

Abstract
Tetranectin is a plasminogen kringle 4 domain-binding protein present in plasma and various tissue locations. Decreased plasma tetranectin or increased tetranectin in stroma of cancers correlates with cancer progression and adverse prognosis. A possible mechanism through which tetranectin could influence cancer progression is by altering activities of plasminogen or the plasminogen fragment, angiostatin. Tetranectin was found to bind to the kringle 1-4 form of angiostatin (ASTK1-4). In addition, tetranectin inhibited binding of plasminogen or ASTK1-4 to extracellular matrix (ECM) deposited by endothelial cells. Finally, tetranectin partially counteracted the ability of ASTK1-4 to inhibit proliferation of endothelial cells. This latter effect of tetranectin was specific for ASTK1-4 since it did not counteract the antiproliferative activities of the kringle 1-3 form of angiostatin (ASTK1-3) or endostatin. These findings suggest that tetranectin may modulate angiogenesis through interactions with AST.
INTRODUCTION
Tetranectin (TN) plays a role in skeletal formation during development since targeted deletion of the protein results in spinal deformity [1]. The function(s) of tetranectin in postnatal life have not been elucidated although there is evidence for roles in tissue remodeling, coagulation, and cancer. Tetranectin was originally isolated as a plasminogen-binding protein that can enhance plasminogen activation in the presence of tissue plasminogen activator [2]. Tetranectin binds to plasminogen through a calcium-sensitive interaction of its C-terminal domain with kringle 4 domain of plasminogen [3, 4]. Tetranectin also has a distinct binding site in its N-terminus that mediates binding to complex sulfated carbohydrates (eg, heparin) [5]. The N-terminus of tetranectin could, therefore, mediate binding to extracellular matrix components.
Plasma levels of tetranectin are approximately 100 nM in healthly adults [6]. However, these levels decline in patients with cancer and rheumatoid arthritis [6, 7, 8]. Tetranectin is also found in a mobilizable set of granules in neutrophils [9], in monocytes [10] and platelets [7], and in various tissue locations like cartilage and the extracellular matrix (ECM) of developing or regenerating muscle [11, 12, 13]. Tetranectin is implicated in the pathogenesis of cancer since decreased plasma levels of tetranectin correlate with cancer progression [6, 14]. In the case of ovarian cancer, decreased plasma levels of tetranectin were a stronger predictor of adverse prognosis than cancer stage [15]. Furthermore, tetranectin is present in the stroma of various cancers (eg, breast, ovary, colon), whereas it is not present in normal tissue from which the cancers arose [16, 17]. Positive staining for tetranectin in cancer stroma has also been strongly correlated with cancer progression [15].
The mechanisms through which tetranectin may participate in cancer progression have not been elucidated. Tetranectin colocalizes with plasminogen in the invasive front of melanoma lesions [18], although how tetranectin affects binding or local activation of plasminogen in cancer stroma has not been determined. This paper explores the hypothesis that tetranectin may interact with angiostatin. Angiostatin is formed in cancer tissues by proteolytic degradation of plasminogen. The predominant form of angiostatin produced in cancer tissues is ASTK1-4 [19, 20, 21]. ASTK1-4 inhibits cancer progression and metastasis by inhibiting cancer-related angiogenesis. We demonstrate that tetranectin binds to ASTK1-4 and reduces its ability to bind to ECM of endothelial cells or to inhibit endothelial cell growth.
METHODS
Reagents
Human plasminogen and an antibody directed against K1-3 domain of plasminogen were purchased from Enzyme Research Labs (South Bend, Ind). Rabbit anti-human tetranectin (with or without horseradish peroxidase attached) was obtained from DAKO Corp (Carpinteria, Calif).
Recombinant angiostatins and endostatin
Recombinant human angiostatins containing kringle domains 1-3 and 1-4 (ASTK1-3 and ASTK1-4), and recombinant human endostatin were graciously provided by Drs Nicolas MacDonald and Kim Lee Sim (EntreMed, Inc, Rockville, Md). ASTK1-4 was produced in Chinese hamster ovary cells and purified as described in [22]. Endostatin and ASTK1-3 were produced in Pichia pastoris [23, 24]. Native ASTK1-4 derived from human plasma was purchased from Angiogenesis Research Industries (Chicago, Ill). The recombinant and native angiostatins had similar endothelial cell growth inhibitory properties (data not shown). The native preparation was used in the endothelial growth inhibition assays (see below).
