

Abstract
Recent studies have suggested that interleukin 1 receptor‐like 1 (ST2) plays a critical role in pathogenesis of several cardiovascular disease conditions. In this study, we examined association of 13 single nucleotide polymorphisms (SNPs) of ST2 gene with essential hypertension (EH) risk in 1151 patients with EH and 1135 controls. Our study showed that variants rs11685424, rs12999364 and rs3821204 are highly associated with an increase in risk of EH, while rs6543116 is associated with a decrease risk of EH. Notably, in silico analyses suggested the G>C change of rs3821204, which located within the 3′UTR of soluble ST2 mRNA, disrupted a putative binding site for miR202‐3p. Functional analyses suggested that miR‐202‐3p significantly decreased soluble ST2‐G mRNA stability and inhibited its endogenous expression. Furthermore, we found increased plasma‐soluble ST2 (sST2) level was highly associated with CC genotype of rs3821204 in vivo. Taken together, our findings provide the first evidence that genetic variants in ST2 gene are associated with EH risk and variant rs3821204 may influence the development of EH by controlling sST2 expression.
Keywords: ST2, miR‐202‐3p, functional polymorphism, essential hypertension
Introduction
Hypertension is a significant public health problem worldwide and affects more than 30% of the population 1. EH is multifactorial disease for which genetic and environmental factors (such as diet rich in sodium, alcohol and smoking) are determinants 2, 3. Thus, identification of susceptibility genes for EH provides insights into the disease pathogenesis 4, 5.
It is well known that the interleukin 33 (IL‐33)/interleukin 1 receptor‐like 1 (ST2) pathway plays an important role in pathogenesis of cardiovascular disease such as atherosclerosis, heart failure and hypertension 6, 7. The ST2 gene is located on chromosome 2q12 and contains 11 exons, a proximal promoter and a distal promoter. This gene encodes an IL‐33 receptor which consists of two main isoforms, a transmembrane ligand (ST2L) and a soluble component (sST2, soluble ST2) 8. IL‐33 and ST2L form a heterodimeric receptor complex with IL‐1R accessory protein (IL‐1RAcP), triggering selective immune and inflammatory responses that result in antihypertrophic and antifibrotic myocardial effects 9, 10, 11, 12. Meanwhile, sST2 binds to IL‐33, and functions as a decoy receptor for IL‐33 signal, resulting in inhibition of cardiovascular disease benefits 11, 13, 14. Clinically, sST2 is associated with hypertension, and increased sST2 levels predict increased systolic blood pressure (SBP) 7, 15. Moreover, sST2 is reported as a novel cardiac biomarker of mechanical strain, which was recently shown to be increased in hypertension 16, 17.
Although higher sST2 concentration has been reported in association with hypertension, association between ST2 polymorphisms and EH has not been systematically investigated. Accordingly, we suggested that genetic variations of the ST2 gene influence EH development by interfering the function or expression of ST2. To investigate this, we performed a genetic association analysis of ST2 and EH risk in a case–control study of the Han Chinese population. Further, we performed functional analyses to investigate the underlying functional role of ST2 variants in EH development.
Materials and methods
Study population
The sample population included 1151 EH patients and 1135 controls. Patients were unrelated to each other and recruited from the Han Chinese population. EH patients were recruited from three hospitals (Union Hospital, Tongji Hospital and Wugang Hospital) in Wuhan City, Hubei province, China 18. Hypertension was diagnosed as average SBP ≥140 mmHg, and/or average diastolic pressure (DBP) ≥90 mmHg, and/or self‐reported current treatment for hypertension with antihypertensive drugs. Control subjects were from the same community as the patients. Control subjects showed SBP <140 mmHg, DBP <90 mmHg and had never been treated for hypertension. Blood pressure was measured according to reported guidelines 2. All patients completed an Inter‐Heart questionnaire and were interviewed by trained interviewers for their demographic data, medical history, history of disease, family history of cardiovascular disease and lifestyle habits (including smoking and alcohol consumption). Six normal tissues (heart, kidney, liver, peripheral blood mononuclear cells (PBMCs), spleen and vein) were obtained at Union Hospital, and immediately stored in liquid nitrogen after surgery until further use. Each tissue was from four different donors. The study was approved by the Ethics Committee of Tongji Medical College, and all participants provided written informed consent and adhered to the principles of the Declaration of Helsinki.
Selection of polymorphisms
TagSNPs were selected based on the HapMap phase I & II database [http://www.hapmap.org; with CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) as the reference set]. According to the criteria of r 2 ≥ 0.8 and minor allele frequency (MAF) ≥0.05, to cover the distal promoter region of ST2, we extended 30 kb into the 5′ and 2000 bp into the 3′ end of ST2, and selected six tagSNPs to be analysed. Another seven SNPs were selected from previous reports of coronary heart disease (CHD) (‐27307T/A and ‐27614C/A), asthma and atopic dermatitis 19, 20, 21. In total, 13 SNPs were selected and genotyped (Table 1).
