

Abstract
The relationships between exposure, biomarkers (vascular endothelial growth factor (VEGF), soluble VEGF receptors (sVEGFR)‐1, ‐2, ‐3, and soluble stem cell factor receptor (sKIT)), tumor sum of longest diameters (SLD), diastolic blood pressure (dBP), and overall survival (OS) were investigated in a modeling framework. The dataset included 64 metastatic renal cell carcinoma patients (mRCC) treated with oral axitinib. Biomarker timecourses were described by indirect response (IDR) models where axitinib inhibits sVEGFR‐1, ‐2, and ‐3 production, and VEGF degradation. No effect was identified on sKIT. A tumor model using sVEGFR‐3 dynamics as driver predicted SLD data well. An IDR model, with axitinib exposure stimulating the response, characterized dBP increase. In a time‐to‐event model the SLD timecourse predicted OS better than exposure, biomarker‐ or dBP‐related metrics. This type of framework can be used to relate pharmacokinetics, efficacy, and safety to long‐term clinical outcome in mRCC patients treated with VEGFR inhibitors. (ClinicalTrial.gov identifier NCT00569946.)
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ A modeling framework in sunitinib‐treated gastrointestinal stromal tumors identified circulating biomarkers and adverse effects as better predictors of overall survival (OS) than tumor size (SLD). Similar relationships may be of value for predicting OS in metastatic renal cell carcinoma (mRCC) patients treated with axitinib.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The relationships between axitinib exposure, biomarkers related to VEGFR inhibition, hypertension (the most common adverse effect for axitinib), SLD, and OS were investigated in axitinib‐treated Japanese mRCC patients.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ Early changes in soluble VEGFR‐3 could forecast tumor response. This analysis is one of the first to demonstrate SLD dynamics as a predictor of OS, which was better than biomarker‐ or hypertension‐related metrics or tumor size change at a specific week.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The modeling framework can be used as a template to leverage data collected during oncology clinical trials when developing new targeted therapies, facilitate identification of predictors for long‐term clinical outcome, and select the most promising dosing schedules.
In metastatic renal cell carcinoma (mRCC) the vascular endothelial growth factor (VEGF) is typically overexpressed and mRCC is predominantly refractory to traditional cytotoxic chemotherapies. Several first‐line treatment alternatives with targeted therapies exist, including the tyrosine kinase inhibitors (TKIs) sunitinib and pazopanib.1 However, patients often develop biological resistance and receive second‐line treatment.2 Axitinib is a potent and selective oral TKI targeting the VEGF receptors (VEGFR) 1, 2, and 3 and primarily displays antiangiogenic activity. The drug is approved in Europe, the United States, Japan, and elsewhere for the treatment of advanced renal cell carcinoma (RCC) after failure of one prior systemic therapy,3 and is currently a preferred choice as second‐line therapy for patients progressing after first‐line therapy.2 Moreover, axitinib has shown clinical activity in first‐line mRCC in recent phase II and III trials.4, 5 Axitinib is approved at a starting dose of 5 mg twice daily (b.i.d.) and dose increase or reduction is recommended based on individual safety and tolerability, including increased blood pressure (BP). Dose titration enables patients with good tolerability at a 5 mg starting dose to reach higher exposures6 and results in a better objective response rate.4
The conventional Response Evaluation Criteria in Solid Tumors (RECIST), which are based on a categorization of the response seen on the sum of longest diameters (SLD), were designed to evaluate therapeutic efficacy of cytotoxic agents.7 However, RECIST may not reflect the clinical benefit of antiangiogenic drugs for which tumor shrinkage may be limited or delayed.8 Increases in blood pressure are common after initiation of anti‐VEGF therapy9 and have been proposed as an independent predictor for overall survival (OS) and progression‐free survival (PFS) in axitinib‐treated mRCC patients10 and axitinib‐ and other TKI‐treated solid tumors,11 including sunitinib‐treated gastrointestinal stromal tumors (GIST).12, 13 Optimal axitinib exposure, leading to best achievable long‐term outcome, may, however, differ among mRCC patients and dose selection cannot likely be solely based on pharmacokinetics (PK) or BP measurements.6 Increases in VEGF and decreases in the soluble fragments of its receptors (sVEGFR‐1, ‐2, and ‐3) have been suggested as biomarkers of angiogenesis inhibition and predictors for clinical response in RCC treated with TKIs,14 including axitinib.15, 16 A better understanding of the relationships between axitinib exposure, plasma biomarkers, BP, SLD, and long‐term clinical outcome can be valuable for identifying robust pharmacodynamic (PD) biomarkers and guide treatment decisions.
By integrating quantitative knowledge on anticancer drugs' safety and efficacy, pharmacometric modeling has shown value in guiding oncology clinical trial design and rational dose selection, thereby optimizing benefit/risk management for cancer patients.17, 18, 19, 20 As an example, in an overarching modeling framework Hansson et al. elucidated the relations between drug exposure, the timecourse of circulating biomarkers (VEGF, sVEGFR‐2, and sVEGFR‐3, and soluble stem cell factor receptor (sKIT)), SLD, adverse effects (fatigue, hand‐foot syndrome, neutropenia, and hypertension), and OS in sunitinib‐treated GIST patients.12, 21 Increased VEGF, decreased sVEGFR‐2, sVEGFR‐3, and sKIT concentrations and diastolic BP (dBP) elevation were dependent on sunitinib exposure and dosing schedule. Sunitinib exposure together with sVEGFR‐3 and sKIT dynamics predicted the SLD timecourse. A smaller baseline SLD and larger sVEGFR‐3 decrease over time were associated with longer OS. Alternatively, hypertension and neutropenia could be used as predictors for OS.
In the present work, the relationships between axitinib exposure, the timecourses of potential biomarkers (VEGF, sVEGFR‐1, ‐2, and ‐3, sKIT), SLD, dBP, and OS in axitinib‐treated Japanese mRCC patients were explored and quantified using pharmacometric models.
METHODS
Patients and data
During the development of axitinib, biomarker data were collected from 64 Japanese cytokine‐refractory mRCC patients involved in a single‐arm, open‐label, multicenter phase II study.22 Axitinib was administered in 4‐week cycles at a starting dose of 5 mg b.i.d. In eligible patients axitinib dose was increased by 2–3 mg b.i.d. up to 10 mg b.i.d. every 2 weeks or more (n = 5), or decreased to 2 mg b.i.d. (n = 41) based on tolerability (BP and other nonhematologic adverse effects) and dosing history was recorded. The major reason for dose reduction or treatment discontinuation/interruption was proteinuria (28%). SLD was measured according to RECIST 1.0. Biomarkers, SLD, dBP, and OS assessment times are summarized in Table 1. This study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation guidelines on Good Clinical Practice, and applicable local regulatory requirements and laws. All participants provided informed consent. The study protocol was approved by an institutional review board at each site.