Enzyme-linked immunoabsorbent assay (ELISA) for binding of angiostatin or plasminogen to tetranectin
Binding of angiostatin or plasminogen to tetranectin was assessed by coating plates initially with wild type or mutant forms of tetranectin. Tetranectin was diluted to a final concentration of 6.8 μg/mL (100 nM) in coating buffer (bicarbonate buffer at pH 9.6), added to 96-well microtitre plates (Costar, Corning Inc, Corning, NY), and incubated overnight at 4°C. The wells were washed and then incubated with either plasminogen (22.5 μg/mL) or angiostatin (50 μg/mL) at room temperature for 1 hour. Bound plasminogen or angiostatin was detected by addition of a 1 : 1000 dilution of antibody directed against the kringle 1-3 domain of plasminogen (Enzyme Research Laboratories, South Bend, Ind) for 1 hour at room temperature. Preliminary studies demonstrated that this antibody recognized angiostatin and plasminogen to a similar extent. A secondary antibody (HRP-labeled donkey anti-mouse IgG; Jackson Research Labs, West Grove, Pa) was then added at 1 : 40 000 dilution for 1 hour at room temperature. Binding was detected using a TMB peroxidase EIA substrate kit (BioRad, Hercules, Calif) and 1N H2SO4. OD 450 readings were measured using a Titertek Multiscan reader. In all experiments, background binding of plasminogen and angiostatin was tested by including additional wells coated with 2.5% BSA only. Note that initial experiments were attempted in which angiostatin or plasminogen was coated onto ELISA plates followed by addition of tetranectin. However, it was found that background binding of tetranectin to BSA-coated plates was too high to reliably assess binding by this method.
Recombinant tetranectins
Recombinant wild-type human tetranectin was produced in E coli, refolded and purified as described in [10]. Mutant tetranectins were generated by site-directed mutagenesis as described in [3, 4].
Assay of binding of plasminogen or angiostatin to ECM of endothelial cells
ECM was prepared from human umbilical vein endothelial cells (HUVECs) grown for 2 days postconfluence as described in [25]. HUVECs were obtained from Clonetics Products, a division of BioWhittaker (San Diego, Calif) and cultured as outlined in the manufacturer's instructions. The subendothelial matrix was recovered by removing cells with 0.5% Triton X-100 in PBS (pH 7.4) followed by incubation with 25 mM NH4OH to remove cytoskeletal elements, and then washed with PBS supplemented with 0.05% tween-20. The adherent ECM was incubated with 1% BSA in PBS to saturate nonspecific protein binding sites. AST (at 0.5, 0.25, and 0.125 μM) was preincubated with TN (at 100 nM) for 30 minutes at 37°C and then added to designated wells of 96-well plate. Bound AST was detected as described above.
Assay of endothelial cell proliferation
HUVECs were seeded overnight in minimum essential medium containing 2.5% fetal bovine serum (FBS) in a 96-well plate (5000 cells/well) at 37°C. The following day, fresh medium supplemented with basic fibroblast growth factor (bFGF; 10 ng/mL) was added. In addition, tetranectin, angiostatin, or endostatin or combinations of these proteins were added to triplicate wells. The cells were incubated for 72 hours by replenishing fresh medium and test substances (bFGF, angiostatin, endostatin, tetranectin) as indicated at 24 hours and 48 hours. Cells were then harvested at 72 hours and counted by hemocytometer.
RESULTS
Plasminogen and angiostatin bind to tetranectin
As expected, plasminogen bound to recombinant wild-type tetranectin (Figure 1). Since the form of angiostatin composed of kringle domains 1-4 of plasminogen (ASTK1-4) contains the kringle 4, we tested its binding to tetranectin in parallel. ASTK1-4 also bound significantly to tetranectin (Figure 1). The mechanism of binding of tetranectin to plasminogen has been determined through the use of tetranectin mutants [3]. Binding is calcium-sensitive (ie, reduced by increasing concentrations of calcium) and is mediated by C-terminal domain of tetranectin. A mutant form of tetranectin in which lysine 148 was replaced with alanine (TNK148A) binds to plasminogen markedly less than wild type tetranectin [4]. In contrast, substitution of threonine 149 with tyrosine (TNT149Y) resulted in increased binding to kringle 4 [4]. Plasminogen and ASTK1-4 bound significantly less to the TNK148A and significantly more to TNT149Y than to wild type TN (Table 1 and Figure 1).
