

ABSTRACT
Hand-foot-and-mouth disease (HFMD), caused by enterovirus, is a threat to public health worldwide. To date, enterovirus 71 (EV71) has been one of the major causative agents of HFMD in the Pacific-Asia region, and outbreaks with EV71 cause millions of infections. However, no drug is currently available for clinical therapeutics. In our previous works, we developed a set of protease inhibitors (PIs) targeting the EV71 3C protease (3Cpro). Among these are NK-1.8k and NK-1.9k, which have various active groups and high potencies and selectivities. In the study described here, we determined the structures of the PI NK-1.8k in complex with wild-type (WT) and drug-resistant EV71 3Cpro. Comparison of these structures with the structure of unliganded EV71 3Cpro and its complex with AG7088 indicated that the mutation of N69 to a serine residue destabilized the S2 pocket. Thus, the mutation influenced the cleavage activity of EV71 3Cpro and the inhibitory activity of NK-1.8k in an in vitro protease assay and highlighted that site 69 is an additional key site for PI design. More information for the optimization of the P1′ to P4 groups of PIs was also obtained from these structures. Together with the results of our previous works, these in-depth results elucidate the inhibitory mechanism of PIs and shed light to develop PIs for the clinical treatment of infections caused by EV71 and other enteroviruses.
KEYWORDS: EV71, protease, inhibitor, crystal structure, mechanism
INTRODUCTION
Hand-foot-and-mouth disease (HFMD) is a common viral illness among infants and young children that causes fever, sore throat, blisters, pharyngitis, mouth ulcers, and a rash on the hands and feet (1, 2). Enterovirus 71 (EV71) has been the main causative agent of HFMD in recent years. Unfortunately, no drugs for the treatment of this disease are available. EV71 is classified as a member of Enterovirus species A within the genus Enterovirus of the Picornaviridae family (3). In the virus replication cycle, the 7.4-kb positive-sense, single-stranded RNA genome of EV71 is translated into a polyprotein which is subdivided into three primary precursors, P1, P2, and P3. Four structural proteins, VP1 to VP4, derived from the P1 portion, form the capsid of the mature virion (4), and the P2 and P3 regions are cleaved by viral proteases, forming seven individual nonstructural proteins, 2A, 2B, 2C, 3A, 3B, 3C, and 3D (5, 6). The correct replication of EV71 is dependent on the effective cleavage of the viral polyprotein by viral 2A protease (2Apro) and 3C protease (3Cpro) (7). Of these two proteases, 3Cpro not only takes major responsibility for polyprotein cleavage, with the exception that 2Apro is responsible for the cleavage of VP1/2A and 3C/3D, but also plays multiple roles in various biological processes (8).
EV71 3Cpro is a classical cysteine protease. Cysteine proteases are a large family containing many members functioning in various physiological processes (9). The dysfunction of cysteine proteases results in inflammatory diseases, neurodegenerative disorders, rheumatoid arthritis, Alzheimer's disease, and other diseases (10). Peptidomimetic inhibitors are important compounds for the discovery of drugs targeting cysteine proteases. All peptidomimetic inhibitors share a conserved chemical structure, i.e., a modified peptide recognition sequence for specific binding to the protease and an electrophilic functional group for anchoring the catalytic cysteine in the active site of the enzyme (9). According to the mode of inhibitor-protein interaction, peptidomimetic inhibitors are subdivided into the reversible inhibitors (e.g., aldehydes, nitriles, α-keto amides) and the irreversible inhibitors (e.g., α,β-unsaturated esters, disulfides), among which the activity of the inhibitors is proportional to the electrophilicity of the reactive group (9). Although these peptidomimetic inhibitors potently inhibit the enzymatic activity, the covalent irreversible modifiers are not considered potential drug candidates, being limited by the poor selectivity resulting from the strongly reactive electrophiles and the potential toxicity caused by the formation of protein adducts (11). Therefore, success with the development of anti-EV71 protease inhibitors (PIs) remains challenging.
In the past several years, we have concentrated on developing and optimizing anti-EV71 PIs. We developed a highly efficient assay (12, 13) and discovered a series of potent anti-EV71 PIs targeting EV71 3Cpro (14–18). Among them, NK-1.8k (a peptidyl aldehyde derivative) and compound 9 and NK-1.9k (cyanohydrin derivatives) have the highest potencies against EV71 infection with good pharmacological properties, but they still exhibit distinct features. For example, compound 9 and NK-1.9k have significantly higher selectivities due to their use of cyanohydrin as the functional group, but the aldehyde group of NK-1.8k has strong electrophilicity and may react with both the thiol group of cysteine proteases and the hydroxyl group of serine proteases. An interesting observation is that NK-1.8k (half-maximal [50%] inhibitory concentration [IC50] = 0.11 ± 0.02 μM) presented a stronger enzyme-inhibitory potency than compound 9 (IC50 = 1.13 ± 0.10 μM) and NK-1.9k (IC50 = 0.31 ± 0.05 μM), but NK-1.8k was observed to have improved antiviral activity compared with that of NK-1.9k (50% effective concentration [EC50] = 24.9 ± 0.2 nM) in a cell-based virus proliferation assay (16). Therefore, it is necessary to obtain insight into the function of these PIs at the molecular level and define the critical target sites for further optimization.
RESULTS
Chemical features of NK-1.8k, NK-1.9k, and AG7088.
AG7088 (rupintrivir) (Fig. 1A) was originally designed to be an inhibitor of human rhinovirus (HRV) 3Cpro and was later found to exhibit broad-spectrum antiviral activity against other members of the Picornaviridae family (19–21). It is also the first compound whose structure in complex with EV71 3Cpro was determined (22). AG7088 represents a typical peptidyl mimic PI, which is composed of a γ-lactam P1 group mimicking a glutamine residue, a P2 group consisting of 4-fluorophenylalanine, a P3 group consisting of valine, a P4 group consisting of 5-methyl-3-isoxazole, and an α,β-unsaturated ester P1′ group as the functional group (Fig. 1A). Although AG7088 was reported to potently inhibit a subset of picornaviruses, this compound is not satisfied by further pharmacological development due to its poor aqueous solubility and low oral bioavailability (17, 30). In our previous works, we first shortened the three-residue mimics (AG7088) to the two-residue peptidyl mimics. Second, δ-lactam was used to replace the γ-lactam in the P1 position. This replacement not only increases the binding of the inhibitor to the target protein but also produces a higher hydrophobicity, which allows the compound to pass through the plasma membrane more easily. Most importantly, we replaced the P1′ group from an α,β-unsaturated ester in AG7088 with an aldehyde group in NK-1.8k or a cyanohydrin group in compound 9 and NK-1.9k. The α,β-unsaturated ester is not preferred by pharmacology and should be excluded in drug design, since it is known to form an irreversible covalent bond with the catalytic residues in all cysteine proteases, thus causing many side effects during the treatment of virus infections. The replacement by an aldehyde group or a cyanohydrin group increases selectivity and stability. To elucidate and compare the precise inhibitory mechanisms of these PIs, we determined the crystal structures of wild-type (WT) and drug-resistant EV71 3Cpro in complex with NK-1.8k. We anticipate the discovery of essential sites for further optimization of anti-EV71 PIs.