Table 1.
SNP locations and allele frequencies
| SNP | Location | Genotype | MAFa | HWE P a |
|---|---|---|---|---|
| rs3755278 | Intron | A/G | 0.067 | 1 |
| rs10206753 | Exon | C/T | 0.133 | 1 |
| rs1041973 | Exon | A/C | 0.156 | 0.527 |
| rs11685424 | Distal promoter | A/G | 0.487 | 0.05 |
| rs6543116 | Distal promoter | A/G | 0.367 | – |
| rs951774 | Distal promoter | A/C | 0.233 | 0.251 |
| rs10515922 | Distal promoter | C/T | 0.151 | 1 |
| rs13006559 | Distal promoter | T/C | 0.07 | 1 |
| ‐27307T/A | Distal promoter | T/A | – | – |
| ‐27614C/A | Distal promoter | C/A | – | – |
| rs12999364 | Between genes | T/C | 0.45 | 0 |
| rs3821204 | 3′‐Flanking region | C/G | 0.358 | 0.403 |
| rs13431828 | 5′‐Flanking region | T/C | 0.089 | 1 |
National Center for Biotechnology Information (NCBI) data; –, data not found in http://www.ncbi.nlm.nih.gov/snp/.
Bioinformatics
Appropriate bioinformatics websites were used to predict the putative binding site for rs3821204: http://snpinfo.niehs.nih.gov/snpinfo/snp func.htm and http://epicenter.ie-freiburg.mpg.de/services/microsniper/.
DNA isolation and genotyping
Fasting venous blood was collected from the peripheral vein. DNA was extracted using a Puregene kit (Gentra Systems, Inc., Minneapolis, MN, USA). Twelve SNPs were genotyped using the Sequenom MassArray system (Sequenom Inc., San Diego, CA, USA). The primers and probes used are listed in Table S1. The remaining SNP, rs3755278, was genotyped by TaqMan SNP allelic discrimination (Applied Biosystems, Foster City, CA, USA). The catalogue number for genotyping rs3755278 was C_27477453_10. TaqMan data collection and analysis were performed with SDS 2.2.1. Overall, all 13 SNPs were successfully genotyped with a call rate >96%. About 10% of the samples were randomly selected for repeat genotyping of the three SNPs for quality control, and the results were 100% concordant.
Cell culture and genotyping
The human umbilical vein cell line, EA.hy926, human embryonic kidney cell line, HEK293, and T‐acute lymphoblastic leukaemia cell line, KOPTK1, were obtained from American Type Culture Collection (ATCC) and cultured according to the manufacturer's protocols.
Genomic DNA was isolated from cells using the Guide Cell and Tissue Genomic DNA Extraction Kit (Tiangen, Beijing, China). The genotypes of cell lines were determined by Taqman genotyping assay.
Biological variable determination
Total cholesterol (TC), triglyceride (TG) and fasting blood glucose (FBG) levels were measured by standard laboratory procedures at the Department of Clinical Laboratory, Union Hospital.
RNA isolation and reverse transcription PCR
Total RNA was extracted using Trizol (Life Technologies, Carlsbad, CA, USA). For reverse transcription PCR (RT‐PCR), cDNA was obtained using the SuperScriptIII® Frist‐Strand Synthesis System (Life Technologies). Gene expression was monitored by PCR assay. The primer sequences were as follows: sST2 forward, 5′‐ GGCACACCGTAAGACTAAGTAG‐3′ and sST2 reverse, 5′‐CAATTTAAGCAGCAGAGAAGCTCC‐3′; β‐actin forward, 5′‐CCCAGCCATGTACGTTGCTAT‐3′ and β‐actin reverse, 5′‐TCACCGGAGTCCATCACGAT ‐3′. Levels of miR‐202‐3p and U6 were monitored by quantitative RT‐PCR using the Bulge‐Loop™ miRNA RT‐PCR primer set (RiboBio Co., Ltd. Guangzhou, China).
UTR cloning and point mutant construction
The 3′‐untranslated region (UTR) fragment of ST2 was amplified with primers: forward, 5′‐GTCTCTAGAATCCCCCACTCCCTCC‐3′ and reverse, 5′‐CAGTCTAGAATCTGTGTTCCTGCCC‐3′. A point mutation (G>C) was introduced by recombinant PCR using the primers: forward, 5′‐GTTTTTCTGGTCATAATGAAC‐3′ and reverse, 5′‐GTGTTCATTATGACCAGAAAAACGTATAGAACGG‐3′. Fragments were digested with XbaI and inserted into the luciferase reporter vector, pGL3‐control (Promega, Madison, WI, USA).
