

Abstract
Cisplatin derivatives can form various types of DNA lesions (DNA‐Pt) and trigger pleiotropic DNA damage responses. Here, we report a strategy to visualize DNA‐Pt with high resolution, taking advantage of a novel azide‐containing derivative of cisplatin we named APPA, a cellular pre‐extraction protocol and the labeling of DNA‐Pt by means of click chemistry in cells. Our investigation revealed that pretreating cells with the histone deacetylase (HDAC) inhibitor SAHA led to detectable clusters of DNA‐Pt that colocalized with the ubiquitin ligase RAD18 and the replication protein PCNA. Consistent with activation of translesion synthesis (TLS) under these conditions, SAHA and cisplatin cotreatment promoted focal accumulation of the low‐fidelity polymerase Polη that also colocalized with PCNA. Remarkably, these cotreatments synergistically triggered mono‐ubiquitination of PCNA and apoptosis in a RAD18‐dependent manner. Our data provide evidence for a role of chromatin in regulating genome targeting with cisplatin derivatives and associated cellular responses.
Keywords: cancer, chromatin, cisplatin, click chemistry, histone deacetylase
Emerging evidence indicates that functional interactions between small molecules and genomic DNA are influenced by chromatin features.1, 2 Cisplatin derivatives represent some of the most commonly employed drugs in the clinical management of cancer.3 These drugs form covalent bonds with purine residues to produce toxic DNA‐Pt including mono‐adducts, inter‐ and intra‐strand crosslinks. Thus, cellular responses to cisplatin are pleiotropic and inherently complex. For example, diverse repair mechanisms including nucleotide excision repair (NER), base excision repair (BER) and DNA crosslink repair involving the Fanconi anemia pathway can process DNA‐Pt, and these lesions can also alter the structure of chromatin or be influenced by chromatin features.4 Alternatively, low‐fidelity DNA polymerases (e.g. Polη) can bypass DNA‐Pt through a mechanism known as translesion synthesis, enabling continued replication in the presence of DNA‐Pt, conferring resistance to platinum (Pt) drugs.5
To study DNA‐Pt in the context of chromatin, we sought to develop a surrogate probe that would allow for the chemical labeling of DNA‐Pt in cells. Visual detection of DNA‐Pt with high resolution at the single‐cell level can potentially provide the means to monitor proteins at sites of lesions and to identify small molecules with a propensity to modulate targeting with cisplatin derivatives in an unbiased manner. Prior expertise in characterizing mechanisms of action of small molecules prompted us to develop an azide‐containing derivative to label DNA‐Pt by means of click chemistry.1, 6 Previous efforts towards the synthesis of clickable cisplatin derivatives suitable for cell imaging led to the development of an iminoacridine‐Pt complex and a flexible alkyl‐azide‐containing derivative.7
With this in mind, we synthesized the cyclic azidoplatinum‐containing derivative we named 2‐aminomethylpyridine (dichloro) PtII azide (APPA, Figure 1 A and Supporting Information).8 The design of APPA was inspired from the structures of cisplatin and picoplatin (Figure 1 A), taking advantage of the aromatic methyl substituent to form a rigid five‐membered ring with Pt. Thus, APPA exhibits a structure that can form bulky DNA lesions, where the ring prevents free rotation of the pyridine core chelated to Pt. This structural distinction was considered important given that the chemical nature of ligands bound to Pt at sites of lesions in cells can potentially affect how these lesions are detected and dealt with.9 Like cisplatin and picoplatin, APPA exhibited anti‐proliferative properties against human osteosarcoma U2OS cells (Figure 1 B). Thus, we evaluated the reactivity of APPA towards DNA and our ability to label DNA‐Pt with strained or terminal alkyne‐containing fluorophores. A 26‐mer hairpin‐forming DNA oligonucleotide containing a single 1,2‐GG dinucleotide, prone to form an intra‐strand crosslink upon treatment with Pt drugs, was incubated with APPA, purified and then incubated with a strained alkyne‐containing Alexa 488 (Figure 1 C).10 The reaction products were then analyzed and characterized by mass spectrometry. We identified three ion peaks corresponding to the free unreacted hairpin along with the unlabeled and fluorescently labeled Pt adducts (Figure 1 D). Next, we performed similar experiments in cells using APPA and the control compound 2‐aminomethylpyridine (dichloro) PtII (APP, Figure 1 E), a structurally related active analogue of APPA devoid of azide functionality and therefore not amenable to labeling. Labeled genomic DNA‐Pt obtained from APPA‐treated cells displayed increased fluorescence compared to equal amounts of DNA collected from APP‐treated cells as monitored by dot blot (Figure 1 E). This data demonstrated that APPA can form DNA‐Pt with genomic DNA in living cells.