Table 1.
Summary of study assessments and available data
| Variable | Per protocol assessment time (study day) | Available data (n; follow‐up duration in days, median [range]) |
|---|---|---|
| VEGF, sVEGFR‐1, ‐2, ‐3, sKIT |
Cycle 1: pre‐dosing Cycle 2‐7: day 1 EoT/discontinuation |
n = 436 for each biomarker; 168 [32‐624]a |
| Sum of longest diameters |
Cycle 1: pre‐dosing Subsequent odd no. cycles: day 1 EoT/discontinuation |
n = 476; 337 [36‐731]b |
| Diastolic blood pressure |
Cycle 1: pre‐dosing, day 8, 15, 22 Cycle 2‐4: day 1, 15 Cycle >4: day 1 EoT/discontinuation |
n = 308; 29 [21‐29]c |
| Overall survival | Until EoT/discontinuation and every 6 months thereafter | 16/48 deaths/censored; 457 [85‐781] |
EoT, end of treatment; n, number of observations included in the analysis; sKIT, soluble stem cell factor receptor; sVEGFR‐1, ‐2, ‐3, soluble vascular endothelial growth factor receptor 1, 2, 3; VEGF, vascular endothelial growth factor.
Summary statistics on follow‐up duration exclude one patient with biomarker data available at baseline only.
Summary statistics on follow‐up duration exclude two patients with tumor data available at baseline only.
Only data from the first month (all visits in Cycle 1 and day 1 in Cycle 2) were modeled.
Model development
Nonlinear mixed effect models were developed using NONMEM software v. 7.3.23 Parameters were estimated using the first‐order conditional estimation method with interaction (FOCEI), and for dropout and OS analysis, the Laplacian estimation method. R v. 3.1.1, the R‐based package Xpose v. 4, Perl‐speaks‐NONMEM (PsN) toolkit v. 4, and Piraña v. 2.9.0 were used for data pre‐ and postprocessing, graphical visualization, and model diagnostics.24
Model selection was based on goodness‐of‐fit plots and the objective function value (OFV, −2·log‐likelihood). A significance level of P < 0.05 as assessed by the OFV difference (dOFV) was used to discriminate between nested models. The predictive performance of the biomarkers, SLD, and dBP models was assessed using (prediction‐corrected) visual predictive checks ((pc)VPCs),25 where 95% confidence intervals (CIs) derived from 500 simulated datasets were compared to the observed data. Kaplan–Meier VPCs, comparing the 95% CI derived from 200 simulations to the observed time‐to‐event (TTE) data, were used to evaluate the dropout and OS model performance. Relative standard errors (RSE) of parameter estimates were obtained from the NONMEM Sandwich matrix for continuous data and from the R matrix for dropout and OS models.
Exponential and additive interindividual variability (IIV) were evaluated as appropriate. Semiparametric distributions were tested when indicated graphically.26 Residual unexplained variability (RUV) was evaluated for all continuous data models using additive, proportional, or combined error models.
Pharmacokinetics
Empirical Bayes estimates (EBEs) of apparent clearance (CL/F) obtained from a published population PK model10 were used to calculate the daily area under the concentration–time curve , where Dosedaily is the daily dose accounting for dose increases and reductions. Since axitinib has a relatively short typical elimination half‐life (3 h in Japanese patients),10 AUCdaily was assumed to be 0 on days off‐therapy.
Biomarker models
Indirect response (IDR) models27 where axitinib inhibits sVEGFR‐1, ‐2, and ‐3 and sKIT production, and VEGF degradation, were investigated. Linear, maximal effect (Emax) and sigmoidal Emax drug effects driven by AUCdaily were evaluated. Linear disease progression functions were explored to describe potential changes in biomarkers in the absence of drug. Models for each biomarker were developed separately before being combined into a joint model to explore correlations between model parameters.
Tumor size model
Tumor models where axitinib induces a decrease in SLD were investigated. Zero‐order and first‐order tumor growth were evaluated. The axitinib effect on SLD was driven either directly by axitinib exposure (AUCdaily), or indirectly by individual model‐predicted changes in the different biomarkers (absolute value, or absolute or relative change from baseline). An approach similar to population PK parameters and data (PPP&D) was adopted,28, 29 i.e., population biomarker parameters were fixed while individual biomarker parameters were predicted simultaneously with SLD parameters based on both biomarker and SLD data. Drivers were tested alone and in combination. An exponential decay in drug effect describing potential tumor regrowth was investigated.
As dropout from tumor measurements may not be completely random, a logistic regression model was developed to mimic varying measurement durations in the SLD simulations. Investigated predictors in the dropout model included the time since start of treatment, AUCdaily, the observed baseline SLD and predicted SLD at the time of evaluation, and progressive disease (PD, yes/no) defined as a 20% increase in SLD from nadir. During simulations, dosing records were imputed based on the last observed dose until the time of last observed tumor assessment in the study.
Diastolic blood pressure model
According to protocol, new or additional antihypertensive treatments would be prescribed to patients having dBP elevation during axitinib therapy. Therefore, only data from the first treatment cycle were modeled. IDR models were investigated with axitinib stimulating the production of dBP response through linear, power, or (sigmoidal) Emax drug effects driven by Dosedaily or AUCdaily.
Overall survival model
Parametric TTE models were developed for OS data. Exponential, Weibull, Gompertz, log‐normal, and log‐logistic distributions were investigated to describe the baseline hazard. Predictors were tested one by one and thereafter in combination. Individual parameters were used to compute the model‐based predictors. Due to model instability their uncertainty could not be accounted for.30 Evaluated baseline predictors included Eastern Cooperative Oncology Group (ECOG) performance status, demographics (age, sex, body weight), and model‐predicted baseline biomarkers, dBP and SLD values. Time‐varying predictors included Dosedaily, AUCdaily, the predicted timecourse, absolute and relative change from baseline in biomarkers and SLD, and the derivative of SLD predicted timecourse. Dose and time‐varying predictors were extrapolated based on the last recorded dose assuming that patients were treated with axitinib until death or censoring, as the protocol supported treatment continuation in case of clinical benefit (no new lesion and SLD smaller than at baseline) despite progressive disease according to RECIST. Additionally, model‐predicted relative changes from baseline in biomarkers at week 4, in dBP at weeks 2 and 4, and in SLD at week 8, the maximum absolute dBP during cycle 1 and a dBP greater than 90 mmHg during cycle 1 (yes/no) were evaluated.