Figure 1.
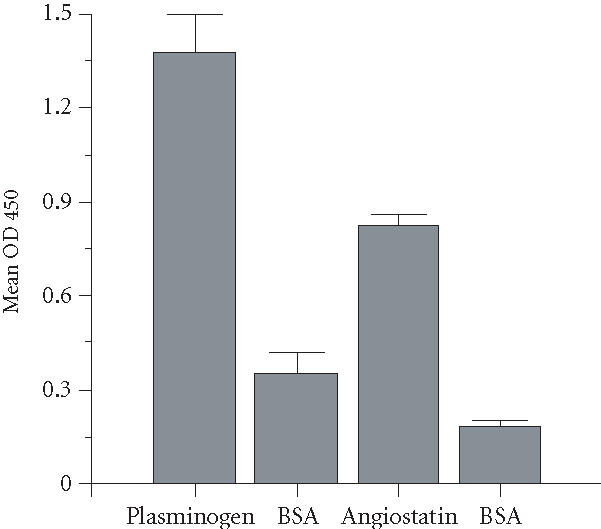
Binding of plasminogen or ASTK1-4 to tetranectin. ELISA plates were coated with recombinant wild-type human tetranectin (100 nM) or BSA, and then treated with plasminogen (22.5 μg/mL) and ASTK1-4 (50 μg/mL). Results shown are mean ± SEM of 5 experiments (each experiment done in triplicate). Binding of plasminogen and ASTK1-4 to tetranectin was significantly greater than binding to BSA-coated plates (). Binding of plasminogen to TN was significantly greater than binding of ASTK1-4 ().
Table 1.
Binding of plasminogen and angiostatin, containing kringles 1-4 or kringles 1-3 (ASTK1-4, ASTK1-3), to mutant tetranectins. Binding of plasminogen, angiostatin K1-4 (ASTK1-4), or angiostatin K1-3 (ASTK1-3) to TN mutants was assessed by ELISA as in Figure 1. Results shown are mean ± SEM of 6 specific binding determinations. Binding of plasminogen or AST to BSA was subtracted from results shown.
| Protein added | TNK148A | TNT149Y |
|---|---|---|
| Plasminogen | 0.02 ± 0.006 | 1.73 ± 0.14 |
| ASTK1-4 | 0.06 ± 0.02 | 1.83 ±0.21 |
| ASTK1-3 | 0.02 ± 0.006 | 0.43 ± 0.05 |
As shown in Figure 1, ASTK1-4 bound to wild-type TN significantly less than plasminogen. However, binding of ASTK1-4 to the TNT149Y form was equivalent to binding of plasminogen. Increased plasminogen and angiostatin binding of TNT149Y could result from the greater affinity of this mutant for kringle 4. TNT149Y is also distinguished from wild-type tetranectin in that it binds to the kringle 2 domain of plasminogen [4], which could be involved in binding to ASTK1-4. This is likely to be the case since ASTK1-3 showed significant binding to TNY149A, whereas binding of wild-type TN to ASTK1-3 was not significantly greater than background binding to BSA (data not shown). Nonetheless, binding of ASTK1-3 to TNT149Y was markedly less than that of ASTK1-4 or plasminogen, indicating that increased affinity of TNT149Y for kringle 4 accounts for most of the increased binding of this mutant to ASTK1-4.
Angiostatin (ASTK1-4) binds to ECM of endothelial cells and tetranectin inhibits this binding
Our next goal was to determine if tetranectin alters functional activities of angiostatin. Plasminogen binds to ECM of endothelial cells [25, 26]. We wanted to determine if angiostatin also binds to ECM of endothelial cells and to determine the effect of tetranectin on this binding. ECM of HUVECs was prepared as described in [25]. As shown in Figure 2, plasminogen did bind to this matrix and this binding was significantly inhibited by pre-incubation of plasminogen with a physiological concentration (100 nM) of wild-type tetranectin. As shown in Figure 3, ASTK1-4 also bound to the ECM and binding was again significantly reduced by tetranectin.
Figure 2.