Figure 1.
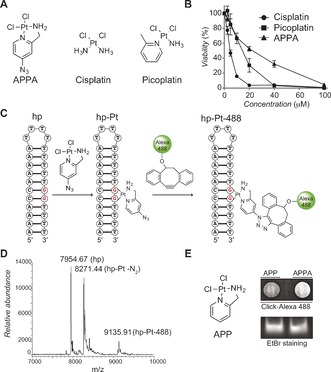
A) Molecular structures of APPA, cisplatin and picoplatin. B) Anti‐proliferative activity of cisplatin derivatives against U2OS cells. C) Schematic representation of a DNA hairpin (hp), a 1,2‐GG intra‐strand DNA crosslink (hp‐Pt) and chemical labeling of DNA‐Pt (hp‐Pt‐488). For clarity, a single regioisomer is shown for hp‐Pt‐488. D) Mass spectrometry detection of a free DNA hairpin, the corresponding DNA‐Pt and its labeled counterpart. hp‐Pt was observed as the molecular ion peak with loss of nitrogen. E) Molecular structure of APP and detection of genomic DNA platination by dot blot. DNA samples were purified from U2OS cells treated with APP (250 μm) or APPA (250 μm) for 3 h. EtBr (Ethidium bromide) stained gel of input DNA used as loading control.
With this methodology in hand, we investigated the localization of DNA‐Pt in cells. To this end, cells were treated with APPA and fixed with formaldehyde prior to labeling DNA‐Pt using copper catalysis. Consistent with previous observations,7, 11 labeled DNA‐Pt exhibited a diffuse cytoplasmic and nuclear staining (Figure 2 A), which likely reflected the targeting of nuclear DNA as well as proteins and RNA. As a means to selectively detect DNA‐Pt, we implemented a pre‐extraction protocol to remove the staining linked to soluble proteins and RNA targets of Pt drugs (Figure 2 B). This protocol allowed for the detection of labeled DNA‐Pt in the nucleus with some sub‐nuclear regions displaying increased fluorescence intensity (Figure 2 C). For instance, labeled DNA‐Pt colocalized with fibrillarin (see Supporting Information), in agreement with the idea that Pt drugs target rRNA in the nucleolus.7b Collectively, these data validated APPA as a functional clickable probe suitable for studying genome targeting with cisplatin derivatives in cells.
Figure 2.
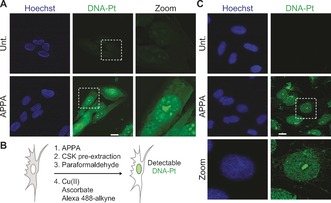
A) Detection of labeled DNA‐Pt in U2OS cells. B) Schematic representation of a strategy for enhancing the detection of DNA‐Pt in cells. C) Detection of labeled DNA‐Pt in U2OS cells subjected to pre‐extraction. Cells were treated with APPA (250 μm for 3 h). Zoomed images are 3×. Scale bar, 20 μm. Unt., untreated.