FIG 1.
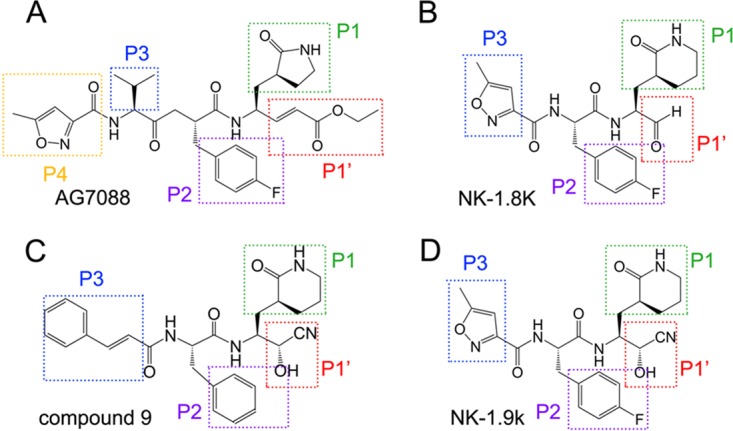
Comparison of the chemical structures of AG7088 (A), NK-1.8k (B), compound 9 (C), and NK-1.9k (D). The P1′ to P3 or P4 positions of each compound are indicated.
Overall structures of EV71 3Cpro in complex with NK-1.8k.
WT 3Cpro and N69S-mutated 3Cpro were first individually crystallized under crystallization conditions similar to those described in a previous report (8). Because the binding of substrate or inhibitor to EV71 3Cpro leads to a conformational shift of the β-ribbon, it is not surprising that the soaking of NK-1.8k to both crystals finally failed. The crystals of WT and N69S-mutated EV71 3Cpro in complex with NK-1.8k obtained by cocrystallization with a protein-to-compound molar ratio of 1:5 and their structures were evaluated by the molecular replacement method (Table 1).
TABLE 1.
Data collection and refinement statistics
| Parametera | Value(s) forb: |
|
|---|---|---|
| WT 3Cpro–NK-1.8k | N69S 3Cpro–NK-1.8k | |
| Data collection statistics | ||
| Space group | P21 | P21 |
| Unit cell dimensions | ||
| a, b, c (Å) | 95.0, 70.7, 94.9 | 94.5, 70.9, 94.9 |
| α, β, γ (°) | 90, 118.2, 90 | 90, 118.2, 90 |
| Wavelength (Å) | 1.5418 | 1.5418 |
| Resolution (Å) | 50.00 (2.64)–2.60 | 50.00 (3.34)–3.20 |
| No. of all reflections | 126,147 (6,298) | 55,551 (2,236) |
| No. of unique reflections | 34,176 (1,702) | 17,977 (860) |
| Completeness (%) | 100.0 (100.0) | 97.6 (93.9) |
| Average I/σI | 10.1 (2.0) | 9.2 (2.1) |
| Rmerge (%) | 9.7 (59.0) | 18.1 (64.4) |
| Refinement statistics | ||
| No. of reflections used (σF > 0) | 32,910 | 15,670 |
| Resolution range (Å) | 50.00–2.60 | 50.00–3.20 |
| Rwork/Rfree (%) | 19.6/32.7 | 22.5/26.7 |
| RMSD | ||
| Bond distance (Å) | 0.009 | 0.010 |
| Bond angle (°) | 1.007 | 1.236 |
| Avg B-factor | ||
| Protein | 38.7 | 48.1 |
| Compound | 45.2 | 57.3 |
| Ramachandran plot (%) | ||
| Allowed region | 99.1 | 96.3 |
| Additionally allowed region | 0.9 | 3.5 |
| Disallowed region | 0 | 0.2 |
I, intensity of a reflection; F, structure factor.
Numbers in parentheses are the corresponding values for the highest-resolution shell.
In the complexes, EV71 3Cpro exhibits a typical compact globular architecture which contains two topologically equivalent antiparallel six-stranded β-barrel domains (Fig. 2A and B). A long shallow groove for substrate binding is located between the two β-barrel domains and is fully occupied by compounds in both complexes (Fig. 2C and D). The structure of EV71 3Cpro has a distinct element, i.e., a β-ribbon, which is located between βB2 and βC2 (residues G123 to H133) (8). In the structure of unliganded EV71 3Cpro, the β-ribbon is in an open conformation and is flipped open with its tip pointing away from the active site of 3Cpro (8). In the structures of EV71 3Cpro in complex with NK-1.8k, this β-ribbon changes to a closed conformation and seals the bottom of the substrate-binding groove (Fig. 2B), which is similar to the conformation when binding substrate or inhibitors (22, 23).
FIG 2.
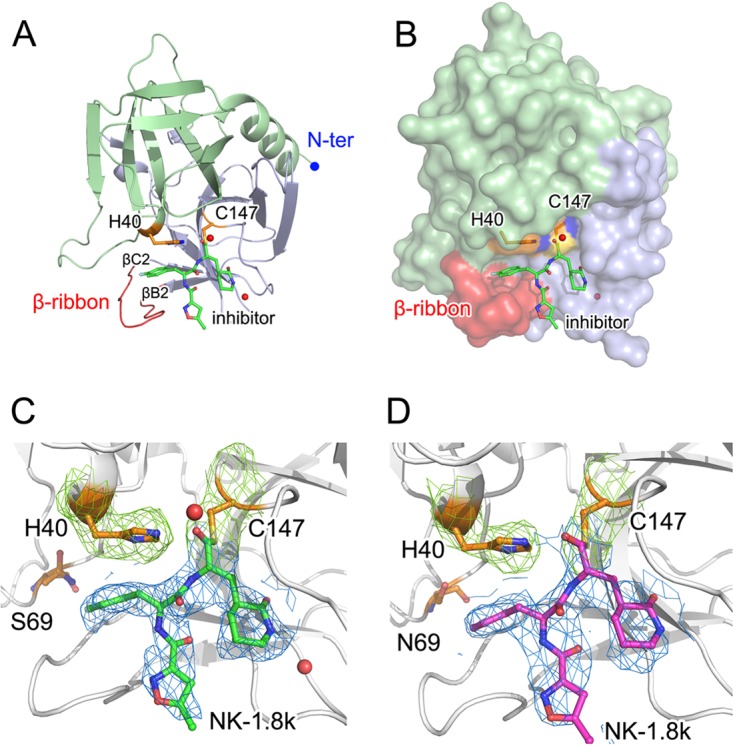
Structures of NK-1.8k bound with WT or N69S-mutated EV71 3Cpro. (A, B) Overall structure of NK-1.8k in complex with EV71 3Cpro. The polypeptide of EV71 3Cpro is shown as a colored cartoon (A) or with a colored surface (B), in which the N-terminal (N-ter) domain is colored blue. The β-ribbon, which is important for substrate or inhibitor binding, is colored red. (C, D) Densities of bound NK-1.8k in complex with WT (C) or N69S-mutated (D) EV71 3Cpro. The polypeptide of EV71 3Cpro is shown as a white cartoon. In WT (C) and N69S-mutated (D) EV71 3Cpro, NK-1.8k is presented as green and magenta sticks, respectively. Catalytic residues H40 and C147, as well as drug resistance-conferring residue N69S, are displayed as gold sticks. Both NK-1.8k molecules are covered by a 2Fo − Fc omit map at 1.1 σ, while H40 and C147 are covered by a 2Fo − Fc map at 1.6 σ (Fo is F observe and Fc is F calculate).