Censoring, defined as loss to follow‐up or nonoccurrence of death at the end of the study, was described by a competing hazard function to account for varying follow‐up durations.
RESULTS
A schematic representation of the final modeling framework is depicted in Figure 1.
Figure 1.
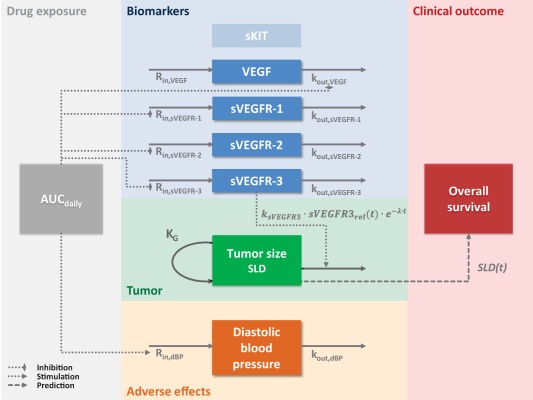
Schematic representation of the modeling framework for axitinib in metastatic renal cell carcinoma (mRCC). Axitinib daily area under the curve (AUCdaily) was used as a driver of the timecourses of biomarkers (the vascular endothelial growth factor VEGF and its soluble receptors sVEGFR‐1, ‐2, and ‐3) and diastolic blood pressure (dBP). Biomarker timecourses were described by indirect response models where axitinib inhibits the loss of VEGF response and the production of sVEGFR‐1, ‐2, and ‐3 responses. sKIT was not affected by axitinib. The model describing tumor size (sum of longest diameters, SLD) included an exponential growth and an effect of the relative change in sVEGFR‐3 from baseline over time (sVEGFR‐3rel(t)) that induces tumor size reduction and washes out over time. The SLD timecourse (SLD(t)) was predictive of overall survival. KG, first‐order growth rate constant; kout, first‐order rate constant for the degradation or loss of response; ksVEGFR‐3, tumor size reduction rate constant related to sVEGFR‐3 response; λ, tumor resistance/regrowth appearance rate constant; Rin, zero‐order rate constant for the production of response. Dashed arrows represent relationships identified as significant.
Patients and data
Patients were treated with axitinib for a median of 51 weeks (range, 1.7–104). Available biomarker, SLD, dBP, and OS data are summarized in Table 1. Four sVEGFR‐1 and four sVEGFR‐3 concentration values (<1%) were below the limit of quantification and omitted from the analysis dataset. Two patients had SLD data available at baseline only. At the end of the follow‐up period, 16 patients had died and 48 patients were censored from OS analysis.
Biomarkers' models
Log‐transformed biomarker data were well described by IDR models where axitinib inhibits VEGF degradation (Eq. 1) and sVEGFR‐1, ‐2, and ‐3 production (Eq. 2) (Figure 1). A linear time‐dependent disease progression component with slope α described a drug‐independent VEGF increase, whereas no disease progression was identified for sVEGFR‐1, ‐2, and ‐3. Drug effects were described by inhibitory Emax (VEGF, sVEGFR‐1 and ‐3) or sigmoidal Emax models (sVEGFR‐2) assuming that maximum inhibition can be achieved (maximum inhibitory effect Imax = 1).
| (1) |
| (2) |
kout is the first‐order rate constant for the biomarker degradation, expressed as , where MRT is the biomarker mean residence time in plasma. Rin is the zero‐order rate constant for biomarker production, calculated as with Base being the baseline biomarker concentration. γ is the Hill coefficient and AUC50 the AUCdaily leading to half Imax. No axitinib drug effect was identified on sKIT and the data were best described by a linear and constant change over time (Supplementary Material). The sKIT model was therefore not included in the joint biomarker model, nor were sKIT‐related predictors tested on SLD or OS.
When VEGF, sVEGFR‐1, ‐2, and ‐3 were modeled jointly, large correlations (80–99%) were identified between individual AUC50 for sVEGFR‐1, ‐2, and ‐3 and the IIV magnitudes were similar; hence, a common IIV term was used. Moreover, the AUC50 could be shared for sVEGFR‐2 and ‐3 without worsening the model fit. Parameter estimates and their uncertainty are reported in Table 2. AUC50 values were in the range of observed AUCdaily (31.95–1,861 μg·h/L), with VEGF being most sensitive to axitinib (AUC50 of 354 vs. 717–1,380 μg·h/L for the other biomarkers). VEGF and sVEGFR‐1 typically displayed fast turnover (MRT of 0.722 and 0.624 days, respectively) compared to sVEGFR‐2 and ‐3 (MRT of 19.7 and 5.76 days, respectively). A common additive RUV term for all four biomarkers was applied to account for that the biomarkers were sampled at the same time. All model parameters were estimated with reasonable uncertainty (<34% RSE), except for MRT of sVEGFR‐1 (69% RSE), for which the 95% CI obtained from sampling importance resampling was 0.0444–1.58 days.31 pcVPCs show a good predictive ability of the joint biomarker model (Figure 2).
Table 2.
Parameter estimates and their uncertainty for the final joint biomarker model
| VEGF | sVEGFR‐1 | sVEGFR‐2 | sVEGFR‐3 | |||||
|---|---|---|---|---|---|---|---|---|
|
Typical value (RSE%) |
IIV %CV (RSE%) |
Typical value (RSE%) |
IIV %CV (RSE%) |
Typical value (RSE%) |
IIV %CV (RSE%) |
Typical value (RSE%) |
IIV %CV (RSE%) |
|
| Base (pg/mL) | 65.0 (7.8) | 43 (12) | 83.5 (2.9) | 17 (12) | 8,850 (2.8) | 15 (12) | 19,500 (6.5) | 49 (15) |
| MRT (days) | 0.722 (25) | — | 0.624 (69)a | — | 19.7 (17) | 75 (22) | 5.76 (12) | — |
| Imax | 1 FIX | — | 1 FIX | — | 1 FIX | — | 1 FIX | — |
| AUC50 (μg·h/L) | 354 (13) | 39 (34) | 1,380 (13) | 45b (17) | 717c (8.6) | 45b (17) | 717c (8.6) | 45b (17) |
| γ | 1 FIX | — | 1 FIX | — | 0.733 (16) | — | 1 FIX | — |
| α (year−1) | 0.650 (28) | 87 (22) | — | — | — | — | — | — |
| RUVd | 0.376 (5.9) | — | 0.193 (5.3) | — | 0.162 (14) | — | 0.263 (6.5) | — |
| Common RUVd | 0.0593 (26)e | — | 0.0593 (26)e | — | 0.0593 (26)e | — | 0.0593 (26)e | — |
VEGF, vascular endothelial growth factor; sVEGFR‐1, 2, 3, soluble vascular endothelial growth factor receptor 1, 2, 3; RSE, relative standard error; IIV, inter‐individual variability; CV, coefficient of variation; Base, baseline biomarker concentration; MRT, mean residence time; Imax, maximal inhibitory effect; AUC50, axitinib area under the concentration‐time curve giving half of the maximal effect; γ, Hill coefficient; α, slope of the disease progression; RUV, residual unexplained variability.