Next, we searched for small molecule modulators of genomic DNA targeting with cisplatin derivatives, using APPA staining as a readout. We screened a defined set of small molecules that either operate at the level of chromatin or that are used in cancer treatment in conjunction with Pt drugs (Figure 3 A and Supporting Information). U2OS cells were cotreated independently with each small molecule and APPA, then subjected to chemical labeling and labeled DNA‐Pt were analyzed by fluorescence microscopy. While most small molecules had no discernable effect on the localization of DNA‐Pt, pre‐treatment with the clinically approved DNA methyl transferase inhibitor azacytidine (5‐Aza)12 and the histone deacetylase (HDAC) inhibitor SAHA13 led to the occurrence of foci of DNA‐Pt, indicating the presence of targeted clusters of purine residues at these sites that can include mono‐adducts, 1,2‐GG and 1,3‐GTG intra‐strand as well as inter‐strand lesions (Figure 3 B and C). Western blotting analysis confirmed that SAHA induced hyperacetylation of histone H4 (see Supporting Information), an established marker of open chromatin.14 Furthermore, the structurally distinct Class I HDAC inhibitor MS‐27515 also led to increased number of DNA‐Pt foci, implicating further the role of HDACs and chromatin in genome targeting with APPA (see Supporting Information). Importantly, the intensity of DNA‐Pt foci was reduced when cells were cotreated with a two‐fold excess of cisplatin acting as a competitor, indicating that both APPA and cisplatin target similar genomic loci (Figure 3 D). These data suggested that chromatin relaxation resulting from SAHA treatment revealed de novo DNA targets of APPA. Interestingly, DNA‐Pt occurring in SAHA‐treated cells did not predominantly colocalize with the centromere and telomere markers CENP‐A and TRF1, respectively, excluding these loci‐containing repetitive sequences rich in 1,2‐purine residues as primary targets of APPA under these conditions (see Supporting Information).
Figure 3.
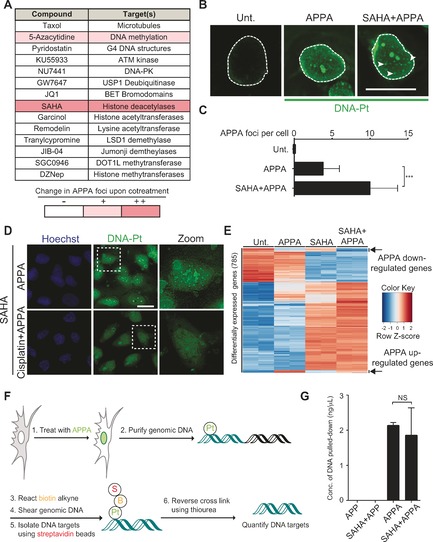
A) Small molecules screened for effects on genome targeting with APPA. B) Fluorescence microscopy detection of labeled DNA‐Pt. Arrows indicate DNA‐Pt foci. Scale bars, 20 μm. C) Quantification of B. >100 cells scored per condition; n=3; Foci were counted by means of visual inspection. D) Cisplatin competes with APPA for genome targeting. Zoomed images are 3×. Scale bars, 20 μm. E) Heatmap of differentially expressed genes in SAHA and APPA cotreated cells. Genes were selected based on differential expression in SAHA and APPA cotreatment compared to untreated. Data from two independent experiments per condition are shown. Gene expression was monitored by RNA‐Seq as described in Supporting Information. F) Scheme of DNA‐Pt pull‐down methodology. G) Quantification of DNA isolated by pull‐down from samples treated as indicated; n=3. All experiments performed in U2OS cells. Cells were treated with APPA (250 μm for 3 h), APP (250 μm for 3 h), SAHA (2.5 μm for 5 h) and cisplatin (500 μm for 5 h). Error bars represent mean ±SD; ***P<0.001, Student's t‐test; NS, not significant.
Given that DNA‐Pt can alter transcription,16 we performed RNA‐Seq analyses to evaluate whether SAHA treatment affected the transcriptional response induced by APPA. Our analysis identified a subset of genes that were up‐ or down‐regulated by APPA compared to untreated cells (Figure 3 E and Supporting Information). Interestingly, the transcriptional changes measured in cells cotreated with SAHA and APPA could be attributed to that observed either in SAHA or APPA independent treatments. This supported the idea that the DNA‐Pt clusters detected in cotreated cells did not trigger additional transcriptional effects that were unique to this cotreatment. Furthermore, qPCR experiments validated that cisplatin induced similar changes in gene expression compared to APPA, providing further evidence that APPA and cisplatin shared genomic targets (see the Supporting Information).