Comparison of overall structure with unliganded EV71 3Cpro.
We superimposed our complex structures with the structure of EV71 3Cpro in an unliganded state. Most regions of 3Cpro presented with high degrees of structural identity in all structures, with the root mean square deviation (RMSD) being less than 0.7 Å. However, several distinct regions could be observed (Fig. 3). Because the discrepancies in the structures of the WT and N69S EV71 3Cpro enzymes in complex with NK-1.8k are located in the catalytic position, we used WT EV71 3Cpro in complex with NK-1.8k for comparison with the unliganded enzyme. First, additional amino acids which belong to the expression vector form an extended α-helical part at the N termini in the unliganded EV71 3Cpro, but these vector residues are completely missing in the EV71 3Cpro–NK-1.8k complex. We speculate that the crystallization conditions, crystal packing, or other environmental factors might be the reason for this discrepancy, and we believe that this is not related to the biological mechanism. The most significant structural shift occurred in the region spanning residues M109 to G148, which covers both the β-ribbon and the catalytic loop. In the complex structure, βB2 (residues M109 to G123), which is a continuous β-strand in the unliganded enzyme, breaks into two separate short β-strands linked by a relaxed loop. The β-ribbon shifts from its unliganded position toward the substrate-binding groove with a distance of 30 Å. As a result, βC2 shifts approximately 10° toward the substrate-binding groove with N139 as the origin. Moreover, the catalytic loop, which spans F140 to G149, also moves a maximum of 4.2 Å to further seal the substrate-binding pocket. Furthermore, the region covering residues L53 to L72 also presented a large conformational change. In the complex, βE1 (residues L53 to V63), which is a continuous β-strand in the unliganded enzyme, breaks into two separate short β-strands linked by a relaxed loop. Furthermore, the loop region connecting βE1 and βF1 moves a maximum of 4.1 Å away from the unliganded position. These structural variations should be caused by the binding of NK-1.8k, and some of them could also be observed in other liganded EV71 3Cpro enzymes.
FIG 3.
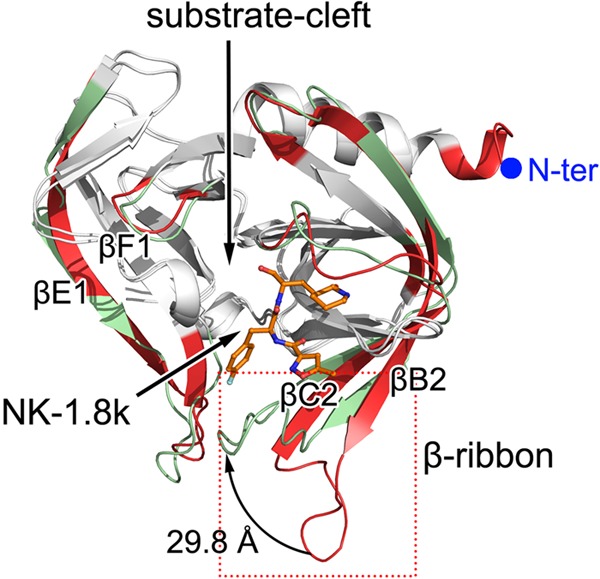
Structural comparison of EV71 3Cpro in the unliganded form and in complex with NK-1.8k. The structure of unliganded EV71 3Cpro (PDB accession no. 3OSY) was superimposed onto that of EV71 3Cpro in complex with NK-1.8k by using the CCP4 program. The polypeptides of EV71 3Cpro in the unliganded form or in complex with NK-1.8k are displayed as red and green cartoons, respectively. The structural elements with RMSDs of less than 0.7 Å are colored white. The bound NK-1.8k is shown as colored sticks.
Comparison of the overall structure with that of AG7088-bound EV71 3Cpro.
We also compared our complex structures with the structure of EV71 3Cpro in complex with AG7088. Most regions of 3Cpro presented high degrees of structural identity in all structures, with RMSDs being less than 0.5 Å. Structural variations were also observed (Fig. 4). Residues G1 to N14 form a canonical α-helix in the EV71 3Cpro–NK-1.8k complex, but the first three residues cannot be modeled in the EV71 3Cpro-AG7088 complex. This is also likely caused by a nonbiological reason, which is similar to the reason in the structure of unliganded EV71 3Cpro. Interestingly, although in both complexes EV71 3Cpro was in complex with inhibitors, βB2 (residues M109 to G123) and βE1 (residues L53 to V63) in the EV71 3Cpro-AG7088 complex were continuous β-strands like the β-strand in unliganded EV71 3Cpro, but all break into two separate short β-strands linked by a relaxed loop. Moreover, although the conformations of the β-ribbons in both structures bound with inhibitors are in a closed form, they still present structural shifts with an RMSD of 0.8 to 1.3 Å for their C-α atoms. Because AG7088 is a three-residue peptidyl mimic but NK-1.8k is a two-residue peptidyl mimic and the β-ribbon is responsible for the interaction with the P4 position of the substrate or inhibitor, this observation is not surprising. Another very interesting structural variation is that the region spanning residues V68 to L72 is a part of βF1 in both unliganded and AG7088-bound EV71 3Cpro but forms a relaxed loop structure bending to the substrate-binding groove in the EV71 3Cpro–NK-1.8k complex. We denote this region site 69 and discuss its relationship with the P1 group and drug resistance later in this report.
FIG 4.
