from 0 to 100% solvent B over 30 min at a flow rate of 1.0 ml/min. The inhibitory potencies of VT-1598 and the clinical antifungal azoles were compared as the percent inhibition of sterol 14α-demethylation in 60-min reactions. The experiments were performed in triplicate, and the results are presented as means ± standard deviations (SDs).
X-ray crystallography.
For crystallographic experiments, the truncated A. fumigatus CYP51B was diluted 10-fold with 20 mM potassium phosphate buffer (pH 7.2) containing 10% (vol/vol) glycerol, 0.1 mM EDTA, and 10 μM VT-1598 (CM-buffer) and applied to a CM-Sepharose column equilibrated with CM-buffer containing 50 mM NaCl. The column was washed with 5 bed volumes of equilibration buffer and then 40 bed volumes of CM-buffer with an increasing linear gradient of NaCl (50 to 200 mM). The protein was eluted with CM-buffer containing 350 mM NaCl, and the eluted proteins were pooled, concentrated to about 500 μM using an Amicon Ultra 50 K concentration device, frozen in liquid nitrogen, and stored at −80°C until use. The crystals were obtained at 16°C by the hanging-drop vapor diffusion technique. Equal volumes of complex solution preincubated with 24.5 mM n-octyl-β-d-glucoside and 5.8 mM tris(carboxyethyl)phosphine (TCEP) were mixed with the mother liquor (20% [wt/vol] polyethylene glycol 3350 and 0.2 M lithium acetate [pH 7.4]) and equilibrated against the reservoir solution. The crystals were cryoprotected by soaking them in the mother liquor with 40% (vol/vol) glycerol and flash-cooled in liquid nitrogen. The data were collected on the 21-ID-F beamline of the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL), at a wavelength of 0.9786 Å using a MAR225 charge-coupled-device detector. The diffraction images were integrated using the Mosflm program (33) and scaled with the Aimless program (CCP4 Program Suite, version 6.3.0 [34]). The structure was determined by molecular replacement in Phaser software (35), using the hemoprotein moiety from the complex of A. fumigatus CYP51B with voriconazole (PDB accession number 4UYM) as the search model. An iterative model of the protein–VT-1598 complex was then built with the Coot program (36) and refined with the Refmac5 program in the CCP4 suite (34). Data collection and refinement statistics are summarized in Table 1. The model of A. fumigatus CYP51A was built as described previously (18). Structural figures were prepared with the Chimera program.
A. fumigatus cellular growth inhibition assay.
The A. fumigatus cellular growth inhibition assay was performed following general procedures for evaluating fungal drug susceptibility as described previously (37). Briefly, A. fumigatus strains 1022 and 32820 (ATCC MP-12) were grown in potato-dextrose broth at 27°C for 48 h. From each broth, 100 μl of culture was diluted to 500 μl in 0.05% Tween 20–1× phosphate-buffered saline (PBS), plated on potato-dextrose agar plate, and incubated at 37°C for 48 h. Conidia from the strains were separated and diluted in 5 ml of 0.05% Tween 20–PBS. Each strain was seeded into a 96-well plate at a concentration of 1 × 103 spores. Various concentrations of VT-1598, VT-1161, and voriconazole ranging from 1 nM to 100 μM were added in triplicate into each well, and DMSO was used as a negative control. The plates were incubated at 37°C for 24 h, and 10% alamarBlue (Thermo Scientific) was added to the cultures for 6 h. Fluorescence was measured at an excitation wavelength of 570 nm and an emission wavelength of 600 nm using a fluorescence microplate reader. Each independent experiment was repeated in triplicate.
Accession number(s).
The coordinates and structure factors have been deposited in the Protein Data Bank (PDB) under accession number 5FRB.
ACKNOWLEDGMENTS
This work was supported by Viamet Pharmaceuticals, Inc. (Durham, NC), and by National Institutes of Health grant R01 GM067871 (to G.I.L.). Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at Sector 21 of the Advanced Photon Source (Argonne, IL). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract DE-AC02-06CH11357. Use of LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (grant 085P1000817).