Then, we developed a protocol to isolate DNA targets of APPA from cells (Figure 3 F). Cells were treated with APPA ± SAHA prior to being subjected to affinity pull‐down as previously performed for other classes of molecules.2, 17 The amount of DNA pulled down from cells was similar in both experimental conditions (Figure 3 G). This result was in line with the idea that SAHA did not solely act by increasing the number of DNA targets per se, but rather induced a redistribution of DNA‐Pt throughout the genome, potentiating DNA targeting at particular sites. This was consistent with the idea that chromatin relaxation induced by SAHA altered accessibility of the genome to APPA, providing additional insights into how HDAC inhibitors can sensitize cells to genotoxic drugs.18
The occurrence of DNA‐Pt foci in SAHA‐treated cells prompted us to determine whether clusters of lesions could act as physical roadblocks, potentially affecting DNA‐templated processes. Strikingly, DNA‐Pt colocalized with RAD18, an E3 ubiquitin ligase that mediates mono‐ubiquitination of the proliferating cell nuclear antigen PCNA in cells cotreated with SAHA and APPA (Figure 4 A). Moreover, clusters of DNA‐Pt also colocalized with PCNA (see Supporting Information). Cotreatment with SAHA and APPA led to mono‐ubiquitination of PCNA (Figure 4 B), a mark that can promote the recruitment of low‐fidelity polymerases involved in TLS,19 and cells cotreated with SAHA and cisplatin or APPA exhibited increased number of RAD18 foci (see Supporting Information). Furthermore, treatment with SAHA and cisplatin led to the focal accumulation of the TLS polymerase Polη20 that colocalized with PCNA (Figure 4 C and Supporting Information). Altogether, these data were consistent with the notion that SAHA and cisplatin derivatives synergistically activated TLS. It is noteworthy that levels of phosphorylated replication protein A (p‐RPA) and CHK1 (p‐CHK1) were similar in APPA‐treated and cotreated cells indicating the absence of additional replication stress (see Supporting Information). This was in agreement with the notion that HDAC inhibitors promoted a redistribution of DNA‐Pt in contrast to a global increase in number of lesions. Remarkably, cotreatment with SAHA and APPA triggered apoptosis signaling as defined by the cleavage of PARP and the activation of caspase 3, in several cancer cell lines including colon carcinoma HCT‐116, osteosarcoma U2OS and ovarian carcinoma A2780 cells (Figure 4 D and Supporting Information). To evaluate whether RAD18 was directly involved in apoptotic signaling under these conditions, we performed similar experiments with matched HCT‐116 RAD18 knockout (KO) cells. Western blotting indicated that cells devoid of RAD18 did not display PCNA mono‐ubiquitination in response to SAHA and APPA cotreatment (Figure 4 D). Additionally, markers of apoptosis were not detected in HCT‐116 RAD18 KO cells even though DNA‐Pt foci formed similarly in WT and RAD18 KO cells cotreated with SAHA and APPA (Figure 4 D and Supporting Information). These results, along with the RAD18‐dependent PCNA mono‐ubiquitination implicated RAD18 in promoting apoptosis signaling in response to SAHA and APPA cotreatment. Comparable results were observed in SAHA and cisplatin cotreated WT and RAD18 KO cells, demonstrating that this response was a common feature of these cisplatin derivatives (Figure 4 E). Although the induction of apoptosis signaling by RAD18 could be potentially counterintuitive owing to the well‐established role of this factor in DNA repair processes and TLS,5 HDAC inhibitors have been shown to re‐sensitize cancer cells refractory to cisplatin.18 Therefore, our results suggested that the higher level of apoptosis signaling observed in RAD18 expressing cells in response to SAHA and cisplatin derivatives is due to the inability of polymerases to efficiently bypass DNA‐Pt clusters. For instance, it is possible that chromatin relaxation mediated by SAHA favored the formation of 1,3‐GTG intra‐strand lesions, which have previously been shown to block TLS polymerases.21 It is also conceivable that HDAC inhibition promoted lesion bypass at the expense of DNA repair,22 thereby leading to detectable clusters of DNA‐Pt prone to trigger apoptosis through other mechanisms.