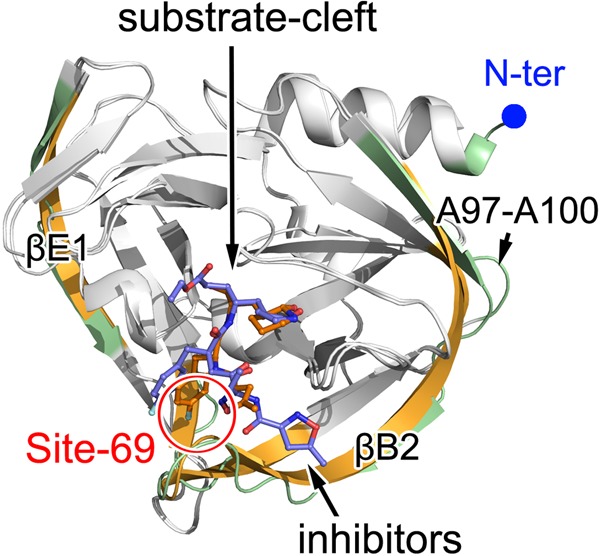
Structural comparison of EV71 3Cpro in complex with AG7088 and NK-1.8k. The structure of EV71 3Cpro bound with AG7088 (PDB accession no. 3SJO) was superimposed onto the structure of EV71 3Cpro in complex with NK-1.8k by using the CCP4 program. The polypeptides of EV71 3Cpro in complex with AG7088 and NK-1.8k are displayed as gold and green cartoons, respectively. The structural elements with RMSDs of less than 0.5 Å are colored white. The bound inhibitors are shown as colored sticks. Site 69 is highlighted.
Binding of NK-1.8k in EV71 3Cpro.
In the structures of both WT and N69S-mutated EV71 3Cpro in complex with NK-1.8k, the aldehyde group is linked to EV71 3Cpro by a covalent bond with the sulfhydryl group of catalytic residue C147 (Fig. 5A and B). The C-terminal ethyl ester of AG7088 (P1′ position) does not attach to the leaving-group side pocket in EV71 3Cpro (Fig. 5C). In contrast, this leaving-group side pocket in HRV 3Cpro is partially occupied by the ethyl ester of AG7088 (22). Because the P1′ position is replaced by an aldehyde group in NK-1.8k or a cyanohydrin group in compound 9, the structural finding that the leaving-group side pocket is in a free state is not surprising (Fig. 5A and C). Since the α,β-unsaturated ester is not pharmacologically preferred and makes no contribution to inhibitor binding, the replacement by an aldehyde group or a cyanohydrin group does not negatively affect enzyme-inhibitor interactions or inhibitory effects. G145 is a key residue for stabilization of the functional group of inhibitors (23). In the structure of EV71 3Cpro-AG7088, the nitrogen atom of G145 forms a hydrogen bond with the O-23 atom in the ester group of AG7088 (Fig. 5D), but such an intermolecular interaction is abolished in EV71 3Cpro–NK-1.8k due to the introduction of a functional group with a smaller molecular size (Fig. 5A and B).
FIG 5.
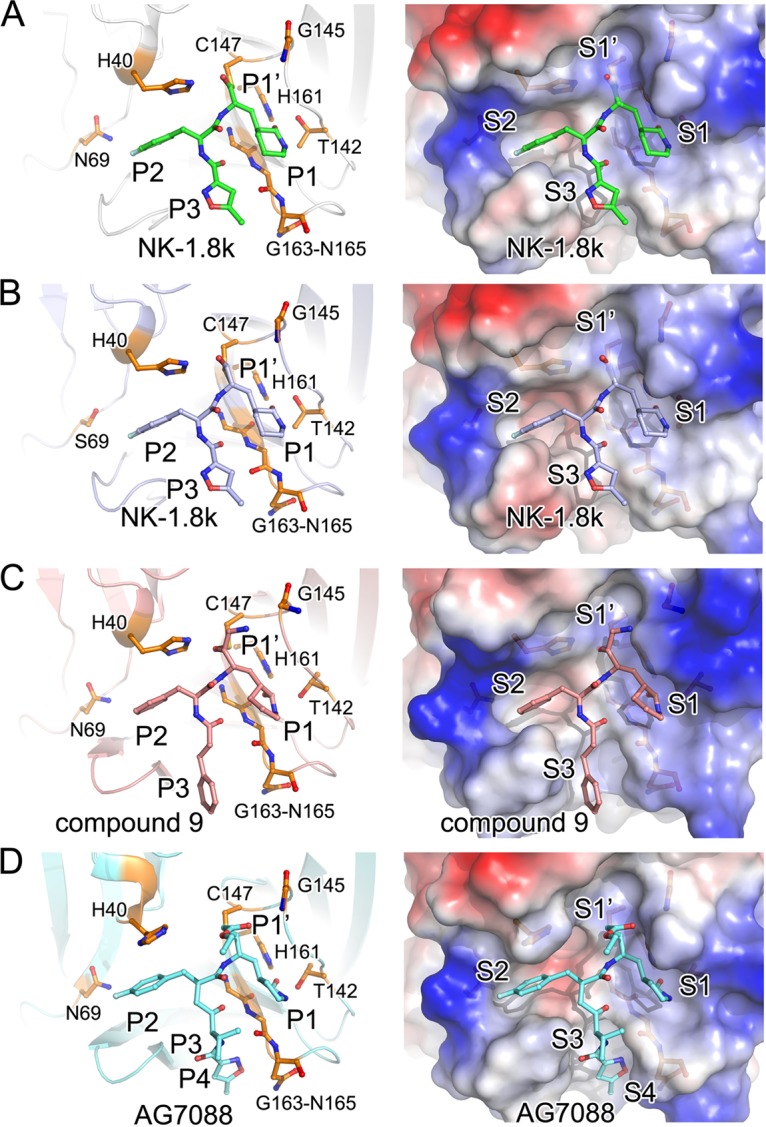
Binding of inhibitors to WT or drug-resistant EV71 3Cpro. The structure of WT (A) or N69S (B) EV71 3Cpro bound to NK-1.8k and the structure of 3Cpro in complex with compound 9 (PDB accession no. 5BPE) (C) or AG7088 (PDB accession no. 3SJO) (D) were superimposed and are shown in the same orientation. (Left) The polypeptides of the enzymes are displayed as cartoon diagrams, while the inhibitors and key residues are represented as colored sticks; (right) the proteins are covered with the electric potential surface. Key residues and subsites for inhibitor binding are labeled.
The difference of NK-1.8k, compound 9, and NK-1.9k from AG7088 is a substitution of the γ-lactam in AG7088 as a δ-lactam (Fig. 1). The bond lengths of the oxygen atom in the δ-lactam with N-ε2 of H161 and O-δ2 of T142 change from 2.7 Å and 2.9 Å, respectively, in EV71 3Cpro-AG7088 to 2.5 Å and 2.8 Å, respectively, in EV71 3Cpro–NK-1.8k. The shorter bonds lead the P1 group to move inside into the S1 site, obtain more burial area, and achieve a higher enzyme-inhibitor affinity. Together with the chemical feature that δ-lactam has a higher hydrophobicity and allows the compound to pass the plasma membrane more easily, the substitution at the P1 position presents a new option for further inhibition optimization.