REFERENCES
- 1.Brown GD, Denning DW, Gow NAR, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv113. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.Denning DW, Bromley MJ. 2015. How to bolster the antifungal pipeline. Science 347:1414–1416. doi: 10.1126/science.aaa6097. [DOI] [PubMed] [Google Scholar]
- 3.Wilson DT, Dimondi VP, Johnson SW, Jones TM, Drew RH. 2016. Role of isavuconazole in the treatment of invasive fungal infections. Ther Clin Risk Manag 12:1197–1206. doi: 10.2147/TCRM.S90335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, Zaoutis TE, Sobel JD. 2016. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis 62:e1–e50. doi: 10.1093/cid/civ933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mathew BP, Nath M. 2009. Recent approaches to antifungal therapy for invasive mycoses. ChemMedChem 4:310–323. doi: 10.1002/cmdc.200800353. [DOI] [PubMed] [Google Scholar]
- 6.Ramana KV, Kandi S, Bharatkumar P, Sharada CHV, Rao R, Mani R, Rao SD. 2013. Invasive fungal infections: a comprehensive review. Am J Infect Dis Microbiol 1:64–69. [Google Scholar]
- 7.Lepesheva GI, Waterman MR. 2007. Sterol 14alpha-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim Biophys Acta 1770:467–477. doi: 10.1016/j.bbagen.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lass-Flörl C. 2011. Triazole antifungal agents in invasive fungal infections. Drugs 71:2405–2419. doi: 10.2165/11596540-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 9.Chapman SW, Sullivan DC, Cleary JD. 2008. In search of the Holy Grail of antifungal therapy. Trans Am Clin Climatol Assoc 119:197–216. [PMC free article] [PubMed] [Google Scholar]
- 10.Garvey EP, Hoekstra WJ, Schotzinger RJ, Sobel JD, Lilly EA, Fidel PL. 2015. Efficacy of the clinical agent VT-1161 against fluconazole-sensitive and -resistant Candida albicans in a murine model of vaginal candidiasis. Antimicrob Agents Chemother 59:5567–5573. doi: 10.1128/AAC.00185-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lockhart SR, Fothergill AW, Iqbal N, Bolden CB, Grossman NT, Garvey EP, Brand SR, Hoekstra WJ, Schotzinger RJ, Ottinger E, Patterson TF, Wiederhold NP. 2016. The investigational fungal CYP51 inhibitor VT-1129 demonstrates potent in vitro activity against Cryptococcus neoformans and Cryptococcus gattii. Antimicrob Agents Chemother 60:2528–2531. doi: 10.1128/AAC.02770-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shubitz LF, Trinh H, Roy M, Garvey EP, Brand SR, Hoekstra WJ, Schotzinger RJ. 2016. Novel CYP51 inhibitors for the treatment of coccidioidomycosis, p 45 Proc 60th Annu Coccidioidomycosis Study Group, Fresno, CA. [Google Scholar]
- 13.Hoekstra WJ, Garvey EP, Moore WR, Rafferty SW, Yates CM, Schotzinger RJ. 2014. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg Med Chem Lett 24:3455–3458. doi: 10.1016/j.bmcl.2014.05.068. [DOI] [PubMed] [Google Scholar]
- 14.Warrilow AGS, Hull CM, Parker JE, Garvey EP, Hoekstra WJ, Moore WR, Schotzinger RJ, Kelly DE, Kelly SL. 2014. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob Agents Chemother 58:7121–7127. doi: 10.1128/AAC.03707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warrilow AGS, Parker JE, Price CL, Nes WD, Garvey EP, Hoekstra WJ, Schotzinger RJ, Kelly DE, Kelly SL. 2016. The investigational drug VT-1129 Is a highly potent inhibitor of Cryptococcus Species CYP51 but only weakly inhibits the human enzyme. Antimicrob Agents Chemother 60:4530–4538. doi: 10.1128/AAC.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schenkman JB, Remmer H, Estabrook RW. 1967. Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol Pharmacol 3:113–123. [PubMed] [Google Scholar]
- 17.Hargrove TY, Wawrzak Z, Alexander PW, Chaplin JH, Keenan M, Charman SA, Waterman MR, Chatelain E, Lepesheva GI. 2013. Complexes of Trypanosoma cruzi sterol 14α-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: structural basis for pathogen selectivity. J Biol Chem 288:31602–31615. doi: 10.1074/jbc.M113.497990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hargrove TY, Wawrzak Z, Lamb DC, Guengerich FP, Lepesheva GI. 2015. Structure-functional characterization of cytochrome P450 sterol 14α-demethylase (CYP51B) from Aspergillus fumigatus and molecular basis for the development of antifungal drugs. J Biol Chem 290:23916–23934. doi: 10.1074/jbc.M115.677310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoekstra WJ, Hargrove TY, Wawrzak Z, da Gama Jaen Batista D, da Silva CF, Nefertiti ASG, Rachakonda G, Schotzinger RJ, Villalta F, Soeiro MDNC, Lepesheva GI. 2016. Clinical candidate VT-1161's antiparasitic effect in vitro, activity in a murine model of Chagas disease, and structural characterization in complex with the target enzyme CYP51 from Trypanosoma cruzi. Antimicrob Agents Chemother 60:1058–1066. doi: 10.1128/AAC.02287-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lepesheva GI, Ott RD, Hargrove TY, Kleshchenko YY, Schuster I, Nes WD, Hill GC, Villalta F, Waterman MR. 2007. Sterol 14 alpha-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem Biol 14:1283–1293. doi: 10.1016/j.chembiol.