Dual‐luciferase reporter gene assay
HEK293 cells were seeded into 24‐well plates at 20,000 cells/well, and incubated at 37°C for 24 hrs. Luciferase constructs (with ~1‐kb sST2 3′‐UTR mRNA) and micro RNA (miRNA) mimics were cotransfected using lipofectamine 2000 (Life Technologies), following the manufacturer's instructions. At 48 hrs after transfection, cells were lysed and luciferase assays performed with the Dual‐Luciferase Reporter Assay System (Promega) on a single automatic injection Mithras luminometer (Berthold Technologies, Bad Wildbad, Germany), following the manufacturer's instructions. Ratios of firefly luciferase to Renilla luciferase readings were calculated.
Plasma sST2 levels determination
Fifty participants were selected from the control group. Plasma sST2 concentrations were measured using a highly sensitive sandwich immunoassay: Pressage ST2 (Critical Diagnostics, San Diego, CA, USA). Test procedures were performed according to the manufacturer's instructions. The lower limit of sST2 detection was 3.1 ng/ml and the upper limit 200 ng/ml.
Statistical analysis
Continuous variables were expressed as mean ± standard deviation (S.D.). Distribution of numerical data was determined by the Kolmogorov–Smirnov normality test. Continuous data with a normal distribution were compared by Student's t‐test and those with unequal variance or without a normal distribution were analysed by Mann–Whitney rank‐sum tests. Chi‐square tests were used to compare categorical variables and Hardy–Weinberg equilibrium (HWE) of SNPs. Unconditional logistic regression analysis was used to determine association between SNPs and hypertension risk, after adjustment for age, sex, smoking, drinking, body mass index (BMI), TG, FBG and family history of hypertension by odds ratios (ORs) and 95% confidence intervals (CIs). Power calculations were performed with the QUANTO software program (Version 1.2.3) (University of Southern California, Los Angeles, CA, USA). One‐way analysis of variance (anova) was used to examine differences in sST2 levels in patients with different rs3821204 genotypes. All other statistical analyses were performed with the statistical analysis software package SPSS 12.0 (SPSS Inc., Chicago, IL, USA). Differences or associations with two‐sided P‐values <0.05 were considered statistically significant.
Results
Characteristics of case and control patients
General participant characteristics are presented in Table 2. There were a total of 2286 participants, including 1151 EH patients and 1135 controls. Male and female numbers in both groups were similar. However, EH patients were older and had greater BMI and higher SBP, DBP, FBG and TG levels than controls. Further, EH patients were more likely to have a history of CHD, diabetes mellitus and a family history of hypertension. TC levels were significantly lower in EH patients than controls, possibly due to use of cholesterol‐lowering medications in the patient group.
Table 2.
General characteristics of the study population
| Variables | Controls (n = 1135) | Cases (n = 1151) | P value |
|---|---|---|---|
| Sex, m/f (%) | 888/247(78.2/21.8) | 899/252(78.1/21.9) | 0.939a |
| Age, years | 58.2 ± 11.7 | 62.3 ± 9.5 | <0.01b |
| Blood pressure, mmHg | |||
| Systolic | 125.5 ± 27.0 | 143.8 ± 24.2 | <0.01b |
| Diastolic | 78.4 ± 11.3 | 86.3 ± 14.1 | <0.01b |
| Body mass index, kg/m2 | 23.4 ± 3.2 | 24.7 ± 3.2 | <0.01b |
| Fasting glucose, mmol/l | 5.6 ± 2.2 | 6.2 ± 3.0 | <0.01b |
| Total cholesterol, mmol/l | 4.59 ± 1.00 | 4.44 ± 1.04 | <0.01b |
| Triglyceride, mmol/l | 1.57 ± 1.26 | 1.72 ± 1.41 | <0.01b |
| Smoking, no/yes (%) | 660/475(58.1/41.9) | 796/353(69.3/30.7) | <0.01a |
| Drinking, no/yes (%) | 766/365(67.7/32.3) | 830/314(72.6/27.4) | 0.012a |
| Past history | |||
| Coronary heart disease, no/yes | 784/351(69.1/30.9) | 361/790(31.4/68.6) | <0.01a |
| Diabetic patient, no/yes | 1041/92(91.9/8.1) | 861/287(75.0/25.0) | <0.01a |
| Family history of Hypertension, no/yes (%) | 862/266(76.4/23.6) | 689/443(60.9/39.1) | <0.01a |
Variables are presented as mean ± S.D. or percentages.
P‐values were calculated using chi‐square tests.
P‐values were calculated using independent‐samples t‐tests.
