

Abstract
Luminal Ca2+ in the endoplasmic and sarcoplasmic reticulum (ER/SR) plays an important role in regulating vital biological processes, including store-operated capacitative Ca2+ entry, Ca2+-induced Ca2+ release, and ER/SR stress-mediated cell death. We report rapid and substantial decreases in luminal [Ca2+], called “Ca2+ blinks,” within nanometer-sized stores (the junctional cisternae of the SR) during elementary Ca2+ release events in heart cells. Blinks mirror small local increases in cytoplasmic Ca2+,orCa2+ sparks, but changes of [Ca2+] in the connected free SR network were below detection. Store microanatomy suggests that diffusional strictures may account for this paradox. Surprisingly, the nadir of the store depletion trails the peak of the spark by about 10 ms, and the refilling of local store occurs with a rate constant of 35 s-1, which is ≈6-fold faster than the recovery of local Ca2+ release after a spark. These data suggest that both local store depletion and some time-dependent inhibitory mechanism contribute to spark termination and refractoriness. Visualization of local store Ca2+ signaling thus broadens our understanding of cardiac store Ca2+ regulation and function and opens the possibility for local regulation of diverse store-dependent functions.
Keywords: calcium-induced calcium release, calcium spark, cardiac myocytes, endoplasmic reticulum, sarcoplasmic reticulum
Local Ca2+ releases from the endoplasmic reticulum (ER) or sarcoplasmic reticulum (SR) in muscle have been shown to underlie neurosecretion, memory encoding, neurite growth, muscle contraction, and apoptosis (1–4). Whereas the ER/SR serves primarily as the intracellular Ca2+ store, luminal Ca2+ plays an active role in many regulatory systems, including store-operated capacitative Ca2+ entry (5, 6), Ca2+-induced Ca2+ release (7–9), and ER/SR stress-mediated cell death (10, 11). Over the last decade, the elementary Ca2+ release events have been directly visualized as Ca2+ “sparks” (12–15), “puffs” (16), “syntillas” (17), or the equivalent (18) in the cytoplasm of both excitable and nonexcitable cells. However, the reciprocal store depletion signals, which were speculated in various models of spark termination (refs. 7 and 19; see ref. 20 for a review), have not been seen experimentally. In theory, a rapid refilling of local store Ca2+ from the bulk of ER/SR might occur and prevent significant local Ca2+ depletion (21, 22). Alternatively, this failing could be due to lack of a means to probe Ca2+ inside this delicate membrane-bound intracellular structure with the required sensitivity, resolution, and speed, given the extremely small release flux involved (≈2·10-19 mol of Ca2+) (12, 17). Using confocal imaging, electron microscopy, and electrophysiological approaches, we investigated dynamic Ca2+ regulation inside nanometer-sized SR structures during elementary Ca2+ release events in intact heart muscle cells. Our results afforded insights into mechanisms underlying spark termination and refractoriness, intra-SR Ca2+ communication, and local regulation of store Ca2+ signaling.
Methods
Electron Microscopy. Rabbit hearts were perfused with 6% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2, at room temperature. The left ventricle trabeculae were dissected out, postfixed in 2% OsO4 in the same buffer, stained en bloc in uranyl acetate, and embedded in Epon. Sections were stained in uranyl acetate and lead salts and examined in a Philips 410 electron microscope. Linear dimensions of SR profiles were obtained by using the measuring tool of photoshop (Adobe Systems, San Jose, CA) and relative volumes of SR compartments, by the point-counting method (23).
Cells. Enzymatically isolated cardiac myocytes from adult New Zealand White rabbits were loaded with 20 μM fluo-5N acetoxymethyl ester (AM) (Molecular Probes) for 2 h at 37°C, as described previously (22, 24). For cytosolic Ca2+ imaging, cells were incubated with 5 μM rhod-2 AM for 15 min. To maintain adequate SR Ca2+ load for the study of Ca2+ sparks and Ca2+ waves, experiments were performed with extracellular Na+ replaced by equimolar Li+, an inhibitor of Na+/Ca2+ exchange, in the Hepes solution: 137 mM LiCl/4.9 mM KCl/1.2 mM MgCl2/1.2 mM NaH2PO4/1 mM CaCl2/15 mM glucose/20 mM Hepes (pH 7.4). Myocytes were perfused at 1 ml/min and field electrical stimulation was delivered at 0.5 Hz.
