

Abstract
Background and Objective
Abiraterone acetate is approved for the treatment of metastatic castration-resistant prostate cancer. The originator abiraterone acetate (OAA) formulation is poorly absorbed and exhibits large pharmacokinetic variability in abiraterone exposure. Abiraterone acetate fine particle (AAFP) is a proprietary formulation (using SoluMatrix Fine Particle Technology™) designed to increase the oral bioavailability of abiraterone acetate. Here, we report on two phase I studies in healthy male subjects aged 18–50 years.
Methods
In Study 101, 20 subjects were randomized in a crossover design to single doses of AAFP 100, 200, or 400 mg or OAA 1000 mg taken orally under fasting conditions. Results suggested that AAFP 500 mg would be bioequivalent to OAA 1000 mg in the fasted state. To confirm the bioequivalence hypothesis and to further expand the AAFP dose range, in Study 102, 36 subjects were randomized in a crossover design to single doses of AAFP 125, 500, or 625 mg or OAA 1000 mg. Both studies included a 7-day washout period between administrations.
Results
Dose-dependent increases in the area under the plasma concentration–time curve and maximum plasma concentration with AAFP were observed in both studies. The AAFP 500-mg bioavailability relative to OAA 1000 mg measured by the geometric mean ratio for area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration was 93.4% (90% confidence interval 85.3–102.4), area under the plasma concentration–time curve from time zero to infinity was 91.0% (90% confidence interval 83.3–99.4), and maximum plasma concentration was 99.8% (90% confidence interval 86.3–115.5). Dose proportionality was seen across all AAFP dose levels (100–625 mg). Abiraterone acetate fine particle was found to be safe and well tolerated in this study.
Conclusion
Abiraterone acetate fine particle 500 mg was demonstrated to be bioequivalent to OAA 1000 mg in healthy volunteers under fasted conditions.
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-017-0536-2) contains supplementary material, which is available to authorized users.
Key Points
| Abiraterone acetate fine particle is a proprietary formulation (using SoluMatrix Fine Particle Technology™) that has enhanced dissolution properties designed to increase the oral bioavailability of abiraterone acetate relative to the originator abiraterone acetate formulation. |
| Abiraterone acetate fine particle displayed dose proportionality over the wide range of doses (100–625 mg) tested under fasted conditions. |
| The 500-mg dose of abiraterone acetate fine particle was bioequivalent to the 1000-mg dose of originator abiraterone acetate based on the 90% confidence interval of the geometric mean ratio of the area under the plasma concentration–time curve and maximum plasma concentration parameters. |
Introduction
Prostate cancer is the second most common cancer in men [1], with an estimated 180,890 new cases of prostate cancer projected for 2016 in USA and 26,120 projected deaths [2]. Androgen activity affects the development of prostate cancer, a finding that has led to therapies that target the synthesis and signaling of these steroid hormones [3]. Once prostate cancer has advanced to metastatic disease, androgen deprivation therapy is employed. However, drug resistance is of serious concern, as further progression of prostate cancer can lead to the lethal phenotype of castration-resistant prostate cancer [4].
Abiraterone is a selective inhibitor of 17α-hydroxylase/C17, 20-lyase, a key enzyme in androgen synthesis, which reduces the level of circulating androgens [5]. Abiraterone acetate is a prodrug for abiraterone. Studies of the originator abiraterone acetate (OAA) tablets (Zytiga®) were conducted in healthy subjects and in patients with prostate cancer. Phase I dose-escalation studies of OAA in prostate cancer patients indicated that it is generally safe and well tolerated and has antitumor activity at tested doses (250–2000 mg) [6]. Abiraterone exposure was observed to approach saturation and not significantly increase beyond the 1000-mg dose [7]. Large variability in abiraterone exposure has also been observed in healthy subjects dosed with single doses of OAA [8].
SoluMatrix Fine Particle Technology™ is intended to increase the in vivo dissolution rate of lipophilic, poorly water-soluble active pharmaceutical ingredients and to improve the low oral bioavailability, high pharmacokinetic variability, and food effects often associated with these types of compounds. In the present study, the pharmacokinetic characteristics and safety profile of abiraterone acetate fine particle (AAFP) tablet formulated using SoluMatrix Fine Particle Technology™ were compared with the reference drug, OAA, in healthy male adults.
Methods
Study Population
Normal healthy male subjects aged 18–50 years with a body mass index between 18 and 30 kg/m2 who met additional inclusion/exclusion criteria were enrolled in the studies. Subjects were in good health based on physical examination and laboratory test results. No concomitant medication use was noted for any subject during the study. Subjects with a recent history (i.e., within the previous year) of mental illness, drug addiction or abuse, or alcoholism, or the use of any drugs known to induce or inhibit hepatic drug metabolism within 30 days of planned dosing were excluded. All enrolled subjects were negative for abused drugs and alcohol at screening and in repeat testing the day prior to dosing of the investigational medical product.