Association between ST2 variants and EH risk
Most of the SNPs conformed to HWE (P > 0.05), except for rs1041973 and rs10515922, which significantly deviated from HWE in EH patients and controls (P < 0.05) (see Table S2). MAFs for ‐27307T/A and ‐27614C/A were 0, and according to the HapMap database, these alleles have not previously been identified in the Chinese Han population. Consequently, we examined association between the remaining nine SNPs and EH risk. To determine association between these SNPs and EH risk, we analysed genotypes using additive and recessive models. Four of these SNPs (rs11685424, rs6543116, rs12999364 and rs3821204) were significantly associated with EH after adjusting for conventional EH risk factors such as age, sex, smoking, drinking, BMI, TG, FBG and family history of hypertension (Table 3).
Table 3.
Genotype frequencies of nine SNPs and their association with essential hypertension risk in the Chinese population
| SNPs | Controlsb | Casesb | Additive modelc | Recessive modeld | ||
|---|---|---|---|---|---|---|
| OR (95% CI)a | P a | OR (95% CI)a | P a | |||
| rs3755278 | 1021/76/4 | 994/99/3 | 1.34(0.94–1.90) | 0.102 | 1.40(0.97–2.02) | 0.071 |
| rs10206753 | 826/257/29 | 841/266/20 | 0.91(0.75–1.11) | 0.339 | 0.93(0.75–1.17) | 0.536 |
| rs11685424 | 307/537/247 | 253/581/282 | 1.25(1.09–1.44) | 0.002 | 1.33(1.06–1.66) | 0.015 |
| rs6543116 | 312/542/252 | 350/566/207 | 0.82(0.71–0.94) | 0.005 | 0.74(0.60–0.92) | 0.007 |
| rs951774 | 626/408/73 | 661/411/55 | 0.92(0.78–1.08) | 0.312 | 0.95(0.78–1.16) | 0.592 |
| rs13006559 | 1052/57/3 | 1051/79/2 | 1.25(0.84–1.88) | 0.278 | 1.36(0.89–2.08) | 0.158 |
| rs12999364 | 437/502/169 | 376/560/190 | 1.26(1.09–1.45) | 0.002 | 1.40(1.14–1.71) | 0.001 |
| rs3821204 | 478/493/134 | 428/540/157 | 1.25(1.08–1.45) | 0.003 | 1.33(1.09–1.63) | 0.005 |
| rs13431828 | 921/177/11 | 933/183/13 | 0.98(0.77–1.23) | 0.845 | 0.97(0.75–1.26) | 0.827 |
It means P‐values <0.05 and these SNPs were significantly associated with EH.
P‐values were calculated by unconditional logistic regression, after adjusting for age, sex, smoking, drinking, BMI, TG, FBG and family history of EH.
Wild‐type homozygote/heterozygote/variant homozygote.
Additive model (wild‐type homozygote versus heterozygote versus variant homozygote).
Recessive model (wild‐type homozygote versus heterozygote + variant homozygote).
Specifically, we found that the A allele of rs11685424 was associated with increased EH risk in both additive (P = 0.002, OR = 1.25, 95% CI = 1.09–1.44) and recessive (P = 0.015, OR = 1.33, 95% CI = 1.06–1.66) models. In contrast, the rs6543116 A allele was associated with decreased EH risk in both additive (P = 0.005, OR = 0.82, 95% CI = 0.71–0.94) and recessive (P = 0.007, OR = 0.74, 95% CI = 0.60–0.92) models. Additionally, there was a dose–response effect for increasing number of rs6543116 A alleles and decreased EH risk. Both rs12999364 and rs3821204 were also associated with EH risk in both additive (P = 0.002, OR = 1.26, 95% CI = 1.09–1.45 and P = 0.003, OR = 1.25, 95% CI = 1.08–1.45, respectively) and recessive (P = 0.001, OR = 1.40, 95% CI = 1.14–1.71 and P = 0.005, OR = 1.33, 95% CI = 1.09–1.63, respectively) models. Overall, our findings suggest increased EH risk in individuals carrying these homozygous ST2 variants. Except for rs11685424 and rs6543116 in the recessive model, P‐values for these SNPs reached significance after Bonferroni correction (P < 0.0055; 0.05/9). There were no significant associations between the remaining five SNPs and EH risk in our population (P > 0.05). Given the frequency of CHD within this case–control sample population, we analysed subgroups without CHD patients to determine whether the effect was influenced by CHD risk. Exclusion of individuals with CHD did not change the results (see Table S3).