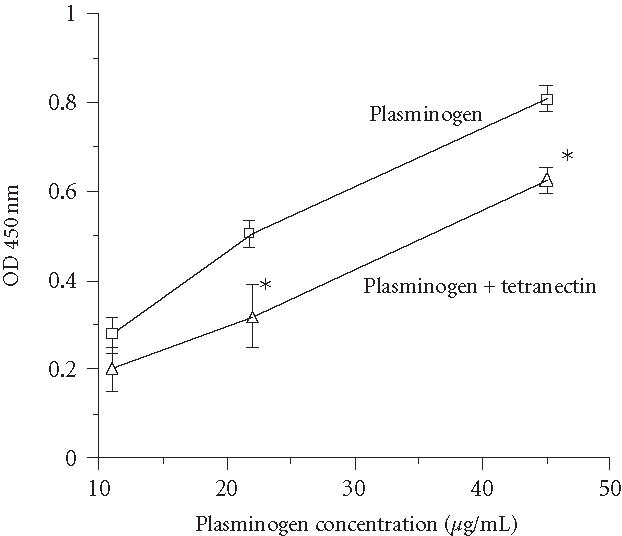
Tetranectin reduces binding of plasminogen to ECM deposited by endothelial cells. ECM of endothelial cells was produced as described in “methods,” and binding of plasminogen was measured using ELISA. The effect of tetranectin (wild type) on binding of plasminogen was assessed by preincubation of plasminogen with tetranectin (100 nM) for 30 minutes at 37°C, followed by addition of these samples to wells coated with ECM. Results shown are mean ± SEM of 4 experiments (each experiment done in triplicate). Binding of plasminogen at 22.5 or 45 μg/mL was significantly reduced by tetranectin (; indicated by *).
Figure 3.
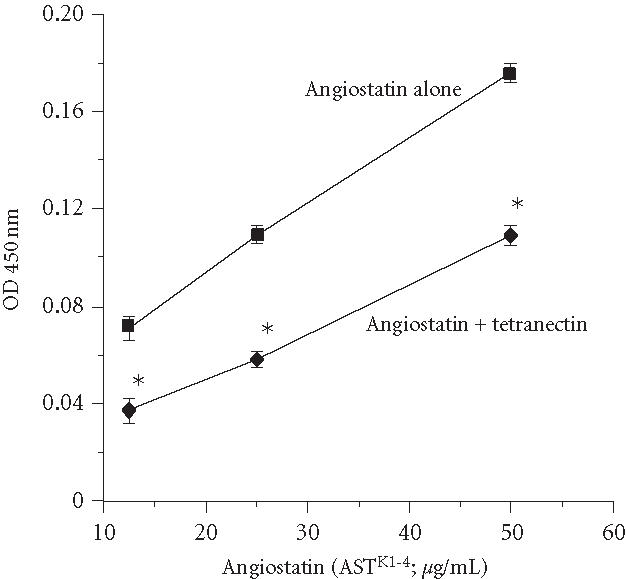
Tetranectin reduces binding of angiostatin to ECM. Binding of angiostatin (ASTK1-4) to ECM of endothelial cells was measured as in Figure 2. At all concentrations tested, binding of ASTK1-4 was significantly reduced by tetranectin (; indicated by *). Results shown are mean ± SEM of 4 experiments (each experiment done in triplicate).
Tetranectin modulates the ability of angiostatin to inhibit endothelial cell proliferation
ASTK1-4 significantly inhibited the bFGF-stimulated growth of HUVECs as expected (Figure 4). Tetranectin alone did not significantly alter proliferation in the presence (Figure 4) or absence (data not shown) of bFGF. However, when HUVECs were treated with both tetranectin and ASTK1-4, proliferation was significantly greater than that with ASTK1-4 alone. Note that tetranectin did not completely reverse the antiproliferative action of ASTK1-4 since there were still significantly fewer cells in cultures treated with the combination of tetranectin and ASTK1-4 than in control cultures.
Figure 4.