Study Design
Study 101 and Study 102 were both single-dose, randomized, open-label, four-period, four-treatment crossover studies performed under fasted conditions, and they were conducted at a single center in USA (PAREXEL International Early Phase Clinical Unit, Baltimore, MD, USA). The test drugs in this study were administered orally. Study 101 was conducted between 11 February, 2014, and 7 April, 2014. Study 102 was conducted between 10 October, 2014, and 9 December, 2014. For Study 101 (n = 20) and Study 102 (n = 36), the subjects met all the eligibility criteria and were subsequently included and randomized in the study. All subjects provided written informed consent and the study was approved by the Aspire Institutional Review Board (Santee, CA, USA).
Study 101 was divided into a screening phase of up to 28 days followed by four sequential 1-day treatment periods, with an intervening 7-day washout period scheduled between administrations. The total study duration was approximately 52 days, including the screening period. The AAFP tablets were given as single doses of 100, 200, and 400 mg, and OAA was given as a single dose of 1000 mg.
Study 102 was divided into a screening period of up to 28 days followed by four sequential 1-day treatment periods, with an intervening 7-day washout period between administrations. The maximum study duration for each subject was approximately 54 days. The AAFP 125-mg tablets were given as single doses of 125, 500, and 625 mg, and OAA was given as a single dose of 1000 mg.
For both studies, enrolled subjects were allocated randomization numbers sequentially and then randomly allocated to sequence groups according to a pre-generated randomization schedule. A single standard dose of OAA 1000 mg (4 × 250-mg tablets) was administered to subjects. All abiraterone acetate doses were given after an overnight fast, and no food was consumed until 4 h after dosing. There was no placebo control group for either study because each subject served as his own control.
The AAFP tablets were manufactured by Mayne Pharma Group Ltd for Churchill Pharmaceuticals LLC. SoluMatrix Fine Particle Technology™ is licensed from iCeutica, Inc. to Churchill Pharmaceuticals LLC for the use of AAFP. The OAA tablets were obtained commercially.
Pharmacokinetic Evaluation Criteria
For the determination of abiraterone concentrations, blood samples were collected prior to dosing (0 h for Study 101, −0.75 h for Study 102) and at 0.25, 0.50, 1.00, 1.50, 2.00, 3.00, 4.00, 6.00, 8.00, 12.00, 24.00, and 48.00 h post-dose. For Study 102, blood samples were also collected at 72.00 h post-dose.
Blood Sample Collection and Bioanalytical Assay
Blood samples (approximately 6 mL) for pharmacokinetic analyses were collected into 6-mL tripotassiumethylenediaminetetraacetic acid-containing tubes. Blood samples were immediately placed on ice. Plasma was separated by centrifugation at 4 °C, transferred to appropriately labeled polypropylene specimen containers, frozen at −20 °C within 1 h of collection, and then stored at −20 °C. Samples were shipped on dry ice to the bioanalytical laboratory for analysis. Plasma samples were analyzed for abiraterone concentrations using a validated bioanalytical assay (Sannova Analytical, Somerset, NJ, USA).
The samples were extracted using the liquid–liquid extraction method and analyzed using reverse-phase liquid chromatography. The analytes were detected using tandem mass spectrometry. The lower limit of quantitation for abiraterone was 0.651 ng/mL. The upper limit of quantitation for abiraterone was 203.519 ng/mL. A calibration curve consisting of two control blanks, two zero standards, and ten non-zero calibration standards covering a concentration range of 0.651–203.519 ng/mL for abiraterone were analyzed with every sample batch. Quality control standards for abiraterone were also analyzed with every sample batch. Internal standard peak area ratio values were used to set up the calibration curve and to determine quality control and unknown sample concentrations. Linear regression for abiraterone was used to obtain the best fit of the data for the calibration curve.
Pharmacokinetic Analysis
The following plasma pharmacokinetic parameters were determined: the area under the plasma concentration–time curve (AUC) from time zero to the time (t) of the last quantifiable concentration (C t) [AUC0–t], as calculated by the linear trapezoidal method; AUC from time zero to infinity (AUC0–∞); maximum measured plasma concentration (C max); time to maximum measured plasma concentration (t max); apparent elimination rate constant (K e) as determined by linear regression of the terminal points of the log-linear concentration–time curve; and apparent elimination half-life (t ½) calculated as loge(2)/K e or 0.693/K e.
Safety Assessments
All subjects who received at least one dose of abiraterone acetate were included in the safety population. Assessments included physical examinations, recording of vital signs, electrocardiography, clinical laboratory testing (hematology, biochemistry, and urinalysis), and adverse event (AE) assessments. Concomitant medications were assessed throughout the clinical study.
Sample Size
For Study 101, previous studies in healthy subjects indicated that following a single 1000-mg dose of OAA, the AUC0–∞ was approximately 500 ng·h/mL with a standard deviation (SD) of approximately 300. It was estimated that 18 subjects would be able to provide a statistical power of 90% to detect a 50% difference in the reference mean between a test and reference dose at the 0.05 level (two-sided) using a paired t test. The clinical study planned to randomize 20 subjects for the four-way crossover design without replacement.
For Study 102, results from Study 101 indicated that the estimated within-subject between-formulation σ (log scale) was about 0.08 for AUC parameters. Assuming that the true AUC mean of a test treatment was within the 95% region of the reference, a sample size of 32 subjects or more with a two-way analysis of variance (ANOVA) model for bioequivalence assessment would have at least 80% power to reject the null hypothesis of bio-inequivalence at the level of 0.05. Taking dropouts into consideration, 36 subjects were planned to be randomized and dosed without replacement.
Statistical Analysis
A parametric general linear model was applied to the pharmacokinetic parameters of AUC, C max, t max, t ½, and K e. An ANOVA for a crossover design was used to examine the differences among the doses (100, 200, and 400 mg AAFP and 1000 mg OAA in Study 101; 125, 500, and 625 mg AAFP and 1000 mg OAA in Study 102) under fasted conditions. The AUC and C max parameters of AAFP 500 and 625 mg and OAA 1000 mg were compared and analyzed on a natural logarithm scale using a three-way ANOVA model with sequence, subject within sequence, treatment, and period as the classification variables to assess relative bioavailability in Study 102 using the bioequivalence approach. To determine bioequivalence, AUC and C max parameters were analyzed on a log scale. The two one-sided t test hypotheses were tested at the 0.05 level by constructing a 90% confidence interval (CI) for the geometric mean ratio. Bioequivalence was concluded if the 90% CIs of the ratios were within 0.80–1.25 for AUC and C max parameters. For dose proportionality, C max and AUC parameters were examined among AAFP doses ranging from 100 to 625 mg for both studies pooled, using a power model, P = a × Doseb, where P represents C max or AUC, and a and b are constants. A value of b approximately equal to 1 was used to establish linearity or dose proportionality over the dose range under study.
Results
Subject Baseline Characteristics and Disposition
Demographic characteristics for both studies are shown in Table 1. For Study 101, 20 subjects met all the eligibility criteria and were subsequently included and randomized to one of the four dose schedules. Seventeen of the randomized subjects (85%) received all four doses, one subject received three doses, and two subjects received two doses of abiraterone acetate.
Table 1.
Summary of demographic and baseline characteristics
| Study 101 | Study 102 | |
|---|---|---|
| Number of subjects | 20 | 36 |
| Mean age (SD), years | 33.4 (8.01) | 34.5 (8.27) |
| Mean weight (SD), kg | 81.24 (11.884) | 82.27 (10.245) |
| Mean height (SD), cm | 177.1 (8.05) | 177.3 (7.55) |
| Mean BMI (SD), kg/m2 | 25.82 (2.691) | 26.14 (2.504) |
| Race, n (%) | ||
| White | 5 (25) | 8 (22.2) |
| African American | 15 (75) | 26 (72.2) |
| Other | 0 (0) | 2 (5.6) |
BMI body mass index, SD standard deviation
For Study 102, 36 subjects met all eligibility criteria and were included and randomized to one of the four dose schedules. Thirty-three randomized subjects (91.7%) received all four doses, one subject received three doses, and two subjects received two doses of abiraterone acetate. Minor protocol deviations were recorded for subjects in both studies during the course of the assessments, but they did not affect the outcomes or interpretations of the study results.
Pharmacokinetic Parameters of AAFP
Study 101
Following administration of AAFP (100, 200, and 400 mg), plasma concentrations of the drug increased rapidly (Fig. 1a, b). Mean peak concentrations were observed at approximately 1.5–2.0 h post-dose, followed by a gradual decline at or below the lower limit of quantification by 48 h post-dose. For OAA 1000 mg, concentrations also peaked at 2.0 h post-dose and were near the lower limit of quantification at 48 h post-dose.
Fig. 1.
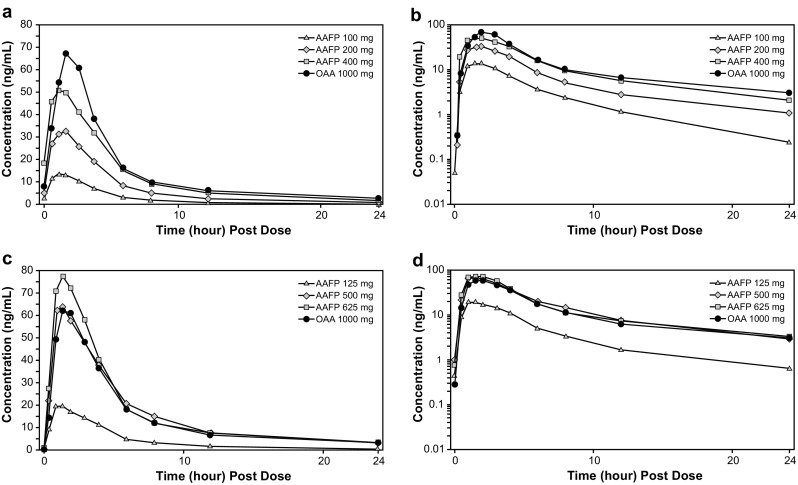
a Linear scale of mean plasma concentration–time plot by treatment for Study 101. b Semi-logarithmic scale of mean plasma concentration–time profile for Study 101. c Linear scale of mean plasma concentration–time plot by treatment for Study 102. d Semi-logarithmic scale of mean plasma concentration–time profile for Study 102. AAFP abiraterone acetate fine particle, OAA originator abiraterone acetate
There was a dose-dependent increase in the AUC and C max (Table 2). For AAFP 100, 200, and 400 mg, AUC0–∞ was 74.5, 183.3, and 319.9 ng·h/mL (421.2 ng·h/mL for OAA); AUC0–t was 67.6, 170.0, and 302.9 ng·h/mL (387.3 ng·h/mL for OAA); and C max was 17.3, 39.1, and 65.4 ng/mL (79.5 ng/mL for OAA), respectively. The median t max for the three AAFP doses ranged from 1.5 to 2.0 h (2.0 h for OAA). The mean t ½ increased with dose for AAFP and was 4.7, 7.8, and 8.8 h for the 100-, 200-, and 400-mg doses, respectively (14.5 h for OAA). Analysis of dose proportionality, using the bioequivalence approach, for AAFP 100- and 400-mg doses compared with the 200-mg dose is shown in Online Resource 1. The drug exposure parameters (AUC and C max) for the highest dose of AAFP (400 mg) were only 18–24% below those for OAA 1000 mg, suggesting that a single 500-mg dose of AAFP should be bioequivalent to a single 1000-mg dose of OAA under fasted conditions.