Confocal Ca2+ Imaging. Linescan images were acquired by using a confocal microscope (LSM510, Zeiss) equipped with a ×63, 1.4 numerical aperture, oil-immersion objective, at sampling rates of 0.75–1.5 ms per line and 50 nm per pixel. To measure SR and cytosol Ca2+ simultaneously, fluo-5N and rhod-2 were excited alternatively by 488- and 543-nm laser lines, respectively (at 667 Hz), and fluorescence was measured at 500–530 and >560 nm, respectively. All experiments were performed at room temperature (22–23°C).
Changes in local SR free [Ca2+] were determined by the formula Δ[Ca2+]SR = Kd[1 + ΔF/(F0 - Fmin)]/[Kd/C0 - ΔF/(F0 - Fmin)] - C0, where F0 and Fmin refer to the fluorescence level at rest and after emptying the SR, respectively; the fluo-5N′s Ca2+ dissociation constant Kd ≈ 400 μM, and the diastolic [Ca2+]SR C0 ≈ 1.0 mM based on previous in situ calibration (22). Fractional changes in calsequestrin-bound Ca2+ store were calculated by Kd,CSQΔ[Ca2+]SR/[C0(Kd,CSQ + [Ca2+]SR)], where Kd,CSQ ≈ 500 μM, referring to the Ca2+ dissociation constant of calsequestrin (25).
Cell-Attached Patch Clamp. Cell-attached patch clamping was established by using an Axopatch 200 B amplifier (Axon Instruments) in loose-seal configuration, as described in ref. 26. Glass patch pipettes of 5–7 MΩ contained the Hepes solution with the L-type Ca2+ channel agonist FPL641176 added at 10 μM. Membrane potential was determined by dividing proportionally the test voltages between the pipette resistance and the seal resistance (20–50 MΩ).
Automated Ca2+ Blink Detection. A computer algorithm used for automated spark detection (27) was modified for the detection of blinks. Specifically, the algorithm first identifies islets of connected pixels that are 0.5 standard deviation (σ) below the mean F0. The signal mass (S, sum of ΔF/F0) was calculated over N pixels in a box outlining an islet (see Fig. 2 A). The criterion for event detection is z = -S/(σ·N1/2) > 6.0, which corresponds to <1 false detection in 100 images (512 × 512) with Gaussian noise characteristics.
Fig. 2.

Ca2+ blink: nanoscopic store Ca2+ depletion. (A)Ca2+ blinks. (i) Raw linescan image of SR Ca2+ with intra-SR indicator fluo-5N. The brief darkening shows a spontaneous jSR Ca2+ release event, a blink. (ii) Normalized image (F/F0, to correct for the nonuniform indicator staining). (iii) Computer-aided automated detection of blinks. (iv) Time course of normalized blink from top. (B) Excitation-activated blinks (arrow). (i) Normalized linescan image showing four blink events. (ii) Time courses of blink events (arrowheads). (iii) Average time course with nonlinear fit (smooth curve; ΔF/F0 = -0.03, time to nadir tnadir = 29 ms, τrecovery = 31 ms). 1 μM nifedipine. (C) Surface plot of averaged blink (upper) with linescan image (lower) (n = 7). (D) Temporal characteristics of a blink (averaged: 36 spontaneous and 22 evoked events). Fit curve shows ΔF/F0 = -0.06, tnadir = 22 ms, and time constant τrecovery = 33 ms. (E) Spatial profile of the blink from D shows full width at half maximum (FWHM) 739 nm.
Data Analysis and Statistics. Fitting of blink time courses and spatial profiles used the nonlinear functions described previously for sparks (28, 29). Blink parametric measurement was based on the fitting results. Blinks or sparks were aligned by the onsets to obtain average time courses, and by the nadirs or peaks to obtain average spatial profiles. For spark–blink pairs, the nonlinear fitting was applied to the spark only for superior signal-to-noise properties. Alignment of the spark–blink pairs was then guided by the onsets of sparks. Computer programs were coded in Interactive Data Language, IDL (Research Systems, Boulder, CO). Data were expressed as mean ± SD. The significance of difference between means was determined, when appropriate, by using the nonparametric Kruskal–Wallis test and the Student t test. A P < 0.05 was considered statistically significant.