The 95% confidence interval obtained from sampling importance resampling was 0.0444–1.58 day.
The IIV in AUC50 for VEGFR‐1, 2, and 3 was quantified using a common variability term.
Common AUC50 parameter for sVEGFR‐2 and 3.
Expressed as standard deviation on log‐scale.
Common RUV for all four biomarkers.
Figure 2.
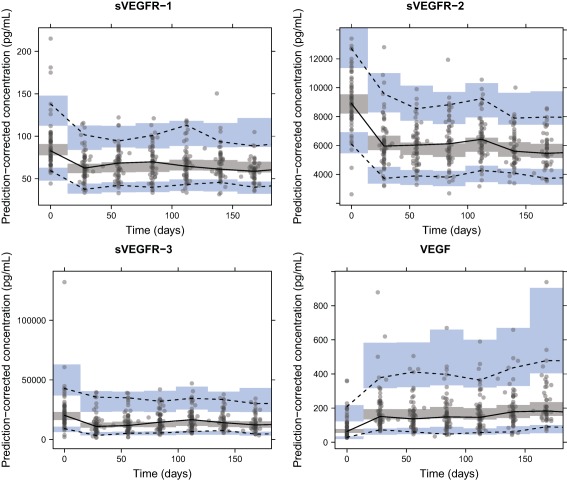
Prediction‐corrected visual predictive checks of the final biomarker models based on 500 simulations. Median (solid line), 5th, and 95th percentiles (dashed lines) of the observed data (solid circles) are compared to the 95% confidence intervals (shaded areas) for the median, 5th, and 95th percentiles of the simulated data. VEGF, vascular endothelial growth factor; sVEGFR‐1, ‐2, ‐3, soluble VEGF receptor 1, 2, 3.
Tumor size model
A tumor size model with an underlying first‐order growth process with rate constant KG best described SLD data (Eq. 3, Figure 1).32 The individual model‐predicted relative change from baseline over time in sVEGFR‐1 (sVEGFR1rel(t), OFV = −339.99) and sVEGFR‐3 (sVEGFR3rel(t), OFV = −339.01) were better drivers of SLD response than all other investigated predictors, including AUCdaily (OFV = −327.06) and sVEGFR‐2 (sVEGFR2rel(t), OFV = −326.38). Despite sVEGFR‐1rel(t) had a one unit lower OFV, sVEGFR‐3rel(t) was chosen to drive the SLD response in the final model, given the large uncertainty in MRT in the sVEGFR‐1 model and for consistency with the published modeling framework in sunitinib‐treated GIST.21 When sVEGFR3rel(t) was included in the model, none of the other predictors further improved the model fit.
| (3) |
ksVEGFR3 is the tumor size reduction rate constant related to sVEGFR‐3 response and λ the tumor resistance/regrowth appearance rate constant. The observed baseline tumor size (SLD(0)) was included as a covariate (i.e., not as a dependent variable), acknowledging the same RUV as for postbaseline observations (B2 method33). The SLD data contained little information on KG, and therefore its value and uncertainty (3.74·10−3 week−1, 6.64% RSE) obtained from a simplified tumor growth inhibition model developed using data from several clinical studies in RCC34 were used as informative prior for KG using the NONMEM $PRIOR subroutine.
The probability of dropping out was estimated to increase with the occurrence of PD ( = 1.22), higher SLD at the time of evaluation ( = 0.00282 mm−1), increasing time since start of study ( = 0.00371 day−1) and decreasing AUCdaily ( = −0.00529 L·h−1·μg−1) (Equations in Supplementary Material). The final SLD and dropout model parameters and their uncertainty are reported in Table 3. The VPCs of the final SLD model accounting for dropout (Figure 3) demonstrate a good predictive performance of the model.
Table 3.
Parameter estimates and their uncertainty for the final tumor size, dropout, diastolic blood pressure, and overall survival models
| Parameter | Estimate (RSE%) | IIV %CV (RSE%) |
|---|---|---|
| Tumor size model | ||
| KG (week−1) | 0.00361 (1.8) | 160 (20) |
| ksVEGFR‐3 (week−1) | −0.174 (15) | — |
| λ (week−1) | 0.101 (18) | 72 (16) |
| RUV (%) | 10.5 (8.2) | 35 (21) |
| Dropout model | ||
| θ0 | −6.11 (7.4) | — |
| θPD | 1.22 (22) | — |
| θSLD (mm−1) | 0.00282 (31) | — |
| θAUC (L·h−1·μg−1) | −0.00529 (18) | — |
| θTime (day−1) | 0.00371 (45) | — |
| Diastolic blood pressure model | ||
| dBP0 (mmHg) | 78.9 (1.4) | 6.7 (12) |
| ShapedBP0 | −5.42 (42) | — |
| MRTdBP (days) | 4.92 (19) | — |
| Emax,dBP | 0.197 (14) | — |
| S0,dBP (L·h−1·μg−1) | 0.00127 (50)a | — |
| RUV (mmHg) | 6.13 (7.0) | — |
| Overall survival model | ||
| β0 | 7.09 (3.2) | — |
| γ | 0.298 (22) | — |
| βSLD (mm−1) | 0.0115 (17) | — |
KG, tumor growth rate constant; ksVEGFR‐3, tumor size reduction rate constant related to soluble vascular endothelial growth factor receptor 3 (sVEGFR‐3) response, which is negative since sVEGFR‐3rel(t) is negative (reduction from baseline); λ, tumor resistance/regrowth appearance rate constant; RUV, residual unexplained variability; θ0, intercept of the logistic regression model; θPD, coefficient for the effect of occurrence of progressive disease; θSLD, coefficient for the effect of sum of longest diameters (SLD) at the time of evaluation; θAUC, coefficient for the effect of axitinib daily area under the curve (AUCdaily); θTime, coefficient for the effect of time since start of study; dBP0, baseline diastolic blood pressure; ShapedBP0, shape parameter in the Box‐Cox transformation of dBP0 random effects; MRTdBP, mean residence time of dBP response; Emax,dBP, maximum axitinib effect on diastolic blood pressure; S0,dBP, slope of the Emax model; β0, scale parameter of the log‐logistic baseline hazard model; γ, shape parameter of the log‐logistic baseline hazard model; βSLD, coefficient for the effect of longitudinal SLD on the hazard.