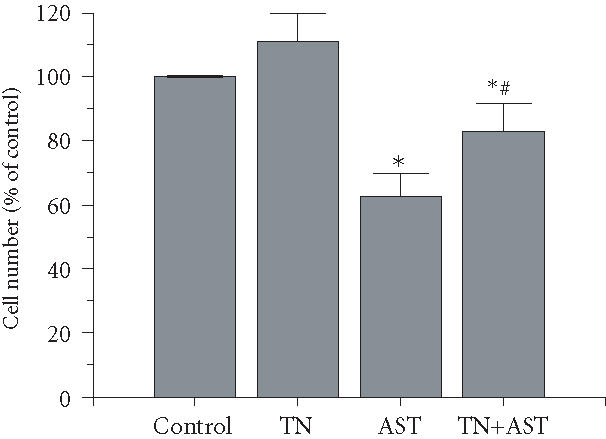
Tetranectin counteracts angiostatin-induced inhibition of proliferation of endothelial cells. HUVECs were seeded overnight in 96-well plates (5000 cells/well). The following day, fresh medium supplemented with basic fibroblast growth factor (bFGF; 10 ng/mL) was added. Where indicated, tetranectin (TN; 100 nM) and/or ASTK1-4 (200 nM) were added to triplicate wells. Cell numbers were counted after 72 hours. ASTK1-4 significantly reduced the number of cells obtained (; indicated by *). There were, however, significantly more cells in cultures treated with the combination of tetranectin and ASTK1-4 than in those treated with ASTK1-4 alone (; indicated by #). Note that cultures treated with AST and tetranectin still have less cells than control cultures ().
In the experiments shown in Figure 4, there was a trend (not statistically significant) toward increased proliferation of endothelial cells in response to tetranectin alone. It was possible, therefore, that the ability of tetranectin to counteract the antiproliferative activity of ASTK1-4 resulted from independent effects of tetranectin on endothelial cell proliferation, rather than from its interaction with ASTK1-4. To study this further, we tested the activity of additional concentrations of tetranectin alone, or tetranectin in combination with endostatin or ASTK1-3 (Table 2). In these experiments, 150 nM concentration of tetranectin alone modestly but significantly increased endothelial cell proliferation. However, a further increase of the concentration of tetranectin to 375 nM resulted in loss of this enhancing activity (ie, endothelial cell counts in cultures treated with 375 nM tetranectin were 38 ± 3 × 103 as compared to 43 ± 1.4 in control; ; ). Both endostatin and ASTK1-3 inhibited the growth of endothelial cells as expected (Table 2). However, tetranectin (150 nM) did not lessen the antiproliferative effect of either ASTK1-3 or endostatin. These results suggest that the ability of tetranectin to counteract the antiproliferative activity of ASTK1-4 is not the result of an independent effect of tetranectin on the endothelial cells.
Table 2.
Effect of tetranectin on endothelial cell proliferation in presence of control buffer, ASTK1-3 or endostatin. Endothelial cell cultures were incubated for 72 hours in presence of the indicated concentrations of recombinant wild-type tetranectin, ASTK1-3, or endostatin alone or combinations of ASTK1-3 or endostatin with tetranectin (final concentration 150 nM). Results represent mean ± SD number of cells/mL (× 1000) in 4–6 experiments (each done in triplicate). All concentrations of ASTK1-3 or endostatin significantly decreased endothelial cells numbers () in presence or absence of tetranectin. Tetranectin alone significantly () increased the number of endothelial cells compared with control buffer alone although it did not alter the effects of any concentration of ASTK1-3 or endostatin.
| Inhibitor added | Control | Tetranectin 150 nM |
|---|---|---|
| Control | 43.4 ± 1.4 | 51 ± 2.9** |
| ASTK1-3 10 nM | 40.5 ± 1.9 | 37.6 ± 5.7 |
| ASTK1-3 50 nM | 33.2 ± 2.2 | 34.1 ± 4 |
| ASTK1-3 1 μM | 32.3 ± 1.6 | 27.7 ± 1.6 |
| Endostatin 125 nM | 32.3 ± 2 | 30.3 ± 2 |
| Endostatin 250 nM | 27.2 ± 1.9 | 23.1 ± 7 |
| Endostatin 500 nM | 25.1 ± 3.5 | 22 ± 3.9 |
DISCUSSION
The important novel findings of this paper are that tetranectin binds to ASTK1-4, inhibits binding of plasminogen and ASTK1-4 to ECM of endothelial cells, and partially counteracts the effects of ASTK1-4 on endothelial cell proliferation. The mechanism of binding of ASTK1-4 to tetranectin is similar to that of plasminogen based on studies using mutant forms of tetranectin with enhanced or reduced ability to bind plasminogen (Table 1). These results also indicate that interactions of ASTK1-4 with tetranectin could be modulated through introduction of discrete modifications of tetranectin's binding site for plasminogen.