Results and Discussions
Microanatomy of the SR Network. The ER/SR is composed of interconnected cisternae and tubules that extend throughout the cytosol (Fig. 1), make signaling contacts with the plasmalemma (30), and are continuous with the nuclear envelope (5). The following description highlights features and morphometrics of cardiac SR in rabbit myocardium that are essential to this work. The SR is divided into two distinct domains: the junctional (jSR) and free (fSR) domains. The jSR forms extended flat cisternae that appear in thin sections as linear profiles, with an average length of 465 ± 200 nm (mean ± SD, n = 147 profiles) and a luminal width of ≈30 nm (Fig. 1 A and B). From this, we estimate an average diameter of 592 nm and a luminal volume of 0.008 fl. The interiors of the cisternae are electron dense (Fig. 1B), because of the Ca2+ binding protein calsequestrin. The pancake-shaped cisternae form junctions with the transverse (T) tubules at the level of the sarcomeric Z lines. SR and T tubules are separated by a narrow (≈12 nm) junctional gap, occupied by feet or cytoplasmic domains of RyRs, the Ca2+ release channels (Fig. 1E). Each SR cisterna bears one or two groups of RyRs containing on the average ≈66 feet. The fSR forms a network most abundant over the A band, with occasional continuities across the Z lines, and contains the majority of the Ca2+ pumps. The jSR and fSR occupy 2.0% and 2.2%, respectively, of the “cytoplasmic” volume (total volume minus mitochondria, nuclei, and SR volumes) in rabbit ventricular myocytes. Because the jSR occupies a narrow band at the level of the Z lines, whereas the fSR is distributed over most of the sarcomere (Fig. 1F), the jSR volume density at the Z line is higher than that of the fSR elsewhere.
Fig. 1.

Microanatomy of SR in rabbit cardiac cells. (A) Longitudinal section of rabbit left ventricular myocyte, showing junctional SR (jSR) cisternae near T tubules (T) and free SR (fSR). Arrowheads indicate continuities between jSR and fSR. (B and C) Details from A. Connections between jSR and fSR (arrowheads in B) were seen in 60 of 136 cisterna profiles in 60-nm-thick sections only at the periphery of the flat jSR cisternae. The average number of connections was therefore estimated to be 4.3 for a 592-nm-diameter jSR cisterna. (D) Tangential view of fSR network: Cross section at the level of the Z line. jSR cisternae (bracketed by arrowheads) are apposed either to T tubules (T) or the surface membrane. Profiles of fSR are seen next to a mitochondrion. (E) The cytoplasmic domains of ryanodine receptors (RyRs) are seen as feet in the gap between jSR and T tubule membranes (between arrows). (F) Diagram showing the relationship between the fSR network, the flat jSR cisternae, T tubules, and feet in rabbit ventricle.
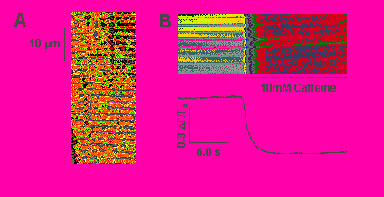
Ca2+ Blinks Inside jSR Cisternae. Dynamic Ca2+ regulation within the SR particularly at the level of the jSR cisternae was visualized with the low-affinity Ca2+ indicator fluo-5N, loaded with a protocol favoring SR retention (Fig. 6, which is published as supporting information on the PNAS web site) (22, 31). Under conditions conducive to spontaneous Ca2+ sparks in rabbit heart cells (see Methods), we detected small, brief, and spatially limited “darkenings” of the fluo-5N signal or Ca2+ blinks (Fig. 2Ai). Blinks occur at the Z lines, where the jSR cisternae are located, and were better resolved in the normalized images (F/F0) (Fig. 2 Aii) and fluorescence time-course plots (Fig. 2 Aiv) as clear dips amid background noise. Computer-automated detection of blinks (Fig. 2 Aiii) assisted the unbiased collection of events and showed that their rate of occurrence was 0.61 ± 0.47 s-1·(100 μm)-1 (n = 15 cells). Motion artifacts could be discounted as a cause, because blinks persisted when tension was inhibited by 5–10 mM 2,3-butanedione monoxime (32) but were completely eliminated by RyR block (10 μM ryanodine for 5 min, n = 5 cells) or inhibition of the SR Ca2+ pump (10 μM thapsigargin for 15 min, n = 6 cells).