Figure 4.
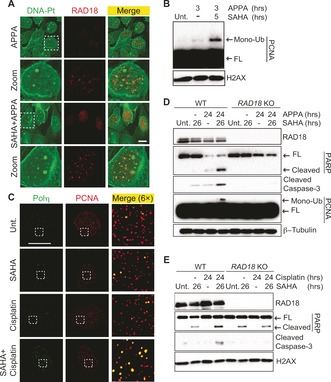
A) Colocalization of labeled DNA‐Pt with RAD18 in U2OS cells. Cells were treated with APPA (250 μm for 3 h) and SAHA (2.5 μm for 5 h). Zoomed images are 3×. Scale bar, 20 μm. B) Western blot analysis of PCNA mono‐ubiquitination in U2OS cells. Cells were treated with APPA (250 μm) and SAHA (2.5 μm) as indicated. C) Focal accumulation of Polη colocalizing with PCNA in U2OS cells. Cells were treated with cisplatin (10 μm for 3 h) and SAHA (2.5 μm for 5 h). Zoomed images are 6×. Scale bar, 20 μm. D) and E) Western blot analysis of apoptotic markers in WT and RAD18 KO HCT‐116 cells. Cells were treated with APPA (250 μm), SAHA (2.5 μm) and cisplatin (10 μm) as indicated. FL, full length.
Engagement of the replication machinery with DNA‐Pt can promote DNA breaks and cell death. However, cells can employ DNA damage tolerance pathways involving mono‐ubiquitinated PCNA and specialized low‐fidelity polymerases to bypass DNA‐Pt.5, 23 These processes play a critical role in resistance to Pt drugs.24 To overcome these mechanisms, cisplatin derivatives containing bulkier ligands, or combination therapies with other drugs have been identified.25 Here, we have developed a versatile strategy to visualize DNA‐Pt in cells with high resolution. This methodology has allowed the unbiased identification of small molecule modulators of genome targeting with cisplatin derivatives. In particular, we have discovered that treating cells with the HDAC inhibitor SAHA and APPA resulted in detectable clusters of DNA‐Pt and activation of a DNA damage response at these sites. While we cannot rule out a putative role of template switching and homologous recombination that could also be mediated by RAD18, the increase of Polη foci in cells cotreated with SAHA and cisplatin is consistent with activation of TLS. Thus, the response observed in these conditions comes in agreement with reports showing that clustered DNA lesions can impair DNA damage response pathways.26 This study has uncovered unanticipated insights into how chromatin alterations can affect the targeting of genomic DNA and sensitization of cancer cells to cisplatin derivatives, establishing a robust experimental platform for basic and translational research relying on chromatin‐targeting small molecules.27
Dedicated to Professor Stuart L. Schreiber
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the PICT‐IBiSA@Pasteur Imaging Facility of the Institut Curie, member of the France‐BioImaging national research infrastructure. Research in the R.R. laboratory is supported by the European Research Council and the Emergence Ville de Paris Program. Results incorporated in this article have received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No [647973]). K.M.M. laboratory is supported by start‐up funds from UT Austin and Cancer Prevention Research Institute of Texas (CPRIT, grant no. R1116) and the NIH National Cancer Institute grants RO1 CA198279 and CA201268. K.M.M. is a CPRIT scholar. The B.X. laboratory is funded by UT Austin start‐up funds and the Welch Foundation (grant no. F1859). This study also made use of the Science Park NGS Facility, supported by the CPRIT Core Facility Support Grant RP120348.