It is of note that although tetranectin bound significantly to ASTK1-4, binding to ASTK1-4 was significantly less than binding to plasminogen. This finding was unexpected since ASTK1-4 contains the principal binding site for tetranectin (ie, kringle 4). It may be that the conformation of kringle 4 in ASTK1-4 differs sufficiently from its conformation in plasminogen to affect binding of tetranectin. This binding difference may be significant in some physiological situations. However, the other results presented in this paper indicate strongly that binding of tetranectin to ASTK1-4 is sufficient to affect other activities of ASTK1-4.
We demonstrate that ASTK1-4, like plasminogen, binds to the ECM of endothelial cells. This finding is novel and of interest since it could relate to localization of angiostatin in vivo. Of more relevance to the aims of this paper, we also found that tetranectin significantly reduced binding of ASTK1-4 to ECM of endothelial cells. The ability of tetranectin to inhibit binding of ASTK1-4 to ECM suggests that it could promote angiogenesis in vivo. We therefore tested whether tetranectin affects the antiangiogenic activity of ASTK1-4. Physiological concentrations of wild-type tetranectin significantly counteracted the effect of ASTK1-4 on endothelial cell proliferation. Tetranectin did not have a similar interaction with ASTK1-3 or endostatin (Table 2), indicating that its ability to counteract the antiangiogenic effects of ASTK1-4 is dependent on binding to the kringle 4 domain and not to some other direct interaction with endothelial cells. Tetranectin alone had a variably enhancing effect on endothelial cell growth at some concentrations. However, this effect was not dose-related and is unlikely to account for the ability of tetranectin to counteract antiproliferative effects of ASTK1-4 based upon results shown in Table 2.
As noted, extensive data derived from the study of clinical samples suggests that increased tetranectin in the stroma of cancer tissues is associated with an adverse prognosis in various cancers. Our findings suggest that tetranectin may promote tumor progression by favoring angiogenesis. Cancer-associated angiogenesis has been quantitated by enumeration of the density of microvessels in tumor stroma. Increased microvessel density is associated with adverse prognosis in many cancers [27]. Future studies could address whether increased microvessel density is associated with stromal tetranectin reactivity. The ability of tetranectin to modify other functional properties of angiostatin should also be examined. One immediate implication of our findings is that ASTK1-4 and ASTK1-3 may have different activities in vivo based on differential binding to tetranectin. This might account for the increased elimination half-life of ASTK1-4 compared with ASTK1-3 in vivo, or the fact that a similar inhibition of cancer metastases was obtained with lower effective in vivo exposure to ASTK1-3 than ASTK1-4 [22].
Angiostatin may inhibit angiogenesis in inflammatory states [28] or after vascular injury [29]. Recent studies demonstrate that biologically active angiostatin is produced by neutrophils [30], and that angiostatin inhibits neutrophil migration and inflammation-induced angiogenesis [31]. Of interest, prior studies demonstrated that tetranectin is contained in a subset of neutrophils, from which it can be released after cell stimulation with various agonists [9]. Hence, interactions of tetranectin and plasminogen or angiostatin may also be involved in inflammatory processes. Whereas angiostatin produced in cancer tissues appears most often to be ASTK1-4 [19, 21], neutrophils produce ASTK1-3 [30]. Therefore, the participation of tetranectin in angiogenesis may vary in different physiological or pathological states depending on which form of angiostatin is produced.
In summary, we demonstrate that tetranectin binds to the form of angiostatin commonly produced in cancer tissues, characterize the mechanism of binding using mutant forms of tetranectin, and show that tetranectin inhibits important functional properties of angiostatin. These findings provide insight into the mechanisms through which tetranectin participates in cancer progression. Furthermore, these findings have implications for therapeutic use of different forms of angiostatin.
ACKNOWLEDGMENT
This work was supported by NIH Grant HL69031 (KLH).