Electrical stimulation-activated sparks summate into global Ca2+ transients (33, 34). To determine whether excitation evokes blinks in parallel to sparks, we examined the excitation-evoked Ca2+ release in the presence of the L-type Ca2+ channel antagonist nifedipine (1 μM, to reduce the density of evoked sparks for resolution of individual events). Linescan image (Fig. 2Bi), individual records (Fig. 2Bii), and averaged signals (Fig. 2Biii) show that depolarization activated synchronized blinks at multiple jSR sites. Such triggered blinks were blocked by the replacement of extracellular Ca2+ with equimolar Ba2+ (n = 6 cells), indicating that they are activated by L-type Ca2+ current by the same mechanism as sparks. These observations suggest that the blinks underlie evoked as well as spontaneous sparks, thus representing a direct real-time measure of intraorganelle Ca2+ in a physiological process.
Blinks Revealing Intra-SR Ca2+ Communication. Important and surprising findings were uncovered with the spatiotemporal analysis of blinks. First, we found that during a blink, local store [Ca2+] falls rapidly, reaching its minimum (“nadir”) in 24 ± 11 ms (n = 36 spontaneous and 22 evoked events), and then recovers in a fast monoexponential process with time constant of 29 ± 20 ms (Fig. 2 C and D). The blinks are thus in sharp contrast to jSR depletions in whole-cell Ca2+ release, namely “Ca2+ scraps” (22), which displayed a time to nadir (tnadir) of 137 ± 44 ms and a half recovery time of 194 ± 92 ms (n = 6) (Fig. 3 C and D). Compared with blinks, the scraps manifest a slower tnadir, for they are the results of many jSR cisternae responding to a common stimulus but not in synchrony. They manifest a slower recovery, because recovery of scraps depends mainly on Ca2+ reuptake globally by the SR Ca2+ pump, whereas the recovery of blinks is presumably dictated by Ca2+ movement within the SR from fSR to jSR. Thus, blink detection unmasks heretofore unappreciated kinetic features of cisternal Ca2+ signaling. Remarkably, tnadir of Ca2+ blinks is significantly longer than the time to peak (tpeak) of sparks as measured by us in the same cells (18 ± 6.2 ms, n = 86; P < 0.001 vs. tnadir) and also as reported for rat and mouse (10 ms) (12, 26, 33, 34). These features suggest that the jSR Ca2+ release flux is a time-dependent declining function, and the termination of release occurs after the peak of the spark.
Fig. 3.

Blinks revealing mechanisms of spark termination. (A) Same-site comparison of jSR depletions in blinks vs. Ca2+ waves. The fractional blink/wave amplitude is 0.61 at the site marked by arrowhead. (B) Statistical analysis of fluo-5N signal (ΔF/F0) for averaged blink from spark–blink pairs (bar 1), individually resolved blinks (bar 2), individual blinks corrected for background contamination (bar 3), full-fledged excitation-evoked release (bar 4), Ca2+ waves (bar 5), and caffeine (10 mM)-elicited Ca2+ release (bar 6). Data are expressed as mean ± SD and n = 6–86. (C) Action potential-elicited store Ca2+ transients or scraps. ΔF/F0 = -0.17 ± 0.05, n = 6. Such [Ca2+] transients correspond to a 65% reduction in the free [Ca2+] and a 38% diminution of the calsequestrin-bound Ca2+ (see Methods). Arrow and dashed line mark the timing of electrical stimulation. (D) Comparison between jSR scraps and blinks. Traces are normalized to the same amplitude. The traces for scraps at jSR and fSR essentially overlap, and the tnadir was 137 ± 44 ms, and the half-recovery time was 194 ± 92 ms (n = 6).