The ST2 polymorphism rs3821204 affects sST2 mRNA stability
In silico analysis showed that the variants, rs11685424, rs12999364, and rs6543116, disrupted transcription factor binding sites, while rs3821204 disrupted a miRNA seeding site (Table S4). Together, this suggests these variants may influence EH susceptibility by controlling their own expression. Previous studies have confirmed this, and shown that rs11685424 and rs6543116 are associated with elevated EH risk and increased sST2 expression 19, 22. As noted, rs3821204 disrupts a putative miRNA binding site; therefore we predicted that this variant controls ST2 expression via a mechanism that is distinct compared with the three other SNPs. To identify this mechanism, we performed a bioinformatics assay using the software, MicroSNiPer, to predict the putative function of rs3821204. We found that the G>C change associated with rs3821204 disrupts a putative binding site for miR‐202‐3p and miR‐548 m (Fig. 1A). Accordingly, we hypothesize that rs3821204 controls sST2 expression in a miRNA‐mediated post‐transcriptional manner.
Figure 1.
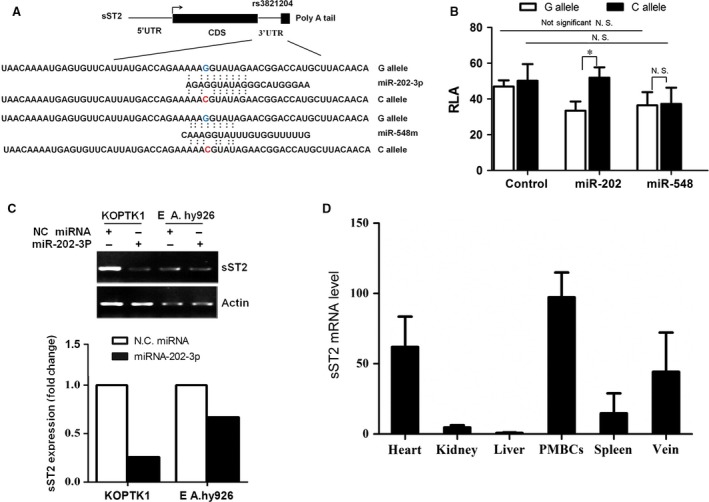
Allelic difference of rs3821204 G>C in sST2 mRNA stability and expression (A) In silico prediction of the miR‐202‐3p binding site in sST2‐G mRNA. Putative rs3821204 function was determined using the SNPinfo database. (B) The effect of miR‐202‐3p using RLA of sST2‐3′‐UTR fusion constructs encompassing different alleles. Constructs containing G or C rs3821204 alleles were cotransfected with miR‐202‐3p or a negative control. Relative luciferase activity was analysed at 48 hrs post‐transfection. Data represent mean ± S.D. from three independent experiments. *P < 0.05. (C) Allelic difference in endogenous sST2 mRNA levels after miRNA‐202‐3p transfection. Genotypes of rs3821204 were determined by Taqman assay in KOPTK1 and EA.hy926 cells. miRNA‐202‐3p mimics were transfected into KOPTK1 (GG) and EA.hy926 (CC) cells. At 48 hrs post‐transfection, cells were harvested to isolate total RNA. sST2 mRNA levels were monitored to determine the effect of miR‐202‐3p on sST2‐G and sST2‐C degradation. (D) miR‐202‐3p expression in different human tissues. Total RNA was isolated from six tissues from four different individuals. miRNA levels were determined by quantitative RT‐PCR using a Bulge‐Loop™ miRNA RT‐PCR primer set. U6 was used as the loading control. miR‐202‐3p in different tissues was expressed relative to liver miR‐202‐3p expression.
As miRNAs often function as negative regulators of gene expression, we performed luciferase assays to determine the effect of rs3821204 on sST2 expression. We found that miR‐202‐3p significantly decreased RLA of ST2‐G but not ST2‐C, while miR‐548 m did not alter RLA of ST2‐G or ST2‐C (Fig. 1B). This is consistent with miR‐202‐3p overexpression, which caused a sharp reduction in endogenous sST2‐G mRNA levels, but not sST2‐C (Fig. 1C).
Additionally, we examined miR‐202‐3p tissue distribution by RT‐PCR. Greatest expression was observed in PBMCs, followed by the heart and vein (Fig. 1D). These findings are coincident with tissue distribution of sST2 protein, suggesting that regulation of sST2 by miR‐202‐3p might be critical to cardiovascular function.
In vivo effect of the ST2 polymorphism rs3821204 on plasma sST2 levels
To examine the effect of rs3821204 on sST2 plasma levels, we performed ELISA and a Taqman genotyping assay in 50 healthy individuals (median [25–75th percentile], 13.28 ng/ml [8.03–16.91 ng/ml]). Plasma sST2 concentrations were significantly lower in patients with GG genotypes compared to those with CC genotypes (P < 0.05) (Fig. 2). These results suggest that rs3821204 G>C is associated with increased plasma sST2 levels in vivo.
Figure 2.