The 95% confidence interval obtained from sampling importance resampling was 0.609–3.14 μg·h/L.
Figure 3.
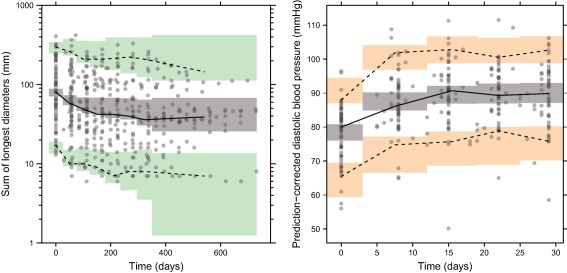
Visual predictive checks of the final sum of longest diameters (SLD, left) and diastolic blood pressure (dBP, right) models based on 500 simulations. Median (solid line), 5th, and 95th percentiles (dashed lines) of the observed data (solid circles) are compared to the 95% CIs (shaded areas) for the median, 5th, and 95th percentiles of the simulated data. Prediction‐correction was used for dBP. For the SLD model, dropout was taken into account in the simulations.
Diastolic BP model
An IDR model with a stimulatory effect of axitinib on response production (Rin,dBP) with an AUCdaily‐driven Emax model, parameterized as a maximal effect Emax,dBP and a slope parameter ( ),35 best described dBP data (Eq. 4, Table 3 , Figure 1). Emax,dBP was estimated to 0.197 and S0,dBP to 0.00127 L·h−1·μg−1, corresponding to an AUC50,dBP of 155 μg·h/L. A Box‐Cox transformation with an estimated shape parameter of −5.42 was applied to baseline dBP (dBP0) IIV to account for the skewed random effects distribution.
| (4) |
The rate constant for the loss of response is defined as , where MRTdBP is the mean turnover time (4.92 days) associated with dBP response, and AUCdaily predicted dBP response better than Dosedaily (dOFV = 7.86). No IIV was identified on drug effect parameters (Emax,dBP and S0,dBP). An additive model best described the RUV. The pcVPC (Figure 3) shows that the model well predicted the overall increase in dBP and the variability during the first month of treatment.
Overall survival model
OS baseline hazard was best described by a log‐logistic distribution with a shape parameter and scale parameter . In the univariate analysis, the best predictor was SLD timecourse (SLD(t), dOFV = −26.9), followed by SLD baseline (dOFV = −13.3). Absolute change in SLD over time, the derivative of SLD predicted timecourse, SLD relative change from baseline at week 8 (i.e., corresponding to tumor size ratio week 8), biomarker‐related predictors (VEGFR‐2(t), VEGF(t), absolute change in sVEGFR‐1(t) and relative change in VEGF from baseline at week 4) and dBP relative change at week 2 also resulted in statistically significant OFV drops; however, these drops were all driven by single individuals. Using both baseline SLD and absolute change in SLD(t) resulted in an OFV drop similar to SLD(t) (dOFV = −27.5) but required one extra parameter. When SLD(t) was included in the model, none of the other predictors further improved model fit. The final OS model is described by Eqs. 5 and 6, and parameter estimates and their uncertainty are reported in Table 3.
| (5) |
| (6) |
was estimated to 0.298, meaning that the hazard, in the absence of changes in SLD(t), initially rises with time before decreasing monotonically ( ). is the coefficient for SLD(t) effect on the hazard, estimated to 0.0115 reflecting a 12% increase in hazard for a 10 mm increase in SLD. A competing log‐logistic function described the hazard of being censored. Kaplan–Meier VPCs for OS (Figure 4) and censoring (Supplementary Material) show adequate predictive properties of the OS model.
Figure 4.
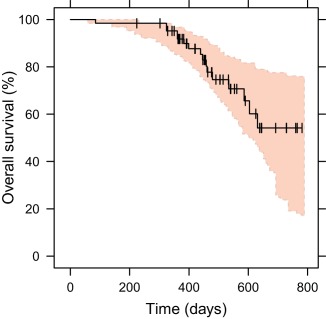
Kaplan–Meier visual predictive checks for the final overall survival model driven by the sum of longest diameters timecourse. The observed Kaplan–Meier curve (black line) is compared to the 95% CI (shaded area) derived from model simulations (200 samples). Vertical black lines represent censored observations.
DISCUSSION
In this pharmacometric framework (Figure 1) we investigated the relationships between drug exposure, soluble biomarkers, tumor size, hypertension, and OS following axitinib treatment in cytokine‐refractory mRCC. Model‐predicted sVEGFR‐3 dynamics predicted tumor size better than axitinib exposure. The SLD timecourse was the best predictor of OS, with an estimated hazard ratio (HR) of 1.12 (95% CI of 1.08–1.17) for every 10 mm SLD increase.
The biomarker timecourses were successfully characterized by IDR models where axitinib inhibited VEGF degradation and sVEGFR‐1, ‐2, and ‐3 production. These model structures are consistent with previously published models in sunitinib‐treated healthy volunteers,36 GIST,21 hepatocellular carcinoma (HCC),37 and metastatic colon cancer (mCC).38 The mechanisms behind these biomarker modulations following VEGFR inhibitor administration have not been fully elucidated. The hypothesis that the VEGF increase observed following VEGFR‐2 inhibition may arise from a reduction in VEGF blood clearance39 is supported by our model. sVEGFR‐2 decrease may result from a ligand‐induced downregulation of VEGFR‐2 from the cell surface, as shown in vitro.40 No axitinib effect was identified on sKIT, confirming previous findings that axitinib has negligible effect on the stem cell factor receptor KIT and acts as a selective VEGFR inhibitor.15, 41, 42 These results differ from those of the study in GIST patients (mostly Caucasian) treated with sunitinib,21 which markedly increases sKIT levels and is less specific to VEGFRs. Results from the joint biomarker model show that in this patient population axitinib more potently inhibited sVEGFR‐2 and ‐3 (AUC50 of 717 μg·h/L) than sVEGFR‐1 (AUC50 of 1,380 μg·h/L). Consistent with the in vitro findings,43 sVEGFR‐2 and ‐3 AUC50 values were lower than for sunitinib in GIST. The estimated typical VEGF and sVEGFR‐2 baseline values were similar to those in healthy volunteers,36 GIST,21 and mCC38, whereas sVEGFR‐3 baseline was about 3‐fold lower than in GIST (63,900 pg/mL) but similar to mCC (21,900 pg/mL). The typical sVEGFR‐3 MRT was shorter in mRCC (5.76 days) than in GIST (16.7 days), denoting a faster turnover rate in mRCC but resulting in similar production rate constant (Rin).