Second, blinks are even more sharply confined in space than sparks, displaying a full width at half maximum (FWHM) of 823 ± 476 nm (n = 58, Fig. 2E) vs. 2.5 ± 0.35 μm (n = 86, P < 0.0001) for sparks. Given the geometry of jSR cisternae (Fig. 1) and confocal volume element (Fig. 7, which is published as supporting information on the PNAS web site), we suggest that a blink is largely restricted to a single jSR cisterna and its contiguous fSR. Indeed, the blink width is only 49% broader than the optical width of the intensely indicator-stained band arising from jSR cisternae (FWHM = 554 ± 130 nm, n = 22). The demonstration of rapid nanoscopic store depletion strongly supports a role of luminal Ca2+ in spark termination and opens the possibility for local regulation of diverse store-dependent biological processes.
Kinetic resolution of blinks also affords a glimpse into intrastore Ca2+ communication. Despite topological continuity with fSR, jSR Ca2+ signaling is compartmentalized during sparks, implicating a substantial resistance to translocation of Ca2+ from the fSR to the release cisternae. We therefore examined the connectivity between jSR and fSR. A direct continuity between the jSR cisterna and the fSR occurs exclusively at the periphery of each jSR cisterna and is seen in fewer than 1/2 of the cisternae profiles (60/136) in thin sections of ≈60-nm thickness (Fig. 1 A and B). From this observation, we estimate that an average jSR cisterna connects to the fSR at four or five sites along its periphery (Fig. 1B) and that smaller cisternae may have fewer connections. The lumen of the connecting tubules was ≈30 nm in diameter. The paucity of connections and their narrow lumen provide the structural basis for the functional compartmentalization of jSR Ca2+ signaling. Quantitatively, we estimated that the fSR-to-jSR Ca2+ transfer occurs with a rate constant of ≈35 s-1. By contrast, the lack of apparent reduction of fSR Ca2+ at a few hundreds of nanometers away from the jSR cisterna (Fig. 2E) suggests that Ca2+ equilibrates relatively rapidly within the fSR, consistent with the network nature of fSR.
Blinks Revealing Spark Termination Mechanisms. SR luminal Ca2+ is thought to regulate RyR sensitivity to cytosolic Ca2+-dependent activation (7, 19, 35–37), such that local store depletion may play a role in the termination of elementary Ca2+ signaling events. Quantifying the degree of jSR Ca2+ depletion in a blink is thus fundamental to understanding the possible involvement of local store depletion in terminating Ca2+ sparks. The blink amplitude was directly measured as ΔF/F0 of -0.08 ± 0.02 (n = 58); measurement of jSR Ca2+ depletion during Ca2+ waves passing the same release sites (Fig. 3A) yielded a blink/wave magnitude ratio of 0.56 ± 0.14 (n = 20). A similar result was observed for the ratio between blink amplitude and the global SR depletion in normal Ca2+ transients (Fig. 3C). However, the apparent blink amplitude may underestimate the true local Ca2+ depletion: our electron microscopy morphometry indicates that a typical jSR cisterna is only 13% of a diffraction-limited confocal volume element (≈0.06 fl). Thus background fluorescence (F0) may be influenced by optically colocalized nonactive cisternae and thereby produce an overestimated F0 and underestimated ΔF/F0. To determine the extent of this possible effect, we carried out serial thin-section electron microscopy to map cisterna organization, and we found that, on average, 1.5 cisternae colocalize to a diffraction-limited confocal volume element (Fig. 7). The corrected blink amplitude in ΔF/F0 is -0.13 (Fig. 3B), and the blink/wave magnitude ratio is 0.86. Based on in situ calibration of the fluo-5N signal (22), the cisternal Ca2+ depletion in a blink corresponds to about 54% reduction of the free Ca2+ and 28% liberation of the calsequestrin-bound Ca2+, which are nearly as substantial as those during full-fledged excitation-evoked releases or Ca2+ waves (Fig. 3B). This result suggests that cisternal Ca2+ signaling operates virtually in an all-or-none fashion, due perhaps to the aforementioned diffusional strictures. The unexpectedly large jSR depletion during a spark further supports the notion for local store depletion to signal the termination of the elementary Ca2+ release events.