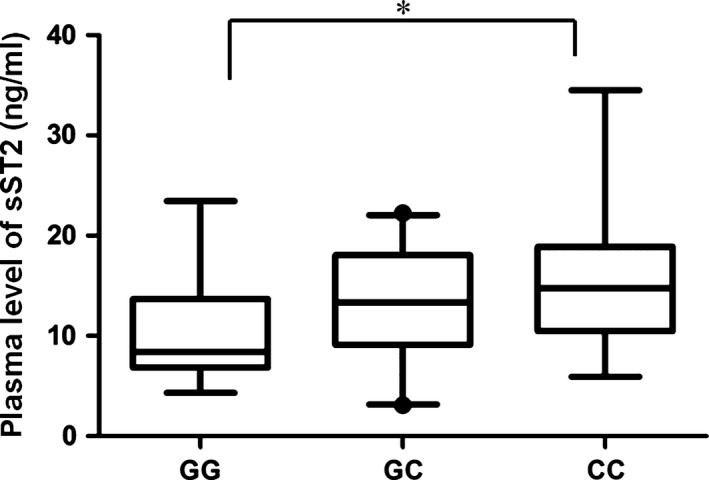
Circulating sST2 levels differ by rs3821204 genotype. Plasma concentrations of sST2 protein were measured by ELISA in 50 healthy donors carrying different rs3821204 genotypes (GG, n = 14; GC, n = 25; and CC, n = 11). Box plots indicate minimum, 10% percentile, maximum and 90% percentile for each group. *P < 0.05.
Discussion
In the present study, we have investigated association between 13 ST2 gene polymorphisms and EH risk. Accordingly, we found four SNPs significantly associated with EH risk. Functional analyses showed the G>C change of rs3821204 disrupted a binding site for miR‐202‐3p and enhance sST2 mRNA expression by blocking miR‐202‐mediated degradation. In addition, patients with the rs3821204 C allele had significantly higher plasma sST2 levels. Taken together, our findings suggest that genetic variation within ST2 modifies EH susceptibility by regulating its own expression.
The IL‐33/ST2 pathway plays an important role in inflammatory and immunity diseases, including asthma, atopic dermatitis and coronary artery disease 23. Recent research reported sST2 as a new prognostic biomarker in patients with hypertension 7, 15. Nonetheless, association between ST2 polymorphisms and EH risk has not been systematically studied. In this case–control study, we found that the rs11685424 and rs6543116 genetic variations within the ST2 promoter region are associated with EH risk. These two variants modify transcriptional activity of the ST2 promoter 19, 22. Thus, it is likely that the biological consequence of these genetic variants may influence EH susceptibility. Recently, Tu and Tsapaki reported association between ST2 variants and CHD 20, 22. To minimize this disturbance, we re‐analysed association between ST2 variants and EH risk in subgroups without CHD. Similar results were obtained, indicating that the four variants are associated with EH independent of CHD status. To the best of our knowledge, this is the first study to systematically investigate association between ST2 genetic variants and EH risk, and provides obvious evidence for their association.
Micro RNAs are key regulators in the pathogenic processes of hypertension, and are involved in endothelial activation and dysfunction, vascular smooth muscle cell proliferation and inflammation 24, 25, 26, 27. Mounting evidence suggests that SNPs located within miRNA binding sites affect hypertension risk by interfering with miRNA function 28, 29, 30, 31. Our findings here demonstrate that the rs3821204 C allele disrupts the miR‐202‐3p binding site within the 3′‐UTR of sST2, and consequently disrupts sST2 mRNA expression and increases circulating sST2 protein levels (Figs. 1B and 2). Furthermore, our findings provide evidence of miR‐202‐3p expression in heart, PBMCs and vein, which is consistent with tissue distribution of sST 32, 33. Taken together, our results suggest that ST2 regulation by miR‐202‐3p is critical for maintaining normal cardiovascular activity, with its disruption having severe consequences.
Previous epidemiological studies have shown that elevated plasma sST2 concentration is associated with risk and outcome of hypertension 7, 15, 16, 17. Consistently, we also found that the rs3821204 C allele is associated with elevated plasma levels of ST2 (Fig. 2), and increased risk of EH (Table 3). Thus, our findings provide further evidence to support the important role of ST2 in EH development. However, the mechanism by which sST2 affects development of EH is poorly understood. Previous studies have shown that IL‐33 signalling via ST2L exhibits cardioprotective effects against various cardiovascular diseases (e.g. myocardial infarction, atherosclerosis, pulmonary arterial hypertension, cardiac hypertrophy and fibrosis) by modulating immune and inflammatory responses 6, 11, 34. Hence, we assume that sST2, an IL‐33 decoy receptor, may function in hypertension risk by disrupting IL33‐ST2L signalling.
In our present study, we had >90% power to detect association between the SNPs and EH risk, at a 0.05 significance level under both additive and recessive models, except for rs11685424, which had 84% power to detect association under the recessive model. Nevertheless, our study has several limitations. First, the study sample was not sufficiently large, especially when CHD patients were excluded. Further studies are needed to replicate our results in a larger sample size population. Second, our sample population was not well matched, but we addressed this using unconditional logistic regression analysis with adjustment for age, sex, smoking, drinking, BMI, TG, FBG and family history of hypertension. Third, all the volunteers recruited in our study were ethnic Han Chinese, and association between these SNPs and EH risk in other ethnic groups needs further validation in larger prospective studies.
In conclusion, we have found significant association between a ST2 polymorphism and risk of EH. Additionally, our findings further implicate rs3821204 as a functional polymorphism that contributes to increase EH risk and enhanced sST2 expression by disruption of a miR‐202‐3p binding site. Thus, our findings imply that ST2 may strongly influence development of EH, further supporting the important role of ST2 in EH, and highlighting ST2 as a valuable target for prevention and treatment of EH.
Conflict of interests
The authors confirm that there are no conflict of interests.
Supporting information
Table S1. Primer sequences for genotyping 12 SNPs from the ST2 gene using the Sequenom platform.
Table S2. Frequencies of 13 ST2 SNPs in the study population.
Table S3. Secondary analyses of four SNPs significantly associated with EH risk in the subgroup without CHD subjects.
Table S4. Functional prediction of ST2 variants.
Acknowledgement
This work was supported by grants from the National Natural Scientific Foundation of China (Grant no. 81172750).
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, et al Executive Summary: Heart Disease and Stroke Statistics‐2016 Update: a Report From the American Heart Association. Circulation. 2016; 133: 447–54. [DOI] [PubMed] [Google Scholar]
- 2. Mancia G, De Backer G, Dominiczak A, et al 2007 Guidelines for the Management of Arterial Hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2007; 25: 1105–87. [DOI] [PubMed] [Google Scholar]
- 3. Simino J, Rao DC, Freedman BI. Novel findings and future directions on the genetics of hypertension. Curr Opin Nephrol Hypertens. 2012; 21: 500–7. [DOI] [PubMed] [Google Scholar]
- 4. Wang Z, Xu Y, Chen S, et al A common missense single nucleotide polymorphism in the E‐selectin gene is significantly associated with essential hypertension in the Han population but only weakly associated in the Uygur population. Hypertens Res. 2012; 35: 413–7. [DOI] [PubMed] [Google Scholar]
- 5. Balam‐Ortiz E, Esquivel‐Villarreal A, Huerta‐Hernandez D, et al Hypercontrols in genotype‐phenotype analysis reveal ancestral haplotypes associated with essential hypertension. Hypertension. 2012; 59: 847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miller AM, Liew FY. The IL‐33/ST2 pathway–A new therapeutic target in cardiovascular disease. Pharmacol Ther. 2011; 131: 179–86. [DOI] [PubMed] [Google Scholar]
- 7. Ho JE, Larson MG, Ghorbani A, et al Soluble ST2 predicts elevated SBP in the community. J Hypertens. 2013; 31: 1431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmitz J, Owyang A, Oldham E, et al IL‐33, an interleukin‐1‐like cytokine that signals via the IL‐1 receptor‐related protein ST2 and induces T helper type 2‐associated cytokines. Immunity. 2005; 23: 479–90. [DOI] [PubMed] [Google Scholar]
- 9. Chackerian AA, Oldham ER, Murphy EE, et al IL‐1 receptor accessory protein and ST2 comprise the IL‐33 receptor complex. J Immunol. 2007; 179: 2551–5. [DOI] [PubMed] [Google Scholar]
- 10. Liu X, Hammel M, He Y, et al Structural insights into the interaction of IL‐33 with its receptors. Proc Natl Acad Sci USA. 2013; 110: 14918–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sanada S, Hakuno D, Higgins LJ, et al IL‐33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest. 2007; 117: 1538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seki K, Sanada S, Kudinova AY, et al Interleukin‐33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail. 2009; 2: 684–91. [DOI] [PubMed] [Google Scholar]
- 13. Demyanets S, Konya V, Kastl SP, et al Interleukin‐33 induces expression of adhesion molecules and inflammatory activation in human endothelial cells and in human atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2011; 31: 2080–9. [DOI] [PubMed] [Google Scholar]