Table 2.
Study 101 summary of plasma pharmacokinetic parameters (pharmacokinetic population)
| Pharmacokinetic parameter | Summary statistics | Treatment (mg)a | |||
|---|---|---|---|---|---|
| AAFP 100 | AAFP 200 | AAFP 400 | OAA 1000 | ||
| N | 19 | 18 | 19 | 19 | |
| AUC0–∞ (ng·h/mL) | Mean | 74.49 | 183.34 | 319.92 | 421.23 |
| SD | 42.22 | 86.70 | 140.74 | 183.83 | |
| CV% | 56.68 | 47.29 | 43.99 | 43.64 | |
| Geometric mean | 64.46 | 166.16 | 290.89 | 383.46 | |
| AUC0–t (ng·h/mL) | Mean | 67.55 | 169.99 | 302.90 | 387.34 |
| SD | 39.37 | 83.73 | 137.17 | 168.67 | |
| CV% | 58.28 | 49.25 | 45.29 | 43.55 | |
| Geometric mean | 57.86 | 152.6 | 273.40 | 352.08 | |
| C max (ng/mL) | Mean | 17.28 | 39.11 | 65.42 | 79.46 |
| SD | 10.41 | 21.69 | 35.58 | 39.56 | |
| CV% | 60.29 | 55.46 | 54.39 | 49.78 | |
| Geometric mean | 14.66 | 33.94 | 56.00 | 70.63 | |
| t max (h) | Mean | 1.55 | 1.78 | 2.32 | 2.16 |
| SD | 0.57 | 0.77 | 1.33 | 0.78 | |
| Median | 1.5 | 1.75 | 2 | 2 | |
| CV% | 37.02 | 43.38 | 57.22 | 36.27 | |
| Geometric mean | 1.45 | 1.64 | 1.99 | 2.03 | |
| t ½ (h) | Mean | 4.72 | 7.83 | 8.84 | 14.48 |
| SD | 2.57 | 3.88 | 2.96 | 5.11 | |
| Median | 3.66 | 6.94 | 8.05 | 14.01 | |
| CV% | 54.35 | 49.51 | 33.45 | 35.32 | |
| Geometric mean | 4.17 | 7.06 | 8.42 | 13.64 | |
| K e (/h) | Mean | 0.18 | 0.11 | 0.09 | 0.05 |
| SD | 0.08 | 0.05 | 0.03 | 0.02 | |
| CV% | 43.38 | 45.27 | 30.80 | 36.98 | |
| Geometric mean | 0.17 | 0.10 | 0.08 | 0.05 | |
AAFP abiraterone acetate fine particle, AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC 0–t area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration, C max maximum plasma concentration, CV% coefficient of variation expressed as percent, K e apparent elimination rate constant, N number of subjects included in the pharmacokinetic population for each treatment, OAA originator abiraterone acetate, SD standard deviation, t ½ elimination half-life, t max time to maximum plasma concentration
aAAFP 100 mg (1 × 100 mg), 200 mg (2 × 100 mg), 400 mg (4 × 100 mg), and OAA 1000 mg (4 × 250 mg) under fasted conditions
Study 102
The dose range in Study 102 was expanded to include AAFP 125-, 500-, and 625-mg doses and compared with OAA 1000 mg as a reference. Following a single-dose administration of each of the three doses of AAFP (125, 500, and 625 mg), plasma concentrations of abiraterone increased with increasing doses (Fig. 1 c, d). The curve for AAFP 500 mg overlapped with the curve for OAA. Peak levels occurred approximately 2 h post-dose for all doses. This pattern was also observed for individual subjects receiving each dose of AAFP.
Table 3 shows the arithmetic means for AUC0–∞, which were 112.1, 438.02, and 473.31 ng·h/mL for AAFP 125, 500, and 625 mg, respectively, compared with 453.2 ng·h/mL for OAA 1000 mg. The AUC0–t means were 102.6, 416.2, and 450.2 ng·h/mL for AAFP 125, 500, and 625 mg, respectively, compared with 415.9 ng·h/mL for OAA. Maximum plasma concentration means were 28.2, 84.2, and 100.8 ng/mL for AAFP 125, 500, and 625 mg, respectively, compared with 83.4 ng/mL for OAA. Comparisons of AUC and C max values for AAFP 125, 500, and 625 mg and OAA 1000 mg are shown graphically in Fig. 2. The median t max was similar at 1.5 h at all AAFP doses compared with 2.0 h for OAA. The median t max for both the AAFP 500- and 625-mg doses (Table 3) compared with OAA was not statistically significant (AAFP 500 mg vs. OAA 1000 mg, P = 0.2301; AAFP 625 mg vs. OAA 1000 mg, P = 0.3041) as determined by the Wilcoxon signed rank test. The elimination half-life for AAFP 500 mg and 625 mg was 14.1 and 14.5 h, respectively, vs. 20.6 h for OAA 1000 mg. The difference in t ½ between AAFP 500 and 625 mg and OAA 1000 mg was statistically significant (P < 0.0001) as determined by the Wilcoxon signed rank test.
Table 3.
Study 102 summary of plasma pharmacokinetic parameters (pharmacokinetic population)
| Pharmacokinetic parameter | Summary statistics | Treatment (mg)a | |||
|---|---|---|---|---|---|
| AAFP 125 | AAFP 500 | AAFP 625 | OAA 1000 | ||
| N | 33 | 34 | 34 | 33 | |
| AUC0–∞ (ng·h/mL) | Mean | 112.12 | 438.02 | 473.31 | 453.18 |
| SD | 65.94 | 249.43 | 247.19 | 219.07 | |
| CV% | 58.81 | 56.94 | 52.23 | 48.34 | |
| Geometric mean | 93.86 | 372.67 | 418.21 | 408.60 | |
| AUC0–t (ng·h/mL) | Mean | 102.55 | 416.23 | 450.19 | 415.91 |
| SD | 63.27 | 245.73 | 241.85 | 210.67 | |
| CV% | 61.70 | 59.04 | 53.72 | 50.65 | |
| Geometric mean | 84.75 | 350.02 | 394.76 | 372.51 | |
| C max (ng/mL) | Mean | 28.22 | 84.16 | 100.76 | 83.40 |
| SD | 16.46 | 44.05 | 63.75 | 57.40 | |
| CV% | 58.34 | 52.34 | 63.27 | 68.83 | |
| Geometric mean | 23.98 | 73.45 | 84.07 | 70.44 | |
| t max (h) | Median | 1.5 | 1.5 | 1.5 | 2 |
| Minimum | 0.5 | 0.5 | 0.5 | 0.5 | |
| Maximum | 4 | 6 | 4 | 8 | |
| t ½ (h) | Mean | 7.20 | 14.12 | 14.54 | 20.64 |
| SD | 3.47 | 6.44 | 5.54 | 9.03 | |
| CV% | 48.28 | 45.61 | 38.07 | 43.75 | |
| Geometric mean | 6.27 | 12.59 | 13.63 | 18.77 | |
| K e (/h) | Mean | 0.13 | 0.06 | 0.05 | 0.04 |
| SD | 0.09 | 0.05 | 0.02 | 0.02 | |
| CV% | 65.70 | 71.26 | 36.10 | 46.69 | |
| Geometric mean | 0.11 | 0.06 | 0.05 | 0.04 | |
AAFP abiraterone acetate fine particle, AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC 0–t area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration, C max maximum plasma concentration, CV% coefficient of variation expressed as percent, K e apparent elimination rate constant, N number of subjects included in the pharmacokinetic population for each treatment, OAA originator abiraterone acetate, SD standard deviation, t ½ elimination half-life, t max time to maximum plasma concentration
aAAFP 125 mg (1 × 125 mg), 500 mg (4 × 125 mg), 625 mg (5 × 125 mg), and OAA 1000 mg (4 × 250 mg) under fasted conditions
Fig. 2.

Mean (standard error) of area under the plasma concentration–time curve and maximum plasma concentration (C max) parameters by treatment for Study 102. AAFP abiraterone acetate fine particle, AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC 0–t area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration, OAA originator abiraterone acetate, PK pharmacokinetic
Bioequivalence analyses of AAFP 500- and 625-mg test doses compared with OAA 1000 mg are shown in Table 4. The relative bioavailability of 500 and 625 mg of AAFP, calculated by the geometric mean ratio of AUC, indicated that a single dose of 500 or 625 mg of AAFP was bioequivalent to a 1000-mg single dose of OAA, based on the predefined bioequivalence criteria of 90% CI within 0.80–1.25. By these same criteria, a single dose of 500 mg of AAFP was also bioequivalent to a single dose of OAA 1000 mg in terms of C max. However, 625 mg of AAFP was not bioequivalent (90% CI 1.02–1.37) for peak (C max) abiraterone exposure.
Table 4.