Once activated, a jSR undergoes refractoriness, as evidenced by the reduced amplitude of the succeeding sparks (Fig. 4A). We reasoned that local refractoriness and spark termination might share some common mechanisms. To delineate possible role of local store depletion in spark refractoriness, we measured the restitution curve for spark amplitude. By using loose-seal patch clamp technique combined with confocal microscopy (26), sparks from subsurface jSR release units were activated by 400-ms patch membrane depolarization to 80 mV above the resting potential. To analyze restitution of local Ca2+ release after spark activation, we acquired recordings with multiple sparks activated in tandem during a single pulse by including the L-type Ca2+ channel agonist FPL641176 (10 μM) in the patch pipette to sustain the trigger Ca2+ influx. The local restitution of Ca2+ release was then indexed by the amplitude of the second spark relative to its corresponding first spark. It is noteworthy that, by using the spark amplitude ratio, this measurement of release recovery from refractoriness should be independent of L-type channel inactivation, because spark morphometrics are known to be unaffected by characteristics of the trigger Ca2+ signal (33, 34). Our results indicate that spark amplitude restitution exhibits a time constant of 187 ms, whereas local store refilling is ≈6-fold faster (Fig. 4B). This kinetic disparity provides important evidence that, in addition to local store depletion, RyR inactivation (38) or “adaptation” (39) or some other time-dependent inhibition occurs during a spark and underlies refractoriness of local Ca2+ release. Coordinated operation of dual mechanisms may confer the reliability, speed, and completeness of release termination in a cardiac cycle.
Fig. 4.

Time courses of refilling of local Ca2+ store and refractoriness of local Ca2+ release. (A) Typical results of a pair of Ca2+ sparks evoked beneath the patch membrane in response to a 80-mV patch depolarization from the resting potential (RP, approximately -75 mV). Note the reduced amplitude of the succeeding spark. (B) The restitution curve for spark amplitude; 167 pairs of sparks obtained by using the protocol in A were grouped by intervals at 20-ms increments. n = 5–12 at each data point. (Inset) Comparison between the fitted blink time course (blue curve, from Fig. 2D) and the fitted restitution curve (red curve, τ = 187 ms).
Spark–Blink Pairs as Complementary Signaling Events. To more rigorously demonstrate that blinks are nanoscopic store Ca2+ signaling events complementary to sparks, we measured simultaneously store and cytosolic Ca2+ by using high-speed dual-indicator confocal microscopy. As shown in Fig. 5A, Ca2+ sparks were well resolved with cytosolic rhod-2 (ΔF/F0 = 0.30, tpeak = 21 ms, τrecovery = 43 ms, FWHM = 2.2 μm). The companion blinks obtained with SR-loaded fluo-5N, however, displayed degraded signal-to-noise property, because spectral separation of fluorescence has curtailed the blink signal (see Methods). To improve the blink measurement, we used signal averaging with sparks as a guide to align the corresponding areas in the rhod-2 and fluo-5N images. Results from 86 events in nine cells uncovered a clear spark-associated blink (Fig. 5 B and C). The average blink closely resembles the blinks in terms of tnadir (32 ms) and τrecovery (23 ms). The apparent blink amplitude, however, was reduced (ΔF/F0 = -0.013) and the width was slightly broadened (FWHM = 1.0 μm). This result was expected because sparks are many times bigger than blinks by spatial size, such that most of the underlying blinks would be out of focus when the companion sparks were captured (Fig. 7). As noted above, the tnadir trails the tpeak by 11 ms. This delay reinforces the idea that the release flux in a spark or blink is a time-dependent declining function, consistent with cisternal Ca2+ depletion and release inactivation.
Fig. 5.

Visualization of complementary spark–blink signal pairs. (A) Simultaneous measurements of sparks and blinks. (Upper) Linescan images of repeated sparks (rhod-2, upper) and blinks (fluo-5N, lower). Dotted ellipses mark corresponding areas in the images. (Lower) Time courses of sparks (black, upper) and blinks (blue, lower). (B) Time courses of the spark (upper) and blink (lower) averaged from 86 event pairs in 9 cells. The fitted smooth curves (red) show sparks (upper) displaying ΔF/F0 = 0.30, tpeak = 21 ms, and τrecovery = 43 ms, and blinks (lower) displaying ΔF/F0 = 0.013, tnadir = 32 ms, and τrecovery = 23 ms. Averaged spark obtained with rhod-2 alone (n = 22 events) displayed ΔF/F0 = 0.41, tpeak = 19 ms, and τrecovery = 31 ms, suggesting that fluo-5N retention in the SR does not alter spark properties. (C) Averaged spatial profiles. Upper: Ca2+ spark (FWHM = 2.2 μm); lower: Ca2+ blink (FWHM = 1.0 μm). (D) Overview of the Ca2+ spark–blink duality with respect to transverse tubule (TT) and SR. LCC, L-type Ca2+ channel..