The SLD reduction, seen in most patients after the start of therapy, was described by a tumor model allowing for axitinib‐induced tumor shrinkage. Tumor resistance was estimated to develop at a faster rate in axitinib‐treated mRCC than in sunitinib‐treated GIST (0.101 vs. 0.0217 week−1). The larger the sVEGFR‐3 decrease, the more profound was the predicted tumor shrinkage. In sunitinib‐treated GIST, sVEGFR‐3 and sKIT reduction, as well as larger sunitinib AUC, were predictive of SLD decreases.21 In sunitinib‐treated HCC, sVEGFR‐2 absolute change from baseline was used as a driver for SLD response in a tumor growth inhibition model.37 It should be noted that sVEGFR‐3 was not evaluated in that study. Our results add evidence to the fact that changes in PD biomarkers, which can easily be measured in plasma, can help to better understand and forecast tumor response in several cancer types treated with VEGFR inhibitors.
BP increases in most patients treated with angiogenesis inhibitors targeting the VEGF pathway, but the underlying pathophysiological mechanisms are not fully understood. Proposed mechanisms include reduction in nitric oxide production, increased prohypertensive agents expression, renin‐angiotensin system activation, microvascular rarefaction, oxidative stress, pressure‐natriuresis system, and arterial stiffness.44 Whereas a BP increase is generally promptly managed by antihypertensive therapies or TKI dose reduction, early dBP elevation after treatment initiation could be an easy‐to‐measure biomarker reflecting effective VEGF inhibition.10 The empirical IDR dBP model presented here identified a nonlinear exposure–response relationship. Chen et al. characterized ambulatory dBP data in mRCC patients monitored over a 24‐h period predosing and 4 and 15 days after axitinib treatment initiation, using an IDR model where two cosine functions described dBP diurnal changes.45 In our dataset, the dBP measurements were performed weekly and diurnal variations could not be accounted for. The Emax estimate in the present analysis was similar to their findings (19.7% vs. 20.8%). The turnover rate (kout=1/MRT) differed, however, with an estimate of 0.203 day−1 in our model vs. 0.254 h−1 in the ambulatory setting. The previous analysis45 implies a time to dBP steady‐state of 14 h that would not explain the current data, where dBP reaches steady‐state after 1–2 weeks. This discrepancy may be explained by differences in study design (e.g., observation frequency).
The pharmacometric framework developed in sunitinib‐treated GIST identified baseline SLD and sVEGFR‐3 dynamics as the best predictors for OS, while here the SLD timecourse was the best predictor. These differences may be due to a discrepancy in tumor dynamics in the two patient populations: more axitinib‐treated mRCC patients achieve complete or partial tumor response compared to sunitinib‐treated GIST, for which stable disease is more frequent. In an alternative model in GIST, absolute neutrophil count (ANC) reduction combined with dBP increase and baseline SLD were predictive of longer OS.12 No strong association between dBP and OS was identified here, which contrasts with previous findings for axitinib‐treated mRCC, where a higher maximum dBP during the first 8 weeks of treatment was related to longer OS.10 The lack of information on antihypertensive therapy in later cycles prohibited a more thorough analysis of dBP‐OS relationships. However, since steady‐state was reached within the first cycle, later changes are primarily expected to be related to changes in dose. Since axitinib rarely induces neutropenia, ANC was not included in our analysis.22, 46
In the statistical analysis of long‐term OS data in axitinib‐treated Japanese mRCC patients, a baseline ECOG of 0 and a greater sVEGFR‐2 reduction were associated with longer OS.16 In our analysis, ECOG or sVEGFR‐2 could not predict OS data, which may be explained by the shorter follow‐up period for OS (maximum 112 vs. ∼285 weeks) used in our analysis to avoid confounding effects of subsequent therapies after axitinib discontinuation. In a population analysis in first‐line or refractory RCC patients treated with temsirolimus, sunitinib, or axitinib, time‐to‐tumor‐growth (TTG) could predict OS.34 However, TTG can suffer from time‐dependent bias.47 Moreover, a large variety of tumor profiles may lead to the same TTG, and TTG ignores the extent of tumor shrinkage.17 For these reasons, TTG was not tested on OS here. Instead, the tumor timecourse was identified to be the best predictor for OS, as previously suggested.48, 49, 50
A potential limitation of our analysis is that it exclusively included data from Japanese patients; validation in a non‐Japanese population may be required. Although no PK differences are expected between Japanese and non‐Japanese patients, ethnic/racial differences in axitinib efficacy and safety may exist.51
In summary, the sVEGFR‐3 relative decrease over time was identified as a driver of tumor dynamics, which in turn was predictive of OS in axitinib‐treated mRCC patients. Together with previous findings in sunitinib‐treated GIST,21 our results support the use of sVEGFR data to better anticipate tumor response in patients treated with VEGF pathway inhibitors. In contrast to sunitinib‐treated GIST, BP and biomarkers dynamics were not as good predictors of OS as SLD timecourse. Using the tumor timecourse is indeed more attractive from a theoretical stand point than summary variables, such as tumor size ratio at a specific day or TTG. This type of overarching pharmacometric framework allows for leveraging clinical trial data and improved understanding of the relationships between drug exposure, potential plasma biomarkers, tumor size, frequently observed adverse effects, and long‐term outcome, and can serve as platforms for identifying safe and efficacious dosing regimens through simulations.
Conflict of Interest
M.A.A. is an employee of Pfizer Ltd. M.O.K. and L.E.F. have acted as paid consultants to Pfizer Ltd. As Deputy Editor‐in‐Chief of CPT: Pharmacometrics & Systems Pharmacology, L.E.F. was not involved in the review or decision process for this article. E.S. declared no conflict of interest.