We could also observe interesting structural features in the S2 position (Fig. 5). H40 is a key residue that not only participates in the catalytic reaction but also structurally forms the ceiling of the S2 site by its imidazole group (22). In the structures of the WT or N69S EV71 3Cpro complex with NK-1.8k and compound 9, the side chain of H40 maintains the same orientation that it maintains in natural substrate-bound or unliganded EV71 3Cpro. In contrast, the side chain of H40 rotates approximately 110° outward from the catalytic center in the structure of 3Cpro complexed with AG7088. From the structures, we observed that the N-ε2 atom of H40 forms a hydrogen bond with the oxygen atoms of the aldehyde group in NK-1.8k, the cyanohydrin group in compound 9, and the carboxyl of the substrate, but a carbon atom of the ethyl ester in AG7088 occupies the position of the oxygen atom that forms a hydrogen bond with H40 and thus pushes the side chain of H40 away from the catalytic center. Again, this structural observation suggests that the α,β-unsaturated ester group may have negative impacts on inhibitor binding.
Moreover, previous studies have suggested that the modification of the P2 residue consisting of 4-fluorophenylalanine is essential for inhibitor binding (22). Beyond the van der Waals contacts of the phenylalanine group with the S2 pocket, the side chain fluoride bonds with the residues at the distal end of the S2 pocket (22), but when the 4-fluorophenylalanine at the P2 residue is replaced by a benzyl group in compound 9, the inhibition of enzymatic activity and virus proliferation clearly changes (16), indicating that the fluoride atom is necessary. Moreover, a previous study also suggested that the structural variabilities at the distal ends of the S2 pockets are pronounced; however, this does not affect the efficacies of the inhibitors (20, 22). All these results indicate that there may exist an additional site that is correlated with the S2 pocket and that plays an essential role in PI function.
Furthermore, it is interesting to find that the condensation of the three-residue peptidyl mimic AG7088 to a two-residue peptidyl mimic by removing the P3 residue directly in NK-1.8k/NK-1.9k and substituting 5-methyl-3-isoxazole with a cinnamoyl group in compound 9 does not have a negative effect on inhibitory activity but, on the contrary, increases the inhibition of enzymatic activity and virus proliferation (16). In the structures, we also noticed that the interactions between the EV71 3Cpro polypeptide and the P3 residues of two compounds largely disappear (Fig. 5). Collectively, the results indicate that the S3 and S4 positions may not be very critical for inhibitor design and more modifications to the P3 residue could be launched in further optimizations to reduce the molecular size or improve the pharmacological properties of inhibitors.
Site 69 is critical for antienterovirus PI development.
A previous study reported that AG7088 inhibits the enzymatic activity of EV71 3Cpro with an IC50 of 2.3 μM and virus proliferation with an EC50 of 1 nM (22). In our results reported here, NK-1.8k inhibited 3Cpro and virus with IC50 of 0.11 μM and an EC50 of 0.108 μM, respectively, while NK-1.9k inhibited 3Cpro and virus with an IC50 of 0.31 μM and an EC50 of 37 nM, respectively. We also documented resistance to NK-1.8k and NK-1.9k caused by a single amino acid substitution of an asparagine residue to a serine residue at position 69 (N69S) within 3Cpro by continuously culturing EV71 in the presence of gradually increasing concentrations of inhibitors (16). Moreover, the N69S mutation helps the virus to escape the effects of treatment with NK-1.8k and NK-1.9k but attenuates the fitness of virus growth (16). Interestingly, a clear structural movement of site 69 could be observed in both WT and N69S-mutated 3Cpro in complex with NK-1.8k (Fig. 3 and 4). Notably, site 69 did not have a structural shift in unliganded and substrate- and AG7088-bound EV71 3Cpro and did not occur in any other investigation of drug resistance; thus, it was not further analyzed in previous studies. This prompted us to obtain insight into site 69 from a structural point of view (Fig. 6A).
FIG 6.
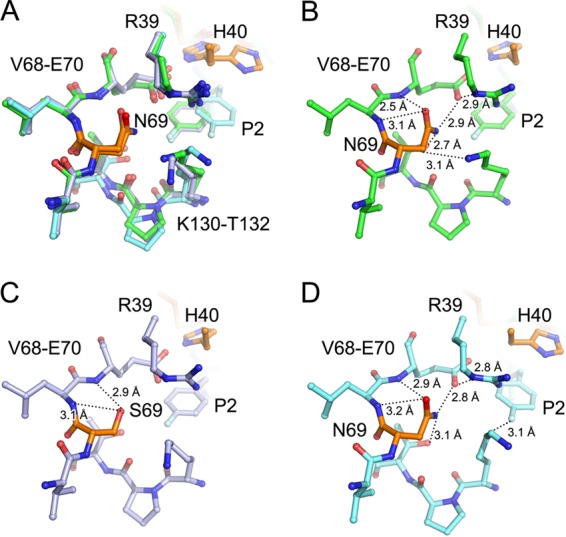
Detailed view of site 69. The structures of WT or N69S-mutated EV71 3Cpro in complex with NK-1.8k and WT EV71 3Cpro in complex with AG7088 were superimposed and are shown together (A) or individually (B to D). Polypeptides of EV71 3Cpro in the WT EV71 3Cpro–NK1.8k complex (B), the N69S EV71 3Cpro–NK1.8k complex (C), or the WT EV71 3Cpro–AG7088 complex (D) are displayed as green, light blue, and cyan sticks, respectively. Site 69 (in which drug resistance is conferred when N69 is mutated to S) and catalytic residue H40 are colored orange. The inhibitor in each complex is presented as sticks with the same color scheme as the residues. Hydrogen bonds are denoted as dotted lines with the measured distance.
N69 is not directly involved in the formation of any site for substrate/inhibitor accommodation but plays an essential role in the stabilization of the substrate-binding groove, particularly the S2 pocket. In the WT EV71 3Cpro–NK-1.8k complex, N69 forms a complicated interaction network to stabilize the S2 pocket. The O-δ1 atom of N69 forms a hydrogen bond with the nitrogen atoms of L70 and E70 with distances of 3.1 Å and 2.5 Å, respectively (Fig. 6B). Moreover, the N-δ1 atom of N69 interacts with the O-γ1 atom of T132 and O-ε1 of E70 with hydrogen bonds at 2.7 Å and 2.9 Å, respectively. In particular, the interaction of N69-E70 stabilizes the conformation of the side chain of E70 and thus results in the formation of a 2.9-Å hydrogen bond between the O-ε1 atom of E70 and the N-ε atom of R39. Moreover, the N-ζ atom of K130 additionally comes into contact with the C-β atom of N69. Because both E70 and R39 play key roles in the formation of the S2 pocket, it is conceivable that the hydrogen-bonded network mediated by N69 is important for substrate/inhibitor binding. In contrast, when the replacement of N69 by a serine residue is introduced under conditions of challenge with NK-1.8k, only the interaction between the O-δ atom of S69 with the nitrogen atoms of L70 and E70 remains, though the distance of S69 O-δ to E70N increases to 2.9 Å (Fig. 6C). Because of the short side chain, the hydroxyl group cannot form an interaction with the side chain of E70, thus resulting in the loss of contact of E70 and R39. This may also make the conformation of the S2 pocket less stable. Consistently, the B-factor of the side chain of R39 is 19 Å2 in WT EV71 3Cpro–NK-1.8k but increases to about 35 Å2 in N69S EV71 3Cpro–NK-1.8k. Because the S2 pocket plays an essential role in the accommodation of the P2 residue of the natural substrate, it is conceivable that the destabilization of the S2 pocket decreases the fitness of virus proliferation. However, the destabilization of the S2 pocket caused by the N69S mutation also has a negative impact on the binding of NK-1.8k and increases the possibility that the natural substrate binds to EV71 3Cpro, thus conferring resistance to NK-1.8k.
Another interesting observation is that the C-β atom of N69 bonds with the N-ζ atom of K130 in WT EV71 3Cpro–NK-1.8k, but this interaction is released in WT EV71 3Cpro-AG7088 (Fig. 6D). This allows the movement of the side chain of K130 and the subsequent formation of a 3.1-Å hydrogen bond between the C-δ atom of K130 and the fluoride in the P2 residue of AG7088. This provides an additional interaction to stabilize the P2 residue of AG7088, which is pushed away from the S2 site by the unusual rotation of the side chain of H40 (Fig. 5D).
Site 69 plays an essential role in inhibitor binding and drug resistance, but this site is not touched by currently reported inhibitors, though it is adjacent to the S2 pocket. The minimum distance between the N69 and P2 residues is still about 4 Å in the complex with NK-1.8k and about 6 Å in the complex with AG7088. It is conceivable that the modifications to the P2 residue with a longer side chain, which may further interact with or stabilize site 69, possibly optimizes the inhibitory effects of PIs. Actually, compound 9 has a benzyl group in the P2 residue, and it inhibits enzymatic activity and virus proliferation with an IC50 and an EC50 of 1.13 μM and 120 nM, respectively, which are worse than the IC50 (0.31 μM) and EC50 (37 nM) of NK-1.9k with a 4-fluorophenylalanine at the P2 residue. This may preliminarily support our speculation, and further investigation is under way.
N69S influences protease activity in vitro.
In order to indicate the influence of the N69S mutation on enzyme activity, the protease assay was performed using the peptide substrate N-methylacridone (NMA)-IEALFQGPPK(DNP)FR with a fluorescence group and a quenching group. The IC50 values of NK-1.8k on WT and N69S proteases were 0.11 μM and 1.15 μM, respectively (Fig. 7A). Moreover, the cleavage activity of the N69S mutant was distinctly reduced in comparison with that of the WT protease (Fig. 7B). Together with these two results, we draw the conclusion that the N69S mutation significantly reduces the activity of EV71 3Cpro.
FIG 7.

In vitro protease activity. (A) IC50 of NK-1.8k for WT and N69S proteases. The percentages of inhibitory activity of NK-1.8k at concentrations ranging from 10 to 0.00975 μM were compared with those in the absence of compound (control). AU, arbitrary units. (B) Cleavage activities of WT and N69S proteases. The intensities of the ODs were read at an excitation λ of 340 nm and an emission λ of 440 nm every 10 min (B).
DISCUSSION
In a series of our previous works (17, 30), we developed a set of anti-EV71 PIs with high potencies and strong selectivity properties. Comparison of these PIs from a structural biology point of view and summarization of their activities would present the inhibitory mechanism of PIs and further the understanding of PIs so that they can be designed and developed to treat EV71 infections. In this study, we determined the structure of EV71 3Cpro in complex with NK-1.8k and also solved the structure of drug-resistant EV71 3Cpro in complex with the inhibitor. Combined with the reported structures of 3Cpro-AG7088 and 3Cpro-compound 9, it was found that the α,β-unsaturated ester of AG7088 not only has obvious pharmacological side effects but also demolishes the 3Cpro-PI interaction, and therefore, replacement of this α,β-unsaturated ester with some other functional group with a smaller molecular size, e.g., aldehyde or cyanohydrin, is necessary for PI development. Moreover, replacement of the P1 residue by a moiety with a larger size and greater hydrophobicity may increase the enzyme-inhibitor interaction. Such a strategy would also provide an additional advantage to allow the compound to pass the plasma membrane more easily.
Most importantly, the structures helped us to define site 69 as an additional key site for PI design. N69 is not directly involved in the formation of any site for substrate/inhibitor accommodation but forms a complicated interaction network to stabilize the S2 pocket. The mutation of N69 to a serine residue abolishes the bond network, destabilizes the S2 pocket, increases the possibility that a natural substrate will bind to EV71 3Cpro in the presence of an inhibitor, and confers drug resistance. Therefore, it is conceivable that a modification of the P2 residue with a longer side chain is possible to optimize the inhibitory effect of PIs.
Additionally, one of the key features of our PIs is that they are two-residue peptidyl mimics but maintain high potency. In the complex structures, we observed that the interactions between the 3Cpro polypeptide and the P3 residues of two compounds largely disappeared. This indicates that the S3 and S4 positions may not be very critical for inhibitor design and more modifications on the P3 residue could be launched in a further optimization to reduce the molecular size or improve the pharmacological properties of PIs.
Except for the modification on each moiety of the inhibitor, another strategy could be learned from the reported structures of PIs for further optimization of NK-1.8k or NK1.9k. For example, the amino bond linking the P2 and P3 residues of PIs is replaced by a ketomethylene structure in AG7088, which significantly increases the stability and pharmacokinetic properties of the compound. We anticipate the use of all these strategies to further develop anti-EV71 PIs for eventual clinical use.
MATERIALS AND METHODS
Protein production.
The original viral strain selected in this study was human EV71 isolate BJ/CHN/2008 (GenBank accession no. HQ615421). The gene for WT 3Cpro was amplified using the primers 3C-forward (5′-GGAATTCCATATGGGCCCGAGCCTTGAC-3′) and 3C-reverse (5′-ATAAGAATGCGGCCGCTTTGCTAGTGAACAATAA-3′). The gene was then subcloned into pET-28a via the NdeI and XhoI restriction sites, which generated a recombinant protein with a 6×His tag at the N terminus. For site-directed mutagenesis, the plasmid harboring WT 3Cpro was used as a template to generate the construct coding for the N69S-mutated enzyme. The N69S mutation was obtained using an Easy Pfu mutation kit (Transgen, China) and primer pair 3C-N69S-forward (5′-GATGAGCAAGGAGTCAGCTTGGAATTAACCCTC-3′) and 3C-N69S-reverse (5′-CTGACTCCTTGCTCATCCACCAATTCAACTGC-3′). All constructs were verified by DNA sequencing.
Expression and purification of the recombinant protein.
The recombinant WT and mutated 3Cpro enzymes were expressed and purified according to the following protocol. The plasmids were transformed into Escherichia coli BL21(DE3) competent cells, and cultures were grown to an optical density (OD) at 600 nm of 0.5 to 0.6 in LB medium at 37°C. Isopropyl-d-1-thiogalactopyranoside was added to a final concentration of 0.5 mM to induce protein expression, and the cultures were grown for an additional 20 h at 18°C. Cells were harvested by centrifugation, resuspended, and homogenized in lysis buffer containing 50 mM Tris-HCl (pH 7.0) and 200 mM NaCl, using a low-temperature ultra-high-pressure cell disrupter (Jnbio, Guangzhou, China). Cell debris was removed by centrifugation at 20,000 × g for 30 min. The resultant supernatant was added to Ni-nitrilotriacetic acid resin (GE Healthcare, USA). The nonspecific contaminants were removed by washing the resin with buffer containing 50 mM Tris-HCl (pH 7.0), 200 mM NaCl, and 50 mM imidazole. The target protein was subsequently eluted with buffer containing 50 mM Tris-HCl (pH 7.0), 200 mM NaCl, and 500 mM imidazole. The eluate was further purified by use of a HiTrap S column (GE Healthcare, USA) with a linear gradient of from 0 mM to 500 mM NaCl with 50 mM Tris-HCl (pH 7.0) and 2 mM dithiothreitol (DTT). The enzyme fractions were pooled and concentrated to 12 mg/ml in a buffer with 20 mM Tris-HCl (pH 7.0), 500 mM NaCl, and 2 mM DTT for storage.
Crystallization, data collection, and structure determination.
NK-1.8k was dissolved in 100% dimethyl sulfoxide to a final concentration of 100 mM and stored at −80°C. The WT–NK-1.8k and N69S–NK-1.8k complexes were prepared at a 1:5 molar ratio of 3Cpro to inhibitor and incubated at 4°C for 2 h before the cocrystallization trials were set up. All the crystals were obtained by the hanging-drop vapor diffusion method at 16°C. The crystals of the WT–NK-1.8k complex appeared and reached their final size within 3 days in a well solution containing 0.1 M Tris-HCl (pH 7.7), 200 mM sodium citrate, and 32% polyethylene glycol (PEG) 3350, and the crystals of N69S–NK-1.9k were yielded in a well solution containing 0.1 M Tris-HCl (pH 7.7), 200 mM sodium citrate, and 16% PEG 3350.
For data collection, a single crystal was mounted on a nylon loop and was flash-cooled with a nitrogen gas stream at 100 K by using 20% (vol/vol) glycerol in the mother liquor as a cryoprotectant. Diffraction data for the crystals of WT–NK-1.8k and N69S–NK-1.8k were collected on an in-house Rigaku RU200 X-ray generator at 1.5418 Å with an R-Axis IV+ image plate. Data were processed and scaled using the HKL2000 package (24). The crystals of two complexes belong to space group P21 and had the following cell parameters: a = 95.0 Å, b = 70.7 Å, c = 94.9 Å, α = γ = 90°, and β = 118.2°. Five complex molecules per asymmetric unit with a Matthews coefficient of 2.50 Å3/Da were present, corresponding to a 50.85% solvent content.
All structures were determined by using the molecular replacement method in the PHASER program (25), with the structure of apo EV71 3Cpro (PDB accession number 3OSY) (8) being the initial search model. Manual model construction and refinement were performed in COOT (26) with the PHENIX (27) program following rigid body refinement, energy minimization, and individual B-factor refinement. The quality of the final refined model was verified by use of the MolProbity web service (28). The final refinement statistics are summarized in Table 1. Structural figures were prepared by use of the PyMOL program (29).
Inhibitory activity of NK-1.8k on WT and N69S proteases.
The IC50s of NK-1.8k were determined with 100-μl samples containing 0.5 μM EV71 WT or N69S mutant protease and a gradient concentration of inhibitor in 50 mM Tris-HCl, pH 7.0, buffer. The reaction solutions were incubated at 30°C for 30 min, and then 20 μM substrate was added to each well. The intensity of the OD for NMA and 2,4-dinitrophenol was read at an excitation (ex) wavelength of 340 nm and an emission (em) wavelength of 440 nm. The IC50s were calculated by the use of GraphPad Prism software.
In vitro protease activity assay.
To demonstrate the influence of the N69S amino acid substitution on enzyme activity, an in vitro protease cleavage assay was carried out. The reaction mixture contained 0.5 μM EV71 WT or N69S mutant protease and 20 μM substrate in 50 mM Tris-HCl, pH 7.0. The solution was then incubated for 2 h at 30°C. The reaction was terminated by addition of acetonitrile every 10 min, and the intensity of the OD was read on a microplate reader (Thermo Scientific).
Accession number(s).
Coordinates and structure factors have been deposited in the Protein Data Bank (PDB) database under accession numbers 5GSO (WT EV71 3C-NK-1.8k) and 5GSW (N69S EV71 3C-NK-1.8k).
ACKNOWLEDGMENTS
This work was supported by the National Major Project (2014CB542800 and 2013CB911100), the National Natural Science Foundation of China (31370733 and 81322023), and the Tsinghua University Initiative Scientific Research Program (20131089228).