In summary, we have investigated Ca2+ signals within nanometer-sized Ca2+ stores in intact cells in real time, by combining measurements of cytosolic Ca2+, nanoscopic store Ca2+, and cellular ultrastructure. We demonstrated that large and rapid nanoscopic store Ca2+ depletions or Ca2+ blinks mirror Ca2+ sparks in the cytosol. Characterization of blinks affords unique insights into Ca2+-induced Ca2+ release termination and refractoriness as well as intraorganelle Ca2+ regulation. The complementary spark–blink signal pairs in heart (Fig. 5D) may provide the prototype for similar reciprocal signals responsible for diverse store-dependent functions ranging from capacitative Ca2+ entry (5, 6) to store Ca2+-dependent apoptotic signaling (10, 11). The highly sensitive measurement of nanoscopic store Ca2+ signal should make it possible to elucidate these signaling pathways with improved molecular and mechanistic resolution.
Supplementary Material
Acknowledgments
We thank Eduardo Rios and Thomas Shannon for valuable discussion on dual indicator imaging, Michael Stern and Edward G. Lakatta for thoughtful reading of the manuscript, Bruce Ziman for technical support, and Nosta Glaser and Jimmy Burril for artwork. This work was supported by National Institute on Aging intramural research funding (to H.C.), National Institutes of Health grants (to W.J.L. and C.F.-A.), and the Chinese Natural Science Foundation and the Major State Basic Science Development Program (to H.C.).
Abbreviations: ER, endoplasmic reticulum; SR, sarcoplasmic reticulum; jSR, junctional SR; fSR, free SR; RyR, ryanodine receptor; FWHM, full width at half maximum.
References
- 1.Neher, E. (1998) Neuron 20, 389-399. [DOI] [PubMed] [Google Scholar]
- 2.Berridge, M. J., Bootman, M. D. & Roderick, H. L. (2003) Nat. Rev. Mol. Cell Biol. 4, 517-529. [DOI] [PubMed] [Google Scholar]
- 3.Bers, D. M. (2002) Nature 415, 198-205. [DOI] [PubMed] [Google Scholar]
- 4.Clapham, D. E. (1995) Cell 80, 259-268. [DOI] [PubMed] [Google Scholar]
- 5.Berridge, M. J. (2002) Cell Calcium 32, 235-249. [DOI] [PubMed] [Google Scholar]
- 6.Putney, J. W., Jr., Broad, L. M., Braun, F. J., Lievremont, J. P. & Bird, G. S. (2001) J. Cell Sci. 114, 2223-2229. [DOI] [PubMed] [Google Scholar]
- 7.Sobie, E. A., Dilly, K. W., dos Santos, C. J., Lederer, W. J. & Jafri, M. S. (2002) Biophys. J. 83, 59-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Terentyev, D., Viatchenko-Karpinski, S. Gyorke, I., Volpe, P., Williams, S. C.,& Gyorke, S. (2003) Proc. Natl. Acad. Sci. USA 100, 11759-11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisner, D. A., Choi, H. S., Diaz, M.E., O'Neill, S. C. & Trafford, A. W. (2000) Circ. Res. 87, 1087-1094. [DOI] [PubMed] [Google Scholar]
- 10.Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B. A. & Yuan, J. (2000) Nature 403, 98-103. [DOI] [PubMed] [Google Scholar]
- 11.Scorrano, L., Oakes, S. A., Opferman, J. T., Cheng, E. H., Sorcinelli, M. D., Pozzan, T. & Korsmeyer, S. J. (2003) Science 300, 135-139. [DOI] [PubMed] [Google Scholar]
- 12.Cheng, H., Lederer, W. J. & Cannell, M. B. (1993) Science 262, 740-744. [DOI] [PubMed] [Google Scholar]
- 13.Klein, M. G. Cheng, H., Santana, L. F., Jiang, Y. H., Lederer, W. J. & Schneider, M. F. (1996) Nature 379, 455-458. [DOI] [PubMed] [Google Scholar]
- 14.Nelson, M. T., Cheng, H., Rubart, M., Santana, L. F., Bonev, A. D., Knot, H. J. & Lederer, W. J. (1995) Science 270, 633-637. [DOI] [PubMed] [Google Scholar]