Author Contributions
E.S., M.A.A., M.O.K., and L.E.F. wrote the article; E.S., M.A.A., M.O.K., and L.E.F. designed the research; E.S., M.A.A., M.O.K., and L.E.F. performed the research; E.S., M.A.A., M.O.K., and L.E.F. analyzed the data.
Supporting information
Additional Supporting Information may be found in the online version of this article.
SUPPLEMENTARY MATERIAL is linked to the online version of the article at http://www.wileyonlinelibrary.com.cpt
Supplementary_material_manuscript_axitinib_1_revised.docx
Title: Supplementary material 1. Final sKIT, Dropout and Censoring models
Figure S1. pcVPC of final sKIT model.
Figure S2: Kaplan‐Meier visual predictive check for the final model for censoring from overall survival (OS) analysis.
Supplementary_material_manuscript_axitinib_2.docx
Example dataset and NONMEM codes
Acknowledgments
The research leading to these results received financial support from the Swedish Cancer Society, Sweden (to LEF and MOK), and from DDMoRe, which is an Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115156, resources of which are composed of financial contributions from the European Union's Seventh Framework Programme (FP7/2007‐2013) and EFPIA companies' in‐kind contribution. The DDMoRe project is also supported by financial contribution from Academic and SME partners. This work does not necessarily represent the view of all DDMoRe partners. The collection of the study data was sponsored by Pfizer.
References
- 1. Ljungberg, B. et al EAU guidelines on renal cell carcinoma: 2014 update. Eur. Urol. 67, 913–924 (2015). [DOI] [PubMed] [Google Scholar]
- 2. Rothermundt, C. et al Second‐line treatment for metastatic clear cell renal cell cancer: experts' consensus algorithms. World J. Urol. 2016. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 3.Pfizer Laboratories Div Pfizer Inc. INLYTA — axitinib tablet, film coated: Full prescribing information. [cited 10/18/2016] Available from: <http://labeling.pfizer.com/ShowLabeling.aspx?id=759> 2012.
- 4. Rini, B.I. et al Axitinib with or without dose titration for first‐line metastatic renal‐cell carcinoma: a randomised double‐blind phase 2 trial. Lancet Oncol. 14, 1233–1242 (2013). 24140184 [Google Scholar]
- 5. Hutson, T.E. et al Axitinib versus sorafenib in first‐line metastatic renal cell carcinoma: overall survival from a randomized phase III trial. Clin. Genitourin. Cancer 15, 72–76 (2017). [DOI] [PubMed] [Google Scholar]
- 6. Rini, B.I. et al Axitinib dose titration: analyses of exposure, blood pressure and clinical response from a randomized phase II study in metastatic renal cell carcinoma. Ann. Oncol. 26, 1372–1377 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Eisenhauer, E.A. et al New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009). [DOI] [PubMed] [Google Scholar]
- 8. Ammari, S. et al Radiological evaluation of response to treatment: application to metastatic renal cancers receiving anti‐angiogenic treatment. Diagn. Interv. Imaging 95, 527–539 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Escalante, C.P. & Zalpour, A. Vascular endothelial growth factor inhibitor‐induced hypertension: basics for primary care providers. Cardiol. Res. Pract. 2011, 816897 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rini, B.I. et al Axitinib in metastatic renal cell carcinoma: results of a pharmacokinetic and pharmacodynamic analysis. J. Clin. Pharmacol. 53, 491–504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rini, B.I. et al Diastolic blood pressure as a biomarker of axitinib efficacy in solid tumors. Clin. Cancer Res. 17, 3841–3849 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Hansson, E.K. et al PKPD modeling of predictors for adverse effects and overall survival in sunitinib‐treated patients with GIST. CPT Pharmacometrics Syst. Pharmacol. 2, e85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. George, S. et al Hypertension as a potential biomarker of efficacy in patients with gastrointestinal stromal tumor treated with sunitinib. Ann. Oncol. 23, 3180–3187 (2012). [DOI] [PubMed] [Google Scholar]
- 14. Zurita, A.J. , Jonasch, E. , Wu, H.K. , Tran, H.T. & Heymach, J.V. Circulating biomarkers for vascular endothelial growth factor inhibitors in renal cell carcinoma. Cancer 115(10 suppl.), 2346–2354 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fujiwara, Y. et al Management of axitinib (AG‐013736)‐induced fatigue and thyroid dysfunction, and predictive biomarkers of axitinib exposure: results from phase I studies in Japanese patients. Investig. New Drugs 30, 1055–1064 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eto, M. et al Overall survival and final efficacy and safety results from a Japanese phase II study of axitinib in cytokine‐refractory metastatic renal cell carcinoma. Cancer Sci. 105, 1576–1583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bender B.C., Schindler, E. & Friberg, L.E. Population pharmacokinetic pharmacodynamic modeling in oncology: a tool for predicting clinical response. Br. J. Clin. Pharmacol. 79, 56–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mould, D.R. , Walz, A.C. , Lave, T. , Gibbs, J.P. & Frame, B. Developing exposure/response models for anticancer drug treatment: special considerations. CPT Pharmacometrics Syst. Pharmacol. 4, e00016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Venkatakrishnan, K. et al Optimizing oncology therapeutics through quantitative translational and clinical pharmacology: challenges and opportunities. Clin. Pharmacol. Ther. 97, 37–54 (2015). [DOI] [PubMed] [Google Scholar]
- 20. Ribba, B. et al A review of mixed‐effects models of tumor growth and effects of anticancer drug treatment used in population analysis. CPT Pharmacometrics Syst. Pharmacol. 3, e113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hansson, E.K. et al PKPD modeling of VEGF, sVEGFR‐2, sVEGFR‐3, and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. CPT Pharmacometrics Syst. Pharmacol. 2, e84 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tomita, Y. et al Key predictive factors of axitinib (AG‐013736)‐induced proteinuria and efficacy: a phase II study in Japanese patients with cytokine‐refractory metastatic renal cell Carcinoma. Eur. J. Cancer 47, 2592–2602 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Beal, S. , Sheiner, L.B. , Boeckmann, A. & Bauer, R.J. NONMEM User's Guides. Icon Development Solutions, Ellicott City, MD; 2009. 1989–2009. [Google Scholar]
- 24. Keizer, R.J. , Karlsson, M.O. & Hooker, A. Modeling and simulation workbench for NONMEM: Tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst. Pharmacol. 2, e50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Petersson, K.J. , Hanze, E. , Savic, R.M. & Karlsson, MO. Semiparametric distributions with estimated shape parameters. Pharm. Res. 26, 2174–2185 (2009). [DOI] [PubMed] [Google Scholar]
- 27. Sharma, A. & Jusko, W.J. Characteristics of indirect pharmacodynamic models and applications to clinical drug responses. Br. J. Clin. Pharmacol. 45, 229–239 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang, L. , Beal, S.L. & Sheiner, L.B. Simultaneous vs. sequential analysis for population PK/PD data I: best‐case performance. J. Pharmacokinet. Pharmacodyn. 30, 387–404 (2003). [DOI] [PubMed] [Google Scholar]
- 29. Wade, J.R. & Karlsson, M.O. Combining PK and PD data during population PK/PD analysis [www.page-meeting.org/?abstract=139]. PAGE 8; 1999.