Bioequivalence analysis of AAFP to OAA
| PK parameters | Average (raw data) | ANOVA model-based least-square mean (log scale) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AAFP 500 mg | OAA 1000 mg | Ratio (%) | AAFP 500 mg | OAA 1000 mg | Mean difference | (90% CI) | Geometric mean ratio | (90% CI of ratio) | |
| AUC0–∞ | 438.023 | 453.179 | 95.8 | 5.906 | 6.000 | −0.094 | (−0.182, −0.006) | 0.910 | (0.833–0.994)a |
| AUC0–t | 416.232 | 415.908 | 98.9 | 5.843 | 5.911 | −0.068 | (−0.160, 0.024) | 0.934 | (0.853–1.024)a |
| C max | 84.159 | 83.398 | 119.5 | 4.260 | 4.262 | −0.002 | (−0.148, 0.144) | 0.998 | (0.863–1.155)a |
| PK parameters | Average (raw data) | ANOVA model-based least-square mean (log scale) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AAFP 625 mg | OAA 1000 mg | Ratio (%) | AAFP 625 mg | OAA 1000 mg | Mean difference | (90% CI) | Geometric mean ratio | (90% CI of ratio) | |
| AUC0–∞ | 473.315 | 453.179 | 107.9 | 6.039 | 6.000 | 0.039 | (−0.049, 0.127) | 1.040 | (0.952–1.136)a |
| AUC0–t | 450.193 | 415.908 | 111.8 | 5.981 | 5.911 | 0.070 | (−0.022, 0.162) | 1.073 | (0.979–1.175)a |
| C max | 100.757 | 83.398 | 138.6 | 4.428 | 4.262 | 0.166 | (0.020, 0.312) | 1.181 | (1.020–1.366) |
| Statistics | AAFP 500 mg | AAFP 625 mg | OAA 1000 mg |
|---|---|---|---|
| N | 34 | 34 | 34 |
| Mean | 1.79 | 1.84 | 2.21 |
| SD | 1.12 | 0.97 | 1.44 |
| Median | 1.50 | 1.50 | 2.00 |
| Minimum | 0.50 | 0.50 | 0.50 |
| Maximum | 6.00 | 4.00 | 8.00 |
| 95% CI (mean) | (1.403, 2.186) | (1.501, 2.176) | (1.703, 2.709) |
| P value (Wilcoxon signed rank) | 0.2301 | 0.3041 |
AAFP abiraterone acetate fine particle, ANOVA analysis of variance, AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC 0–t area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration, CI confidence interval, C max maximum plasma concentration, OAA originator abiraterone acetate, PK pharmacokinetic, SD standard deviation
aThe 90% CI fell within the recommended 0.800–1.250 limits of bioequivalence when analyzed on a log scale
Analysis of dose proportionality of AAFP, using the bioequivalence approach, is shown in Table 5. Area under the plasma concentration–time curve parameters for AAFP 125 vs. 500 mg fell within the recommended 90% CI (0.80–1.25 limit) of the geometric mean ratios for bioequivalence, but C max did not. However, for AAFP 625 mg vs. 500 mg, the 90% CIs of the geometric mean ratios for both AUC and C max were within the limits of bioequivalence.
Table 5.
Bioequivalence analysis of dose proportionality for AAFP 125 and 625 mg after dose normalization to 500 mg
| PK parameters | Average (raw data) | ANOVA model-based least-square mean (log scale) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AAFP 125 mg | AAFP 500 mg | Ratio (%) | AAFP 125 mg | AAFP 500 mg | Mean difference | (90% CI) | Geometric mean ratio | (90% CI of ratio) | |
| AUC0–∞ | 448.472 | 436.075 | 107.4 | 5.970 | 5.946 | 0.024 | (−0.077, 0.125) | 1.024 | (0.926–1.133)a |
| AUC0–t | 410.206 | 414.087 | 104.0 | 5.869 | 5.886 | −0.017 | (−0.125, 0.091) | 0.983 | (0.882–1.096)a |
| C max | 112.884 | 82.452 | 155.2 | 4.583 | 4.303 | 0.28 | (0.113, 0.447) | 1.323 | (1.119–1.564) |
| PK parameters | Average (raw data) | ANOVA model-based least-square mean (log scale) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| AAFP 625 mg | AAFP 500 mg | Ratio (%) | AAFP 625 mg | AAFP 500 mg | Mean difference | (90% CI) | Geometric mean ratio | (90% CI of ratio) | |
| AUC0–∞ | 394.271 | 438.023 | 96.9 | 5.881 | 5.946 | −0.065 | (−0.165, 0.035) | 0.937 | (0.848–1.035)a |
| AUC0–t | 375.011 | 416.232 | 97.9 | 5.826 | 5.886 | −0.060 | (−0.167, 0.047) | 0.942 | (0.846–1.048)a |
| C max | 83.931 | 84.159 | 105.6 | 4.276 | 4.303 | −0.027 | (−0.192, 0.138) | 0.973 | (0.826–1.148)a |
AAFP abiraterone acetate fine particle, ANOVA analysis of variance, AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC 0–t area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration, CI confidence interval, C max maximum plasma concentration, PK pharmacokinetic
aThe 90% CI fell within the recommended 0.800–1.250 limits of bioequivalence when analyzed on a log scale
AAFP Dose Proportionality
The dose proportionality of AAFP 100-, 125-, 200-, 400-, 500-, and 625-mg doses across Study 101 and Study 102 was evaluated using a power model (P = a × Doseb) of linearity analysis. The results are shown in Table 6. The findings indicate that constant b was 0.986 (95% CI 0.864–1.109) for AUC0–∞, 1.015 (95% CI 0.888–1.142) for AUC0–t, and 0.865 (95% CI 0.735–0.995) for C max, indicating a high degree of linearity (b value of approximately 1), which suggests dose proportionality of abiraterone in the range of 100–625 mg of AAFP.