References
- 1.Iba K, Durkin M.E, Johnsen L, et al. Mice with a targeted deletion of the tetranectin gene exhibit a spinal deformity. Mol Cell Biol. 2001;21(22):7817–7825. doi: 10.1128/MCB.21.22.7817-7825.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clemmensen I, Petersen L.C, Kluft C. Purification and characterization of a novel, oligomeric, plasminogen kringle 4 binding protein from human plasma: tetranectin. Eur J Biochem. 1986;156(2):327–333. doi: 10.1111/j.1432-1033.1986.tb09586.x. [DOI] [PubMed] [Google Scholar]
- 3.Graversen J.H, Lorentsen R.H, Jacobsen C, et al. The plasminogen binding site of the C-type lectin tetranectin is located in the carbohydrate recognition domain, and binding is sensitive to both calcium and lysine. J Biol Chem. 1998;273(44):29241–29246. doi: 10.1074/jbc.273.44.29241. [DOI] [PubMed] [Google Scholar]
- 4.Graversen J.H, Jacobsen C, Sigurskjold B.W, et al. Mutational analysis of affinity and selectivity of kringle-tetranectin interaction. Grafting novel kringle affinity onto the tetranectin lectin scaffold. J Biol Chem. 2000;275(48):37390–37396. doi: 10.1074/jbc.M004873200. [DOI] [PubMed] [Google Scholar]
- 5.Lorentsen R.H, Graversen J.H, Caterer N.R, Thogersen H.C, Etzerodt M. The heparin-binding site in tetranectin is located in the N-terminal region and binding does not involve the carbohydrate recognition domain. Biochem J. 2000;347(pt 1):83–87. [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen B.A, Clemmensen I. Plasma tetranectin is reduced in cancer and related to metastasia. Cancer. 1988;62(5):869–872. doi: 10.1002/1097-0142(19880901)62:5<869::aid-cncr2820620503>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 7.Christensen L. The distribution of fibronectin, laminin and tetranectin in human breast cancer with special attention to the extracellular matrix. APMIS Suppl. 1992;26:1–39. [PubMed] [Google Scholar]
- 8.Kamper E.F, Kopeikina L.T, Koutsoukos V, Stavridis J. Plasma tetranectin levels and disease activity in patients with rheumatoid arthritis. J Rheumatol. 1997;24(2):262–268. [PubMed] [Google Scholar]
- 9.Borregaard N, Christensen L, Bejerrum O.W, Birgens H.S, Clemmensen I. Identification of a highly mobilizable subset of human neutrophil intracellular vesicles that contains tetranectin and latent alkaline phosphatase. J Clin Invest. 1990;85(2):408–416. doi: 10.1172/JCI114453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nielsen H, Clemmensen I, Kharazmi A. Tetranectin: a novel secretory protein from human monocytes. Scand J Immunol. 1993;37(1):39–42. doi: 10.1111/j.1365-3083.1993.tb01662.x. [DOI] [PubMed] [Google Scholar]
- 11.Wewer U.M, Ibaraki K, Schjorring P, Durkin M.E, Young M.F, Albrechtsen R. A potential role for tetranectin in mineralization during osteogenesis. J Cell Biol. 1994;127(6):1767–1775. doi: 10.1083/jcb.127.6.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemmensen I, Lund L.R, Christensen L, Andreasen P.A. A tetranectin-related protein is produced and deposited in extracellular matrix by human embryonal fibroblasts. Eur J Biochem. 1991;195(3):735–741. doi: 10.1111/j.1432-1033.1991.tb15761.x. [DOI] [PubMed] [Google Scholar]
- 13.Wewer U.M, Iba K, Durkin M.E, et al. Tetranectin is a novel marker for myogenesis during embryonic development, muscle regeneration, and muscle cell differentiation in vitro. Dev Biol. 1998;200(2):247–259. doi: 10.1006/dbio.1998.8962. [DOI] [PubMed] [Google Scholar]
- 14.Hogdall C.K, Christiansen M, Norgaard-Pedersen B, Bentzen S.M, Kronborg O, Clemmensen I. Plasma tetranectin and colorectal cancer. Eur J Cancer. 1995;31A(6):888–894. doi: 10.1016/0959-8049(94)00520-6. [DOI] [PubMed] [Google Scholar]
- 15.Hogdall C.K, Hogdall E.V, Hording U, Clemmensen I, Norgaard-Pedersen B, Toftager-Larsen K. Pre-operative plasma tetranectin as a prognostic marker in ovarian cancer patients. Scand J Clin Lab Invest. 1993;53(7):741–674. doi: 10.3109/00365519309092579. [DOI] [PubMed] [Google Scholar]