REFERENCES
- 1.Chan KP, Goh KT, Chong CY, Teo ES, Lau G, Ling AE. 2003. Epidemic hand, foot and mouth disease caused by human enterovirus 71, Singapore. Emerg Infect Dis 9:78–85. doi: 10.3201/eid1301.020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen KT, Chang HL, Wang ST, Cheng YT, Yang JY. 2007. Epidemiologic features of hand-foot-mouth disease and herpangina caused by enterovirus 71 in Taiwan, 1998-2005. Pediatrics 120:e244–e252. doi: 10.1542/peds.2006-3331. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Qing J, Sun Y, Rao Z. 2014. Suramin inhibits EV71 infection. Antiviral Res 103:1–6. doi: 10.1016/j.antiviral.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Plevka P, Perera R, Cardosa J, Kuhn RJ, Rossmann MG. 2012. Crystal structure of human enterovirus 71. Science 336:1274. doi: 10.1126/science.1218713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen C, Wang Y, Shan C, Sun Y, Xu P, Zhou H, Yang C, Shi PY, Rao Z, Zhang B, Lou Z. 2013. Crystal structure of enterovirus 71 RNA-dependent RNA polymerase complexed with its protein primer VPg: implication for a trans mechanism of VPg uridylylation. J Virol 87:5755–5768. doi: 10.1128/JVI.02733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun Y, Wang Y, Shan C, Chen C, Xu P, Song M, Zhou H, Yang C, Xu W, Shi PY, Zhang B, Lou Z. 2012. Enterovirus 71 VPg uridylation uses a two-molecular mechanism of 3D polymerase. J Virol 86:13662–13671. doi: 10.1128/JVI.01712-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu YC, Hsu JT, Huang JH, Ho MS, Ho YC. 2003. Formation of enterovirus-like particle aggregates by recombinant baculoviruses co-expressing P1 and 3CD in insect cells. Biotechnol Lett 25:919–925. doi: 10.1023/A:1024071514438. [DOI] [PubMed] [Google Scholar]
- 8.Cui S, Wang J, Fan T, Qin B, Guo L, Lei X, Wang J, Wang M, Jin Q. 2011. Crystal structure of human enterovirus 71 3C protease. J Mol Biol 408:449–461. doi: 10.1016/j.jmb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otto HH, Schirmeister T. 1997. Cysteine proteases and their inhibitors. Chem Rev 97:133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- 10.Leung-Toung R, Zhao Y, Li W, Tam TF, Karimian K, Spino M. 2006. Thiol proteases: inhibitors and potential therapeutic targets. Curr Med Chem 13:547–581. doi: 10.2174/092986706776055733. [DOI] [PubMed] [Google Scholar]
- 11.Shah F, Mukherjee P, Gut J, Legac J, Rosenthal PJ, Tekwani BL, Avery MA. 2011. Identification of novel malarial cysteine protease inhibitors using structure-based virtual screening of a focused cysteine protease inhibitor library. J Chem Infect Model 51:852–864. doi: 10.1021/ci200029y. [DOI] [PubMed] [Google Scholar]
- 12.Cao L, Zhu S, Wang Y, Lou Z, Sun Y. 2015. A comprehensive procedure for antiviral inhibitor discovery using EV71 as an example. Biophys Rep 1:81–89. doi: 10.1007/s41048-015-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shang L, Zhang S, Yang X, Sun J, Li L, Cui Z, He Q, Guo Y, Sun Y, Yin Z. 2015. Biochemical characterization of recombinant enterovirus 71 3C protease with fluorogenic model peptide substrates and development of a biochemical assay. Antimicrob Agents Chemother 59:1827–1836. doi: 10.1128/AAC.04698-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo Y, Wang Y, Cao L, Wang P, Qing J, Zheng Q, Shang L, Yin Z, Sun Y. 2016. A conserved inhibitory mechanism of a lycorine derivative against enterovirus and hepatitis C virus. Antimicrob Agents Chemother 60:913–924. doi: 10.1128/AAC.02274-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shang L, Wang Y, Qing J, Shu B, Cao L, Lou Z, Gong P, Sun Y, Yin Z. 2014. An adenosine nucleoside analogue NITD008 inhibits EV71 proliferation. Antiviral Res 112:47–58. doi: 10.1016/j.antiviral.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Yang B, Zhai Y, Yin Z, Sun Y, Rao Z. 2015. Peptidyl aldehyde NK-1.8k suppresses enterovirus 71 and enterovirus 68 infection by targeting protease 3C. Antimicrob Agents Chemother 59:2636–2646. doi: 10.1128/AAC.00049-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhai Y, Ma Y, Ma F, Nie Q, Ren X, Wang Y, Shang L, Yin Z. 2016. Structure-activity relationship study of peptidomimetic aldehydes as enterovirus 71 3C protease inhibitors. Eur J Med Chem 124:559–573. doi: 10.1016/j.ejmech.2016.08.064. [DOI] [PubMed] [Google Scholar]
- 18.Zhai Y, Zhao X, Cui Z, Wang M, Wang Y, Li L, Sun Q, Yang X, Zeng D, Liu Y, Sun Y, Lou Z, Shang L, Yin Z. 2015. Cyanohydrin as an anchoring group for potent and selective inhibitors of enterovirus 71 3C protease. J Med Chem 58:9414–9420. doi: 10.1021/acs.jmedchem.5b01013. [DOI] [PubMed] [Google Scholar]
- 19.Kuo CJ, Shie JJ, Fang JM, Yen GR, Hsu JT, Liu HG, Tseng SN, Chang SC, Lee CY, Shih SR, Liang PH. 2008. Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg Med Chem 16:7388–7398. doi: 10.1016/j.bmc.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews DA, Dragovich PS, Webber SE, Fuhrman SA, Patick AK, Zalman LS, Hendrickson TF, Love RA, Prins TJ, Marakovits JT, Zhou R, Tikhe J, Ford CE, Meador JW, Ferre RA, Brown EL, Binford SL, Brothers MA, DeLisle DM, Worland ST. 1999. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc Natl Acad Sci U S A 96:11000–11007. doi: 10.1073/pnas.96.20.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patick AK, Binford SL, Brothers MA, Jackson RL, Ford CE, Diem MD, Maldonado F, Dragovich PS, Zhou R, Prins TJ, Fuhrman SA, Meador JW, Zalman LS, Matthews DA, Worland ST. 1999. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob Agents Chemother 43:2444–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Fan T, Yao X, Wu Z, Guo L, Lei X, Wang M, Jin Q, Cui S. 2011. Crystal structures of enterovirus 71 3C protease complexed with rupintrivir reveal the roles of catalytically important residues. J Virol 85:10021–10030. doi: 10.1128/JVI.05107-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu G, Qi J, Chen Z, Xu X, Gao F, Lin D, Qian W, Liu H, Jiang H, Yan J, Gao GF. 2011. Enterovirus 71 and coxsackievirus A16 3C proteases: binding to rupintrivir and their substrates and anti-hand, foot, and mouth disease virus drug design. J Virol 85:10319–10331. doi: 10.1128/JVI.00787-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 25.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. 2007. Phaser crystallographic software. J Appl Crystallogr 40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Emsley P, Cowtan K. 2004. COOT: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 27.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 28.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeLano W. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 30.Zeng D, Ma Y, Zhang R, Nie Q, Cui Z, Wang Y, Shang L, Yin Z. 2016. Synthesis and structure-activity relationship of α-keto amides as enterovirus 71 3C protease inhibitors. Bioorg Med Chem Lett 26:1762–1766. doi: 10.1016/j.bmcl.2016.02.039. [DOI] [PubMed] [Google Scholar]