E. Zacharioudakis, P. Agarwal, A. Bartoli, N. Abell, L. Kunalingam, V. Bergoglio, B. Xhemalce, K. M. Miller, R. Rodriguez, Angew. Chem. Int. Ed. 2017, 56, 6483.
Contributor Information
Dr. Kyle M. Miller, Email: kyle.miller@austin.utexas.edu
Dr. Raphaël Rodriguez, Email: raphael.rodriguez@curie.fr.
References
- 1. Rodriguez R., Miller K. M., Forment J. V., Bradshaw C. R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S. P., Nat. Chem. Biol. 2012, 8, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rodriguez R., Miller K. M., Nat. Rev. Genet. 2014, 15, 783–796. [DOI] [PubMed] [Google Scholar]
- 3. Wang D., Lippard S. J., Nat. Rev. Drug Discovery 2005, 4, 307–320. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Danford A. J., Wang D., Wang Q., Tullius T. D., Lippard S. J., Proc. Natl. Acad. Sci. USA 2005, 102, 12311–12316; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Wu B., Droge P., Davey C. A., Nat. Chem. Biol. 2008, 4, 110–112. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Damsma G. E., Alt A., Brueckner F., Carell T., Cramer P., Nat. Struct. Mol. Biol. 2007, 14, 1127–1133; [DOI] [PubMed] [Google Scholar]
- 5b. Alt A., Lammens K., Chiocchini C., Lammens A., Pieck J. C., Kuch D., Hopfner K. P., Carell T., Science 2007, 318, 967–970; [DOI] [PubMed] [Google Scholar]
- 5c. Sale J. E., J. Cell Sci. 2012, 125, 1633–1643; [DOI] [PubMed] [Google Scholar]
- 5d. Sale J. E., Cold Spring Harbor Perspect. Biol. 2013, 5, a012708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Mailand N., Gibbs-Seymour I., Bekker-Jensen S., Nat. Rev. Mol. Cell Biol. 2013, 14, 269–282. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Larrieu D., Britton S., Demir M., Rodriguez R., Jackson S. P., Science 2014, 344, 527–532; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Abell N., Mercado M., Cañeque T., Rodriguez R., Xhemalce B., J. Am. Chem. Soc. 2017, 139, 1400–1403. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Ding S., Qiao X., Suryadi J., Marrs G. S., Kucera G. L., Bierbach U., Angew. Chem. Int. Ed. 2013, 52, 3350–3354; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3434–3438; [Google Scholar]
- 7b. Wirth R., White J. D., Moghaddam A. D., Ginzburg A. L., Zakharov L. N., Haley M. M., DeRose V. J., J. Am. Chem. Soc. 2015, 137, 15169–15175. [DOI] [PubMed] [Google Scholar]
- 8.A patent application (number PCT/EP2016/081166) on clickable cisplatin derivatives and protocols for the visual detection of DNA-Pt in cells and use as diagnostic tools or to identify synergistic treatments has been filed.