Table 6.
Power-model analysis of dose proportionality for AAFP doses (100, 125, 200, 400, 500, and 625 mg) for pooled data from Study 101 and Study 102
| PK parameters | Statistics | Power model: P = a × Doseb | |
|---|---|---|---|
| a | b | ||
| AUC0–∞ | Parameter estimates | 0.7810 | 0.9864 |
| 95% CI-lower limit | 0.3891 | 0.8640 | |
| 95% CI-upper limit | 1.5677 | 1.1089 | |
| P value | 0.4845 | <0.0001 | |
| AUC0–t | Parameter estimates | 0.6163 | 1.0146 |
| 95% CI-lower limit | 0.2994 | 0.8877 | |
| 95% CI-upper limit | 1.2688 | 1.1415 | |
| P value | 0.1874 | <0.0001 | |
| C max | Parameter estimates | 0.3304 | 0.8648 |
| 95% CI-lower limit | 0.1574 | 0.7345 | |
| 95% CI-upper limit | 0.6937 | 0.9951 | |
| P value | 0.0037 | <0.0001 | |
AAFP abiraterone acetate fine particle, AUC 0–∞ area under the plasma concentration–time curve from time zero to infinity, AUC 0–t area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration, CI confidence interval, C max maximum plasma concentration, PK pharmacokinetic
Safety
Treatment-emergent AEs related to either AAFP or OAA were mostly mild in intensity (grade 1) for both studies. However, three subjects experienced moderate-intensity (grade 2) AEs (upper respiratory tract infection, headache, and increased transaminase). All treatment-emergent AEs were resolved by the end of the studies, with the exception of one subject who was discontinued from the study because of neutropenia following a dose of the OAA. The outcome for this subject was unknown. There were no serious AEs or deaths during the studies.
Discussion
The main finding in this report was that a single dose of AAFP 500 mg was bioequivalent to the recommended dose of OAA 1000 mg [7], thus demonstrating that a 50% lower dose of AAFP produces equivalent pharmacokinetics as OAA. The AAFP formulation displayed dose proportionality over the wide range of doses (100–625 mg) that were tested. With regard to drug exposure, the relative bioavailability of a single AAFP 500-mg dose under fasted conditions was approximately 90–100% compared with a single dose of OAA 1000 mg. Furthermore, the AAFP 500-mg dose was bioequivalent to OAA based on the 90% CI of the geometric mean ratio of AUC and C max parameters. Pooled pharmacokinetic values for OAA 1000 mg [AUC0–∞ 503 ng·h/mL (SD 299); C max 94 ng/mL (SD 58.6)] [9] from previous clinical trials in healthy subjects were within the range of values for OAA 1000 mg and AAFP 500 mg in this study. Although a large proportion of the study sample was healthy African-American volunteers, racial differences in drug metabolism should not impact our findings because in this crossover study the same subjects received the different dose levels of AAFP and the 1000-mg dose of the reference drug (OAA). Therefore, we do not believe that the comparisons between OAA and the various AAFP doses were affected. In addition, AAFP was safe and well tolerated at all doses tested (100–625 mg), under the conditions of these studies.
Following a single dose of AAFP from 125 to 625 mg under fasted conditions, the median t max was approximately 1.5 h post-dose, compared with a median of 2.0 h for OAA 1000 mg, and was in agreement with previously reported t max for OAA 1000 mg [9]. The mean t ½ of abiraterone across the previous clinical studies was 15.2 h, with across-study variation from 12.7 to 19 h [9]. In healthy subjects under fasted conditions in Study 102, the t ½ was 14.1 h for AAFP 500 mg vs. 20.6 h for OAA 1000 mg, indicating that OAA 1000 mg takes more time to be completely eliminated from the body than AAFP 500 mg. At present, the mechanistic basis for the differences in t ½ between AAFP and OAA is unknown, because the same healthy subjects within each study crossed over to the different AAFP dose levels and the 1000-mg dose of OAA (the same subjects were given AAFP and OAA).
Absorption is a function of many factors including dissolution rate, site of absorption, gastrointestinal transit time, concentration gradient, presence of food, and dose [10]. Reduced particle size leads to an enhanced surface area, and this in turn leads to an enhanced rate of dissolution and absorption [11, 12]. The absorption of OAA is reported to be nearly saturated at a dose of 1000 mg [7, 8]. In contrast, the results with AAFP in the current study show continued increased absorption above a single AAFP 500-mg dose; i.e., at AAFP 625 mg. Namely, a single dose of 625 mg of AAFP delivered a higher C max and AUC than 1000 mg of OAA. Higher levels of absorption with AAFP have the potential for clinical benefit in patients with poor absorption or low or no response to OAA. This warrants further exploration.