- 14. Chen WY, Hong J, Gannon J, et al Myocardial pressure overload induces systemic inflammation through endothelial cell IL‐33. Proc Natl Acad Sci USA. 2015; 112: 7249–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coglianese EE, Larson MG, Vasan RS, et al Distribution and clinical correlates of the interleukin receptor family member soluble ST2 in the Framingham Heart Study. Clin Chem. 2012; 58: 1673–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ojji DB, Opie LH, Lecour S, et al Relationship between left ventricular geometry and soluble ST2 in a cohort of hypertensive patients. J Clin Hypertens (Greenwich). 2013; 15: 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ojji DB, Opie LH, Lecour S, et al The effect of left ventricular remodelling on soluble ST2 in a cohort of hypertensive subjects. J Hum Hypertens. 2014; 28: 432–7. [DOI] [PubMed] [Google Scholar]
- 18. Shi Y1, Zhou L, Huang LH, et al Plasma ferritin levels, genetic variations in HFE gene, and coronary heart disease in Chinese: a case‐control study. Atherosclerosis. 2011; 218: 386–90. [DOI] [PubMed] [Google Scholar]
- 19. Shimizu M1, Matsuda A, Yanagisawa K, et al Functional SNPs in the distal promoter of the ST2 gene are associated with atopic dermatitis. Hum Mol Genet. 2005; 14: 2919–27. [DOI] [PubMed] [Google Scholar]
- 20. Tsapaki A, Zaravinos A, Apostolakis S, et al Genetic variability of the distal promoter of the ST2 gene is associated with angiographic severity of coronary artery disease. J Thromb Thrombolysis. 2010; 30: 365–71. [DOI] [PubMed] [Google Scholar]
- 21. Akhabir L, Sandford A. Genetics of interleukin 1 receptor‐like 1 in immune and inflammatory diseases. Curr Genomics. 2010; 11: 591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tu X, Nie S, Liao Y, et al The IL‐33‐ST2L pathway is associated with coronary artery disease in a Chinese Han population. Am J Hum Genet. 2013; 93: 652–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kakkar R, Lee RT. The IL‐33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008; 7: 827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gray C, Li M, Patel R, et al Let‐7 miRNA profiles are associated with the reversal of left ventricular hypertrophy and hypertension in adult male offspring from mothers undernourished during pregnancy after preweaning growth hormone treatment. Endocrinology. 2014; 155: 4808–17. [DOI] [PubMed] [Google Scholar]
- 25. Li C, Mpollo MS, Gonsalves CS, et al Peroxisome proliferator‐activated receptor‐alpha‐mediated transcription of miR‐199a2 attenuates endothelin‐1 expression via hypoxia‐inducible factor‐1alpha. J Biol Chem. 2014; 289: 36031–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lacolley P, Regnault V, Nicoletti A, et al The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res. 2012; 95: 194–204. [DOI] [PubMed] [Google Scholar]
- 27. Cengiz M, Yavuzer S, Kılıçkıran Avcı B, et al Circulating miR‐21 and eNOS in subclinical atherosclerosis in patients with hypertension. Clin Exp Hypertens. 2015; 37: 643–9. [DOI] [PubMed] [Google Scholar]
- 28. Yang Z, Kaye DM. Mechanistic insights into the link between a polymorphism of the 3′UTR of the SLC7A1 gene and hypertension. Hum Mutat. 2009; 30: 328–33. [DOI] [PubMed] [Google Scholar]
- 29. Yang S, Gao Y, Liu G, et al The human ATF1 rs11169571 polymorphism increases essential hypertension risk through modifying miRNA binding. FEBS Lett. 2015; 589: 2087–93. [DOI] [PubMed] [Google Scholar]
- 30. Mopi devi B, Ponnala M, Kumar A. Human angiotensinogen +11525 C/A polymorphism modulates its gene expression through microRNA binding. Physiol Genomics. 2013; 45: 901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gong J, Tian J, Lou J, et al A functional polymorphism in lnc‐LAMC2‐1:1 confers risk of colorectal cancer by affecting miRNA binding. Carcinogenesis. 2016; 37: 443–51. [DOI] [PubMed] [Google Scholar]
- 32. Demyanets S1, Kaun C, Pentz R, et al Components of the interleukin‐33/ST2 system are differentially expressed and regulated in human cardiac cells and in cells of the cardiac vasculature. J Mol Cell Cardiol. 2013; 60: 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mildner M1, Storka A, Lichtenauer M, et al Primary sources and immunological prerequisites for sST2 secretion in humans. Cardiovasc Res. 2010; 87: 769–77. [DOI] [PubMed] [Google Scholar]
- 34. Kunes P, Holubcova Z, Kolackova M, et al Interleukin‐33, a novel member of the IL‐1/IL‐18 cytokine family, in cardiology and cardiac surgery. Thorac Cardiovasc Surg. 2010; 58: 443–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences for genotyping 12 SNPs from the ST2 gene using the Sequenom platform.
Table S2. Frequencies of 13 ST2 SNPs in the study population.
Table S3. Secondary analyses of four SNPs significantly associated with EH risk in the subgroup without CHD subjects.
Table S4. Functional prediction of ST2 variants.