- 30. Lacroix, B.D. , Friberg, L.E. & Karlsson, M.O. Evaluation of IPPSE, an alternative method for sequential population PKPD analysis. J. Pharmacokinet. Pharmacodyn. 39, 177–193 (2012). [DOI] [PubMed] [Google Scholar]
- 31. Dosne, A.G. , Bergstrand, M. , Harling, K. & Karlsson, M.O. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J. Pharmacokinet. Pharmacodyn. 43, 583–596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Claret, L. et al Model‐based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J. Clin. Oncol. 27, 4103–4108 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Dansirikul, C. , Silber, H.E. & Karlsson, M.O. Approaches to handling pharmacodynamic baseline responses. J. Pharmacokinet. Pharmacodyn. 35, 269–283 (2008). [DOI] [PubMed] [Google Scholar]
- 34. Claret, L. , Mercier, F. , Houk, B.E. , Milligan, P.A. & Bruno, R. Modeling and simulations relating overall survival to tumor growth inhibition in renal cell carcinoma patients. Cancer Chemother. Pharmacol. 76, 567–573 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Schoemaker, R.C. , van Gerven, J.M. & Cohen, A.F. Estimating potency for the Emax‐model without attaining maximal effects. J. Pharmacokinet. Biopharm. 26, 581–593 (1998). [DOI] [PubMed] [Google Scholar]
- 36. Lindauer, A. et al Pharmacokinetic/pharmacodynamic modeling of biomarker response to sunitinib in healthy volunteers. Clin. Pharmacol. Ther. 87, 601–608 (2010). [DOI] [PubMed] [Google Scholar]
- 37. Ait‐Oudhia, S. et al Bridging sunitinib exposure to time‐to‐tumor progression in hepatocellular carcinoma patients with mathematical modeling of an angiogenic biomarker. CPT Pharmacometrics Syst. Pharmacol. 5, 297–304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanefendt, F. et al Modeling Sunitinib and Biomarker Response as potential Predictors of Time to Progression in Patients with Metastatic Colorectal Cancer. Annual Meeting of the Population Approach Group in Europe (PAGE) 2012. [Google Scholar]
- 39. Bocci, G. et al Increased plasma vascular endothelial growth factor (VEGF) as a surrogate marker for optimal therapeutic dosing of VEGF receptor‐2 monoclonal antibodies. Cancer Res. 64, 6616–6625 (2004). [DOI] [PubMed] [Google Scholar]
- 40. Ebos, J.M. et al Vascular endothelial growth factor‐mediated decrease in plasma soluble vascular endothelial growth factor receptor‐2 levels as a surrogate biomarker for tumor growth. Cancer Res. 68, 521–529 (2008). [DOI] [PubMed] [Google Scholar]
- 41. Cohen, E.E. et al A phase II trial of axitinib in patients with various histologic subtypes of advanced thyroid cancer: long‐term outcomes and pharmacokinetic/pharmacodynamic analyses. Cancer Chemother. Pharmacol. 74, 1261–1270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hu‐Lowe, D.D. et al Nonclinical antiangiogenesis and antitumor activities of axitinib (AG‐013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 14, 7272–7283 (2008). [DOI] [PubMed] [Google Scholar]
- 43. Gross‐Goupil, M. , Francois, L. , Quivy, A. & Ravaud, A. Axitinib: a review of its safety and efficacy in the treatment of adults with advanced renal cell carcinoma. Clin. Med. Insights Oncol. 7, 269–277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wasserstrum, Y. et al Hypertension in cancer patients treated with anti‐angiogenic based regimens. Cardio‐Oncology 1, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen, Y. , Rini, B.I. , Bair, A.H. , Mugundu, G.M. & Pithavala, Y.K. Population pharmacokinetic‐pharmacodynamic modeling of 24‐h diastolic ambulatory blood pressure changes mediated by axitinib in patients with metastatic renal cell carcinoma. Clin. Pharmacokinet. 54, 397–407 (2015). [DOI] [PubMed] [Google Scholar]
- 46. Rini, B.I. et al Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma: a randomised phase 3 trial. Lancet 378, 1931–1939 (2011). [DOI] [PubMed] [Google Scholar]
- 47. Mistry, H.B. Time‐dependent bias of tumour growth rate and time to tumour re‐growth. CPT Pharmacometrics Syst. Pharmacol. 5, 587 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bender, B.C. et al A population pharmacokinetic/pharmacodynamic model of thrombocytopenia characterizing the effect of trastuzumab emtansine (T‐DM1) on platelet counts in patients with HER2‐positive metastatic breast cancer. Cancer Chemother. Pharmacol. 70, 591–601 (2012). [DOI] [PubMed] [Google Scholar]
- 49. Holford, N. A time to event tutorial for pharmacometricians. CPT Pharmacometrics Syst. Pharmacol. 2, e43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ribba, B. , Holford, N. & Mentre, F. The use of model‐based tumor‐size metrics to predict survival. Clin. Pharmacol. Ther. 96, 133–135 (2014). [DOI] [PubMed] [Google Scholar]
- 51. Chen, Y. et al Axitinib plasma pharmacokinetics and ethnic differences. Investig. New Drugs 33, 521–532 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article.
SUPPLEMENTARY MATERIAL is linked to the online version of the article at http://www.wileyonlinelibrary.com.cpt
Supplementary_material_manuscript_axitinib_1_revised.docx
Title: Supplementary material 1. Final sKIT, Dropout and Censoring models
Figure S1. pcVPC of final sKIT model.
Figure S2: Kaplan‐Meier visual predictive check for the final model for censoring from overall survival (OS) analysis.
Supplementary_material_manuscript_axitinib_2.docx
Example dataset and NONMEM codes