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lepesheva GI, Park HW, Hargrove TY, Vanhollebeke B, Wawrzak Z, Harp JM, Sundaramoorthy M, Nes WD, Pays E, Chaudhuri M, Villalta F, Waterman MR. 2010. Crystal structures of Trypanosoma brucei sterol 14 alpha-demethylase and implications for selective treatment of human infections. J Biol Chem 285:1773–1780. doi: 10.1074/jbc.M109.067470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hargrove TY, Friggeri L, Wawrzak Z, Qi A, Hoekstra WJ, Schotzinger RJ, York JD, Guengerich FP, Lepesheva GI. 2017. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J Biol Chem 292:6728–6743. doi: 10.1074/jbc.M117.778308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotoh O. 1992. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem 267:83–90. [PubMed] [Google Scholar]
- 24.Cherkesova TS, Hargrove TY, Vanrell MC, Ges I, Usanov SA, Romano PS, Lepesheva GI. 2014. Sequence variation in CYP51A from the Y strain of Trypanosoma cruzi alters its sensitivity to inhibition. FEBS Lett 588:3878–3885. doi: 10.1016/j.febslet.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mellado E, Diaz-Guerra TM, Cuenca-Estrella M, Rodriguez-Tudela JL. 2001. Identification of two different 14-{alpha} sterol demethylase-related genes (cyp51A and cyp51B) in Aspergillus fumigatus and other Aspergillus species. J Clin Microbiol 39:2431–2438. doi: 10.1128/JCM.39.7.2431-2438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fan J, Urban M, Parker JE, Brewer HC, Kelly SL, Hammond-Kosack KE, Fraaije BA, Liu X, Cools HJ. 2013. Characterization of the sterol 14α-demethylases of Fusarium graminearum identifies a novel genus-specific CYP51 function. New Phytol 198:821–835. doi: 10.1111/nph.12193. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins NJ, Cools HJ, Sierotzki H, Shaw MW, Knogge W, Kelly SL, Kelly DE, Fraaije BA. 2014. Paralog re-emergence: a novel, historically contingent mechanism in the evolution of antimicrobial resistance. Mol Biol Evol 31:1793–1802. doi: 10.1093/molbev/msu134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolwijck E, van der Hoeven H, de Sévaux RG, ten Oever J, Rijstenberg LL, van der Lee HA, Zoll J, Melchers WJG, Verweij PE. 2016. Voriconazole-susceptible and voriconazole-resistant Aspergillus fumigatus coinfection. Am J Respir Crit Care Med 193:927–929. doi: 10.1164/rccm.201510-2104LE. [DOI] [PubMed] [Google Scholar]
- 29.Ashbee HR, Barnes RA, Johnson EM, Richardson MD, Gorton R, Hope WW. 2014. Therapeutic drug monitoring (TDM) of antifungal agents: guidelines from the British Society for Medical Mycology. J Antimicrob Chemother 69:1162–1176. doi: 10.1093/jac/dkt508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lepesheva GI, Hargrove TY, Rachakonda G, Wawrzak Z, Pomel S, Cojean S, Nde PN, Nes WD, Locuson CW, Calcutt MW, Waterman MR, Daniels JS, Loiseau PM, Villalta F. 2015. VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J Infect Dis 212:1439–1448. doi: 10.1093/infdis/jiv228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omura T, Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J Biol Chem 239:2370–2378. [PubMed] [Google Scholar]
- 32.Lepesheva GI, Zaitseva NG, Nes WD, Zhou W, Arase M, Liu J, Hill GC, Waterman MR. 2006. CYP51 from Trypanosoma cruzi: a phyla-specific residue in the B′ helix defines substrate preferences of sterol 14alpha-demethylase. J Biol Chem 281:3577–3585. doi: 10.1074/jbc.M510317200. [DOI] [PubMed] [Google Scholar]
- 33.Leslie A. 2006. The integration of macromolecular diffraction data. Acta Crystallogr D Biol Crystallogr 62:48–57. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- 34.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaguchi H, Uchida K, Nagino K, Matsunaga T. 2002. Usefulness of a colorimetric method for testing antifungal drug susceptibilities of Aspergillus species to voriconazole. J Infect Chemother 8:374–377. doi: 10.1007/s10156-002-0201-Y. [DOI] [PubMed] [Google Scholar]