SoluMatrix Fine Particle Technology™ produces drug particles 200–800 nm in diameter, which increases the drug particle surface area relative to its mass [13]. The manufacturing process, which does not alter the active therapeutic ingredient, was previously shown to promote rapid absorption of new formulations of diclofenac and meloxicam [11, 12]. The present study in healthy subjects indicates that the use of SoluMatrix Fine Particle Technology™ enhances the bioavailability of abiraterone acetate. An ongoing study (ClinicalTrials.gov Identifier: NCT02737332) will assess the pharmacokinetics and pharmacodynamics of AAFP 500 mg vs. OAA 1000 mg in patients with metastatic castration-resistant prostate cancer.
The limitations of the study include the single dosing of AAFP and OAA, and the use of healthy volunteers for the analyses. In addition, AAFP and OAA were tested under fasted conditions. A subsequent study will analyze the bioavailability of AAFP under fed vs. fasted conditions.
Conclusion
A single AAFP 500-mg dose was bioequivalent to OAA 1000 mg in healthy subjects under fasting conditions. Thus, the proprietary AAFP formulation using SoluMatrix Fine Particle Technology™ allows for the same systemic exposure to be achieved with 50% less drug relative to the recommended dose of OAA 1000 mg.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Yuxin Zhang (Xtiers Consulting, Inc., Potomac, MD, USA) for conducting the statistical analyses and Sharon Reinhard for help with clinical operations. The authors also thank the study participants and the site staff who made this study possible. All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Editorial support (assembling tables and figures, collating author comments, copyediting, fact checking, and referencing) and graphic services were provided by Gautam Bijur, Ph.D., and Nancy Price, Ph.D., of AOI Communications, L.P. and were funded by Churchill Pharmaceuticals LLC.
Compliance with Ethical Standards
Funding
This study was funded by Churchill Pharmaceuticals LLC.
Conflict of interest
Bill Bosch and Paul Nemeth are employees of Churchill Pharmaceuticals LLC. Azra Hussaini and Ronald Goldwater are employees of PAREXEL Early Phase Clinical Unit, which received funding from Churchill Pharmaceuticals LLC to conduct the clinical study.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The Dose Finding Study 101 and Bioavailability Study 102 were conducted in compliance with the International Conference on Harmonisation Good Clinical Practice guidelines. The study was approved by the Aspire Institutional Review Board (Santee, CA, USA).
Consent to participate
All subjects provided written informed consent before study procedures were initiated.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-017-0536-2) contains supplementary material, which is available to authorized users.
References
- 1.International Agency for Research on Cancer. GLOBOCAN cancer fact sheets: prostate cancer. 2016. http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed 23 Sept 2016.
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015;95:80–87. doi: 10.1016/j.steroids.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nussbaum N, George DJ, Abernethy AP, et al. Patient experience in the treatment of metastatic castration-resistant prostate cancer: state of the science. Prostate Cancer Prostatic Dis. 2016;19(2):111–121. doi: 10.1038/pcan.2015.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reid AH, Attard G, Barrie E, de Bono JS. CYP17 inhibition as a hormonal strategy for prostate cancer. Nat Clin Pract Urol. 2008;5(11):610–620. doi: 10.1038/ncpuro1237. [DOI] [PubMed] [Google Scholar]
- 6.Attard G, Reid AH, Yap TA, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26(28):4563–4571. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 7.Janssen Biotech Inc. Zytiga® (abiraterone acetate) tablets. 2016. https://www.zytigahcp.com/shared/product/zytiga/zytiga-prescribing-information.pdf. Accessed 16 Sept 2016.
- 8.European Medicines Agency. Assessment report for Zytiga (abiraterone). 2011. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002321/WC500112860.pdf. Accessed 16 Sept 2016.
- 9.Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review(s): application number: 202379Orig1s000. Zytiga. 2010. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000ClinPharmR.pdf. Accessed 16 Sept 2016.
- 10.Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42(6):620–643. doi: 10.1177/00970002042006005. [DOI] [PubMed] [Google Scholar]
- 11.Desjardins PJ, Olugemo K, Solorio D, Young CL. Pharmacokinetic properties and tolerability of low-dose SoluMatrix diclofenac. Clin Ther. 2015;37(2):448–461. doi: 10.1016/j.clinthera.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 12.Hussaini A, Solorio D, Young C. Pharmacokinetic properties of low-dose SoluMatrix meloxicam in healthy adults. Clin Rheumatol. 2016;35(4):1099–1104. doi: 10.1007/s10067-015-3121-9. [DOI] [PubMed] [Google Scholar]
- 13.Altman R, Bosch B, Brune K, et al. Advances in NSAID development: evolution of diclofenac products using pharmaceutical technology. Drugs. 2015;75(8):859–877. doi: 10.1007/s40265-015-0392-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.