- 15.Tsugorka, A., Rios, E. & Blatter, L. A. (1995) Science 269, 1723-1726. [DOI] [PubMed] [Google Scholar]
- 16.Yao, Y., Choi, J. & Parker, I. (1995) J. Physiol 482, 533-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Crescenzo, V., ZhuGe, R., Velazquez-Marrero, C., Lifshitz, L. M., Custer, E., Carmichael, J., Lai, F. A., Tuft, R. A., Fogarty, K. E., Lemos. J. R. & Walsh, J. V., Jr. (2004) J. Neurosci. 24, 1226-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koizumi, S., Bootman, M. D., Bobanovic, L. K., Schell, M. J., Berridge, M. J. & Lipp, P. (1999) Neuron 22, 125-137. [DOI] [PubMed] [Google Scholar]
- 19.Gyorke, I. & Gyorke, S. (1998) Biophys. J. 75, 2801-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stern, M. D. & Cheng, H. (2004) Cell Calcium 35, 591-601. [DOI] [PubMed] [Google Scholar]
- 21.DelPrincipe, F., Egger, M. & Niggli, E. (1999) Nat. Cell Biol. 1, 323-329. [DOI] [PubMed] [Google Scholar]
- 22.Shannon, T. R., Guo, T. & Bers, D. M. (2003) Circ. Res. 93, 40-45. [DOI] [PubMed] [Google Scholar]
- 23.Weibel, E. R. (1979) Bull. Eur. Physiopathol. Respir. 15, 999-1013. [PubMed] [Google Scholar]
- 24.Capogrossi, M. C., Kort, A. A., Spurgeon, H. A. & Lakatta, E. G. (1986) J. Gen. Physiol. 88, 589-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell, R. D., Simmerman, H. K. & Jones, L. R. (1988) J. Biol. Chem. 263, 1376-1381. [PubMed] [Google Scholar]
- 26.Wang, S. Q., Song, L. S., Lakatta, E. G. & Cheng, H. (2001) Nature 410, 592-596. [DOI] [PubMed] [Google Scholar]
- 27.Cheng, H., Song, L. S., Shirokova, N., Gonzalez, A., Lakatta, E. G., Rios, E. & Stern, M. D. (1999) Biophys. J. 76, 606-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hollingworth, S., Peet, J., Chandler, W. K. & Baylor, S. M. (2001) J. Gen. Physiol. 118, 653-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lacampagne, A., Klein, M. G., Ward, C. W. & Schneider, M. F. (2000) Proc. Natl. Acad. Sci. USA 97, 7823-7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Franzini-Armstrong, C., Protasi, F. & Ramesh, V. (1999) Biophys. J. 77, 1528-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kabbara, A. A. & Allen, D. G. (2001) J. Physiol. 534, 87-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perreault, C. L. Mulieri, L. A., Alpert, N. R., Ransil, B. J., Allen, P. D. & Morgan, J. P. (1992) Am. J. Physiol. 263, H503-H510. [DOI] [PubMed] [Google Scholar]
- 33.Cannell, M. B., Cheng, H. & Lederer, W. J. (1995) Science 268, 1045-1049. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Lopez, J. R., Shacklock, P. S., Balke, C. W. & Wier, W. G. (1995) Science 268, 1042-1045. [DOI] [PubMed] [Google Scholar]
- 35.Tripathy, A. & Meissner, G. (1996) Biophys. J. 70, 2600-2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shannon, T. R., Ginsburg, K. S. & Bers, D. M. (2000) Biophys. J. 78, 334-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trafford, A. W., Diaz, M. E., Sibbring, G. C. & Eisner, D. A. (2000) J. Physiol. 522, 259-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sham, J. S. Song, L. S., Deng, L. H., Chen-Izu, Y., Lakatta, E. G., Stern, M. D. & Cheng, H. (1998) Proc. Natl. Acad. Sci. USA 95, 15096-15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gyorke, S. & Fill, M. (1993) Science 260, 807-809. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}