- 16.Wewer U.M, Albrechtsen R. Tetranectin, a plasminogen kringle 4-binding protein. Cloning and gene expression pattern in human colon cancer. Lab Invest. 1992;67(2):253–262. [PubMed] [Google Scholar]
- 17.Christensen L, Clemmensen I. Differences in tetranectin immunoreactivity between benign and malignant breast tissue. Histochemistry. 1991;95(5):427–433. doi: 10.1007/BF00315737. [DOI] [PubMed] [Google Scholar]
- 18.De Vries T.J, De Wit P.E, Clemmensen I, et al. Tetranectin and plasmin/plasminogen are similarly distributed at the invasive front of cutaneous melanoma lesions. J Pathol. 1996;179(3):260–265. doi: 10.1002/(SICI)1096-9896(199607)179:3<260::AID-PATH586>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 19.O'Reilly M.S, Holmgren L, Shing Y, et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79(2):315–328. doi: 10.1016/0092-8674(94)90200-3. [DOI] [PubMed] [Google Scholar]
- 20.Gately S, Twardowski P, Stack M.S, et al. The mechanism of cancer-mediated conversion of plasminogen to the angiogenesis inhibitor angiostatin. Proc Natl Acad Sci USA. 1997;94(20):10868–10872. doi: 10.1073/pnas.94.20.10868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richardson M, Gunawan J, Hatton M.W, Seidlitz E, Hirte H.W, Singh G. Malignant ascites fluid (MAF), including ovarian-cancer-associated MAF, contains angiostatin and other factor(s) which inhibit angiogenesis. Gynecol Oncol. 2002;86(2):279–287. doi: 10.1006/gyno.2002.6760. [DOI] [PubMed] [Google Scholar]
- 22.MacDonald N.J, Murad A.C, Fogler W.E, Lu Y, Sim B.K. The tumor-suppressing activity of angiostatin protein resides within kringles 1 to 3. Biochem Biophys Res Commun. 1999;264(2):469–477. doi: 10.1006/bbrc.1999.1486. [DOI] [PubMed] [Google Scholar]
- 23.Sim B.K, O'Reilly M.S, Liang H, et al. A recombinant human angiostatin protein inhibits experimental primary and metastatic cancer. Cancer Res. 1997;57(7):1329–1334. [PubMed] [Google Scholar]
- 24.Sim B.K, Fogler W.E, Zhou X.H, et al. Zinc ligand-disrupted recombinant human Endostatin: potent inhibition of tumor growth, safety and pharmacokinetic profile. Angiogenesis. 1999;3(1):41–51. doi: 10.1023/a:1009058931769. [DOI] [PubMed] [Google Scholar]
- 25.Knudsen B.S, Silverstein R.L, Leung L.L, Harpel P.C, Nachman R.L. Binding of plasminogen to extracellular matrix. J Biol Chem. 1986;261(23):10765–10771. [PubMed] [Google Scholar]
- 26.Stack M.S, Gately S, Bafetti L.M, Enghild J.J, Soff G.A. Angiostatin inhibits endothelial and melanoma cellular invasion by blocking matrix-enhanced plasminogen activation. Biochem J. 1999;340(pt 1):77–84.. [PMC free article] [PubMed] [Google Scholar]
- 27.Hlatky L, Hahnfeldt P, Folkman J. Clinical application of antiangiogenic therapy: microvessel density, what it does and doesn't tell us. J Natl Cancer Inst. 2002;94(12):883–893. doi: 10.1093/jnci/94.12.883. [DOI] [PubMed] [Google Scholar]
- 28.Hamacher J, Lucas R, Lijnen H.R, et al. Tumor necrosis factor-alpha and angiostatin are mediators of endothelial cytotoxicity in bronchoalveolar lavages of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2002;166(5):651–656. doi: 10.1164/rccm.2109004. [DOI] [PubMed] [Google Scholar]
- 29.Matsunaga T, Weihrauch D.W, Moniz M.C, Tessmer J, Warltier D.C, Chilian W.M. Angiostatin inhibits coronary angiogenesis during impaired production of nitric oxide. Circulation. 2002;105(18):2185–2191. doi: 10.1161/01.cir.0000015856.84385.e9. [DOI] [PubMed] [Google Scholar]
- 30.Scapini P, Nesi L, Morini M, et al. Generation of biologically active angiostatin kringle 1-3 by activated human neutrophils. J Immunol. 2002;168(11):5798–5804. doi: 10.4049/jimmunol.168.11.5798. [DOI] [PubMed] [Google Scholar]
- 31.Benelli R, Morini M, Carrozzino F, et al. Neutrophils as a key cellular target for angiostatin: implications for regulation of angiogenesis and inflammation. FASEB J. 2002;16(2):267–269. doi: 10.1096/fj.01-0651fje. [DOI] [PubMed] [Google Scholar]