- 9. Vaisman A., Lim S. E., Patrick S. M., Copeland W. C., Hinkle D. C., Turchi J. J., Chaney S. G., Biochemistry 1999, 38, 11026–11039. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Baskin J. M., Prescher J. A., Laughlin S. T., Agard N. J., Chang P. V., Miller I. A., Lo A., Codelli J. A., Bertozzi C. R., Proc. Natl. Acad. Sci. USA 2007, 104, 16793–16797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Jewett J. C., Bertozzi C. R., Chem. Soc. Rev. 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Liang X. J., Shen D. W., Chen K. G., Wincovitch S. M., Garfield S. H., Gottesman M. M., J. Cell. Physiol. 2005, 202, 635–641; [DOI] [PubMed] [Google Scholar]
- 11b. Qiao X., Ding S., Liu F., Kucera G. L., Bierbach U., J. Biol. Inorg. Chem. 2014, 19, 415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christman J. K., Oncogene 2002, 21, 5483–5495. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Taunton J., Hassig C. A., Schreiber S. L., Science 1996, 272, 408–411; [DOI] [PubMed] [Google Scholar]
- 13b. Marks P. A., Breslow R., Nat. Biotechnol. 2007, 25, 84–90. [DOI] [PubMed] [Google Scholar]
- 14. Shahbazian M. D., Grunstein M., Annu. Rev. Biochem. 2007, 76, 75–100. [DOI] [PubMed] [Google Scholar]
- 15. Rosato R. R., Almenara J. A., Grant S., Cancer Res. 2003, 63, 3637–3645. [PubMed] [Google Scholar]
- 16. Todd R. C., Lippard S. J., Metallomics 2009, 1, 280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Müller S., Kumari S., Rodriguez R., Balasubramanian S., Nat. Chem. 2010, 2, 1095–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Kim M. S., Blake M., Baek J. H., Kohlhagen G., Pommier Y., Carrier F., Cancer Res. 2003, 63, 7291–7300; [PubMed] [Google Scholar]
- 18b. Sharma S. V., Lee D. Y., Li B., Quinlan M. P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M. A., Wong K. K., Brandstetter K., Wittner B., Ramaswamy S., Classon M., Settleman J., Cell 2010, 141, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hendel A., Krijger P. H., Diamant N., Goren Z., Langerak P., Kim J., Reissner T., Lee K. Y., Geacintov N. E., Carell T., Myung K., Tateishi S., D'Andrea A., Jacobs H., Livneh Z., PLoS Genet. 2011, 7, e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hicks J. K., Chute C. L., Paulsen M. T., Ragland R. L., Howlett N. G., Gueranger Q., Glover T. W., Canman C. E., Mol. Cell. Biol. 2010, 30, 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Schneider S., Reissner T., Ziv O., Livneh Z., Carell T., ChemBioChem 2010, 11, 1521–1524; [DOI] [PubMed] [Google Scholar]
- 21b. Reissner T., Schneider S., Schorr S., Carell T., Angew. Chem. Int. Ed. 2010, 49, 3077–3080; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3142–3145. [Google Scholar]
- 22. Koch S. C., Kuper J., Gasteiger K. L., Simon N., Strasser R., Eisen D., Geiger S., Schneider S., Kisker C., Carell T., Proc. Natl. Acad. Sci. USA 2015, 112, 8272–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghosal G., Chen J., Trans. Cancer Res. 2013, 2, 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Vaisman A., Masutani C., Hanaoka F., Chaney S. G., Biochemistry 2000, 39, 4575–4580; [DOI] [PubMed] [Google Scholar]
- 24b. Bassett E., King N. M., Bryant M. F., Hector S., Pendyala L., Chaney S. G., Cordeiro-Stone M., Cancer Res. 2004, 64, 6469–6475; [DOI] [PubMed] [Google Scholar]
- 24c. Doles J., Oliver T. G., Cameron E. R., Hsu G., Jacks T., Walker G. C., Hemann M. T., Proc. Natl. Acad. Sci. USA 2010, 107, 20786–20791; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24d. Xie K., Doles J., Hemann M. T., Walker G. C., Proc. Natl. Acad. Sci. USA 2010, 107, 20792–20797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24e. Zhao Y., Biertumpfel C., Gregory M. T., Hua Y. J., Hanaoka F., Yang W., Proc. Natl. Acad. Sci. USA 2012, 109, 7269–7274; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24f. Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G., Oncogene 2012, 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- 25. Dasari S., Tchounwou P. B., Eur. J. Pharmacol. 2014, 740, 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Waters L. S., Minesinger B. K., Wiltrout M. E., D'Souza S., Woodruff R. V., Walker G. C., Microbiol. Mol. Biol. Rev. 2009, 73, 134–154; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Yuan B. F., Jiang Y., Wang Y. S., Wang Y. S., Chem. Res. Toxicol. 2010, 23, 11–19; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26c. Sage E., Harrison L., Mutat. Res. 2011, 711, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Baylin S. B., Jones P. A., Nat. Rev. Cancer 2011, 11, 726–734; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Helin K., Dhanak D., Nature 2013, 502, 480–488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary