

Abstract
Patients admitted for acute heart failure (AHF) experience high rates of in‐hospital and post‐discharge morbidity and mortality despite current therapies. Serelaxin is recombinant human relaxin‐2, a hormone with vasodilatory and end‐organ protective effects believed to play a central role in the cardiovascular and renal adaptations of human pregnancy. In the phase 3 RELAX‐AHF trial, serelaxin met its primary endpoint of improving dyspnoea through day 5 in patients admitted for AHF. Compared to placebo, serelaxin also reduced worsening heart failure (WHF) by 47% through day 5 and both all‐cause and cardiovascular mortality by 37% through day 180. RELAX‐AHF‐2 ( ClinicalTrials.gov NCT01870778) is designed to confirm serelaxin's effect on these clinical outcomes. RELAX‐AHF‐2 is a multicentre, randomized, double‐blind, placebo‐controlled, event‐driven, phase 3 trial enrolling ∼6800 patients hospitalized for AHF with dyspnoea, congestion on chest radiograph, increased natriuretic peptide levels, mild‐to‐moderate renal insufficiency, and systolic blood pressure ≥125 mmHg. Patients are randomized within 16 h of presentation to 48 h intravenous infusions of serelaxin (30 µg/kg/day) or placebo, both in addition to standard of care treatments. The primary objectives are to demonstrate that serelaxin is superior to placebo in reducing: (i) 180 day cardiovascular death, and (ii) occurrence of WHF through day 5. Key secondary endpoints include 180 day all‐cause mortality, composite of 180 day combined cardiovascular mortality or heart failure/renal failure rehospitalization, and in‐hospital length of stay during index AHF. The results from RELAX‐AHF‐2 will provide data on the potential beneficial effect of serelaxin on cardiovascular mortality and WHF in selected patients with AHF.
Keywords: Acute heart failure, Serelaxin, Worsening heart failure, Mortality, Phase 3 trial
Introduction
Acute heart failure (AHF) is the most common cause of hospitalization in patients 65 years and older.1, 2 In part due to the ageing of the population and more effective treatment of chronic heart failure (HF), its prevalence is expected to increase by 25% over the next 20 years3 and the problem has expanded worldwide.4, 5 Patients hospitalized for HF have a 40–50% rate of HF exacerbation, of which 10–15% is in‐hospital worsening heart failure (WHF)6, 7, 8, 9, 10 and 30–40% is rehospitalization, within the first 6 months after discharge as well as a 10–15% mortality rate.11 Compared with ambulatory patients with stable chronic HF, patients hospitalized for AHF have a dramatic increase in their risk of death, similar or worse than that after a hospitalization for acute myocardial infarction or stroke.12 Although this increased risk falls rapidly after discharge, it remains 5‐ to 10‐fold higher than in ambulatory patients even months after the initial episode.13, 14, 15 Potential mechanisms of increased mortality and WHF are outlined in Figure 1. No evidence of efficacy in reducing morbidity and mortality for any new treatment for patients hospitalized for HF has been found; hence no change in either treatment or prognosis has occurred in recent decades.
Figure 1.
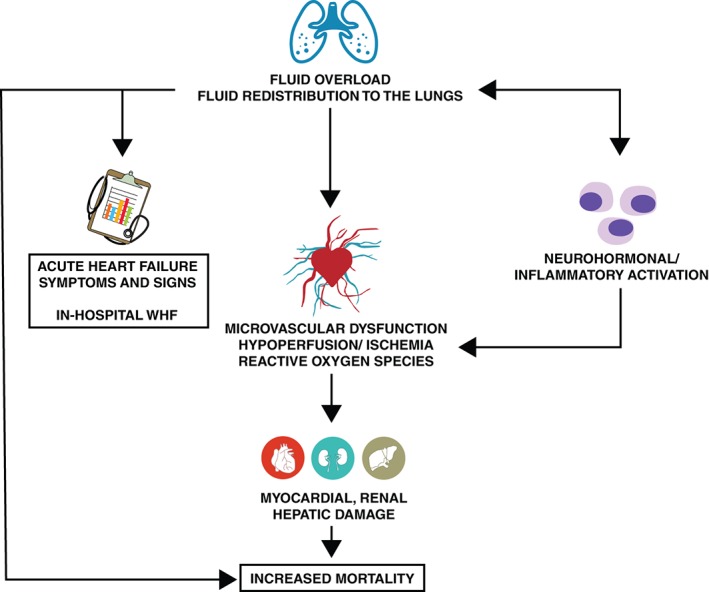
Mechanisms of increased mortality and worsening heart failure (WHF) in acute heart failure.
Serelaxin is a recombinant protein identical in amino acid sequence and structure to the naturally occurring human peptide hormone relaxin‐2, which is associated with many of the maternal haemodynamic and renovascular changes that occur in response to pregnancy, such as systemic and renal vasodilation and increases in global arterial compliance.16, 17 Serelaxin's activity is initiated by binding to its cognate receptor, serelaxin family peptide receptor 1 (RXFP1), which is present in the systemic and renal vasculature as well as in the human heart. Nitric oxide, endothelial endothelin type B receptor, vascular endothelial growth factor, and cAMP act as mediators for the vasodilatory as well as anti‐fibrotic and anti‐inflammatory effects of serelaxin.16 With these pleiotropic effects (Figure 2), serelaxin may benefit AHF patients18 not only through its favourable haemodynamic effects, but also via its protective effects on the heart, kidney, and other organs, leading to potential mortality benefits19 as suggested by the data from RELAX‐AHF.20
Figure 2.

Potential mechanisms of beneficial effect of serelaxin in patients with acute heart failure. ET, endothelin; MMP, matrix metalloproteinase; NO, nitric oxide; NOS, nitric oxide synthetase; TGF, transforming growth factor; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor.
The efficacy and safety of serelaxin as a continuous intravenous (i.v.) infusion for up to 48 h in AHF patients have been evaluated in two multicentre, randomized, double‐blind, placebo‐controlled trials: (i) the dose‐finding phase 2 study Pre‐RELAX‐AHF,21 and (ii) the phase 2 registration study RELAX‐AHF.19 In both of these trials, patients were admitted for acute heart failure with persistent dyspnoea despite i.v. diuretics with normal‐to‐elevated systolic blood pressure (SBP >125 mmHg), congestion on chest radiograph, elevated natriuretic peptides, and mild‐to‐moderate renal insufficiency, and enrolled within 16 h of presentation. The 234 patients enrolled in Pre‐RELAX‐AHF were randomized to four doses of serelaxin ranging from 10 to 250 µg/kg/day or matching placebo. While each dose suggested some clinical benefit, patients assigned to serelaxin 30 µg/kg/day had the greatest overall improvement in signs and symptoms of HF and trends toward improved long‐term outcomes with minimal adverse effects. In the RELAX‐AHF study, 1161 patients admitted for AHF were randomized to receive either serelaxin (n = 581) or matching placebo (n = 580), both in addition to standard‐of‐care AHF treatment. The 48 h i.v. infusion of serelaxin at the dose of 30 µg/kg/day produced dyspnoea relief as demonstrated by a 19.4% treatment improvement compared to placebo measured over 5 days by visual analogue scale, representing one of the two primary efficacy endpoints in the study. However, there was no significant effect on the other primary endpoint of dyspnoea relief through 24 h measured by a Likert scale. Serelaxin treatment significantly reduced the incidence of WHF through day 5 which was the main component of the improvement in dyspnoea and contributed to the reduced length of index hospital stay. These clinical effects were associated with significant improvements in biomarkers suggestive of less end‐organ damage and dysfunction in serelaxin‐treated patients.20 Analysis of all‐cause mortality through day 180 revealed fewer deaths with serelaxin compared to placebo with a total of 42 [Kaplan–Meier (K–M) estimate, 7.3%] deaths in the serelaxin group compared to 65 (K–M estimate, 11.3%) in the placebo group [hazard ratio (HR) 0.63; 95% confidence intervals (CI) 0.43–0.93; P = 0.020]. The mortality difference was largely driven by cardiovascular (CV) death through day 180 (K–M estimates 9.6% and 6.1% in placebo and serelaxin groups, respectively, HR 0.63; 95% CI 0.41–0.96; P = 0.028). Including all patients treated with serelaxin 30 µg/kg/day from the two trials, CV mortality was reduced by 44% (Figure 3; HR 0.56; 95% CI 0.37–0.86; P = 0.007). These findings support a sustained benefit of serelaxin beyond the initial 48 h of administration. Better relief of congestion and protection from damage to the myocardium, kidneys, and liver seem the most likely mechanisms for these long‐term beneficial effects.19, 20 These results were also consistent with a reduction in WHF episodes in serelaxin‐treated patients. Worsening HF is associated with poorer outcomes independently of AHF severity.20
Figure 3.
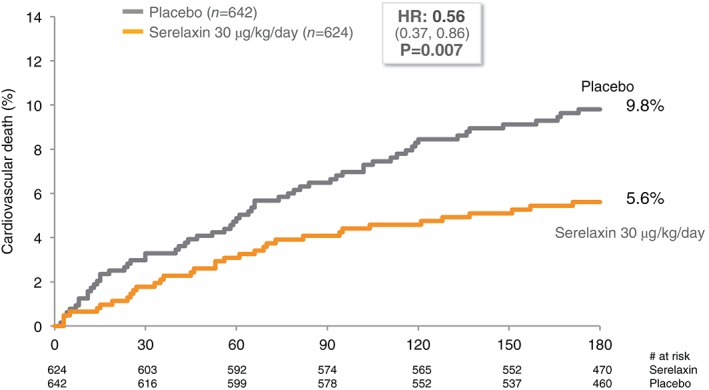
Cardiovascular mortality of patients with acute heart failure in the serelaxin programme treated with serelaxin 30 µg/kg/day compared to placebo.
Given that mortality was not a primary efficacy endpoint of the RELAX‐AHF trial, a global, phase 3 trial designed to evaluate these findings has been initiated. The goal of the second RELAXin in Acute Heart Failure (RELAX‐AHF‐2) trial is to confirm the beneficial effect of serelaxin on 180 day CV death and WHF through day 5, as well as other relevant clinical outcomes.
Study design and methods
The RELAX‐AHF‐2 trial (Figure 4) is a multicentre, randomized, double‐blind, placebo‐controlled trial to evaluate the efficacy, safety and tolerability of i.v. infusion of 30 µg/kg/day serelaxin for 48 h, when added to standard therapy. The trial is event‐driven: ∼6800 AHF patients in 545 centres in 34 countries will be enrolled to obtain at least 547 confirmed CV deaths. Primary efficacy will be determined based on the relative risk reductions in CV death through day 180 and in WHF through day 5. Secondary efficacy endpoints include 180 day all‐cause mortality, length of hospital stay, and CV death or HF/renal failure rehospitalization through day 180.
Figure 4.
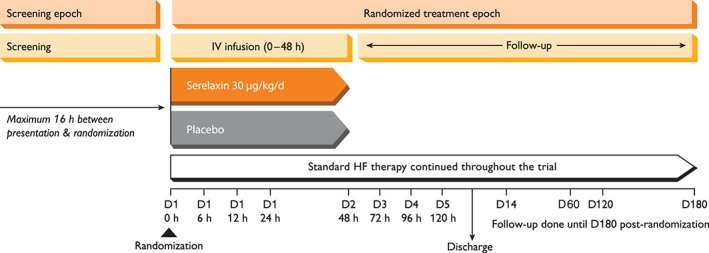
Schematic diagram of study design for RELAX‐AHF‐2 trial. HF, heart failure; IV, intravenous.
The RELAX‐AHF‐2 Executive Committee (Appendix) designed the trial and wrote the study protocol in collaboration with the clinical team from Novartis. The protocol must be approved by the Ethics Review Committee/Institutional Review Board affiliated with each centre and is being conducted in accordance with Good Clinical Practice and the 2002 Declaration of Helsinki. All participants provide written informed consent. The trial is registered on ClinicalTrials.gov, NCT01870778). The protocol was amended five times (see Supplementary material online, Appendix S1).
Study population
The study population includes male and female patients (≥18 years old) admitted to the hospital for AHF with dyspnoea, congestion on chest radiograph, elevated BNP or NT‐proBNP, normal‐to‐elevated SBP ≥125 mmHg, and mild‐to‐moderate renal impairment [estimated glomerular filtration rate ≥25 and ≤75 mL/min/1.73 m2, calculated using the standardized Modification of Diet in Renal Disease (sMDRD) equation] who are anticipated to require i.v. therapy for at least 48 h (Table 1). Patients must remain symptomatic after initial treatment with at least 40 mg i.v. loop diuretic. Patients receiving i.v. nitrates at a dose ≤0.1 mg/kg/h with a blood pressure >150 mmHg are eligible. There is no ejection fraction criterion, such that both HF patients with preserved (HFpEF) or reduced ejection fraction (HFrEF) are enrolled. Patients are randomized within 16 h of the earlier of first administration of i.v. loop diuretic or hospital presentation for the AHF episode in a 1:1 ratio into the two study arms.
Table 1.
Key inclusion and exclusion criteria in RELAX‐AHF‐2
| Key inclusion criteria | Key exclusion criteria |
|---|---|
|
Any other medical condition(s) that may put the patient at risk or influence study results in the investigator's opinion, or that the investigator deems unsuitable for the study, including drug or alcohol abuse or psychiatric, behavioural or cognitive disorders, sufficient to interfere with the patient's ability to understand and comply with the protocol instructions or follow‐up procedures. |
Assessed based on local laboratory.
Presentation starts as the earliest of (i) time of presentation at either the emergency room/department, intensive/cardiac care unit or ward (excludes emergency medical service or other pre‐hospital care); or (ii) time of first i.v. loop diuretic prior to arrival at the hospital (this includes outpatient clinic, ambulance, or hospital including emergency department) for the current AHF episode.
AHF, acute heart failure; COPD, chronic obstructive pulmonary disease; CRT, cardiac resynchronization therapy; eGFR, estimated glomerular filtration rate; GI, gastrointestinal; HF, heart failure; ICD, implantable cardioverter defibrillator; i.v., intravenous; sMDRD, standardized Modification of Diet in Renal Disease.
Study treatment
Serelaxin or matching placebo is administered as an i.v. infusion beginning no more than 4 h after randomization. Infusion continues for up to 48 h according to a weight range‐adjusted dosing regimen at the nominal dose of 30 µg/kg/day. Similar to the protocol adopted in the previous Pre‐RELAX‐AHF and RELAX‐AHF trials, blood pressure is monitored frequently during study drug administration. If at any time during the study drug administration the patient's SBP decreases by >40 mmHg from baseline but the absolute SBP is ≥100 mmHg in two consecutive measurements 15 min apart, the study drug infusion rate is decreased by 50% for the remainder of the infusion period. If the patient's SBP falls to <100 mmHg in two consecutive measurements 15 min apart, the study drug is permanently discontinued. Randomized patients are required to receive standard‐of‐care background HF management during both the index hospitalization and the follow‐up period of 180 days. After randomization, the investigator may prescribe any additional medications dictated by the patients' condition, including i.v. loop diuretics and vasoactive medications.
Study assessments
Patients are assessed daily while hospitalized through day 5 or discharge, whichever comes first. They are also assessed at days 14, 60, 120 (phone contact), and 180 (Table 2). Heart failure signs and symptoms are assessed through day 60, and local haematology and clinical chemistry tests performed through day 5. Adverse events are reported from signing of the informed consent form through day 5 for non‐serious adverse events and through day 14 for serious adverse events. All deaths and hospitalizations reported through day 180 are adjudicated by a Clinical Events Committee (Appendix). Rehospitalization is defined as an unplanned hospitalization (including admission to a hospital or any attendance in an acute care setting, e.g. emergency department, or in another health care facility) of 24 h or greater, regardless of whether the patient was admitted to the hospital.
Table 2.
Assessment schedule
| Time points | Randomized treatment epoch | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Screening epoch | Baseline Hour 0 (Day 1) | Study drug infusion | Post‐treatment daily assessments | Follow‐up | ||||||||||
| Screen Hour ‐16 | Hour 6 (Day 1) | Hour 12 (Day 1) | Hour 24(Day 1) | Hour 48(Day 2) | Hour 72 (Day 3) | Hour 96 (Day 4) | Hour 120 (Day 5) | Discharge | Day 14 | Day 60 | Day 120 | Day 180/PSW | ||
| Screening procedures | X | |||||||||||||
| ECGa | X | X | X | |||||||||||
| ECG substudyb | X | X | ||||||||||||
| Body weight | X | X | X | X | X | X | X | X | X | X | ||||
| Echocardiogramc | X | |||||||||||||
| Physical examination with vital signsd | X | X | X | X | X | X | X | X | X | X | X | |||
| BP and HR measurementse | X | X | X | X | X | X | X | X | X | X | ||||
| Evaluate for systolic BP decrease event | X | X | X | X | X | |||||||||
| Index HF forms | X | X | ||||||||||||
| Health economics—tests/procedures/treatments | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Physician assessment of HF signs and symptomsf | X | X | Xs | Xs | Xs | Xs | Xs | X | X | X | ||||
| Chemistry/haematologyg | X | X | X | X | X | X | X | |||||||
| Laboratory substudyh | X | X | X | X | X | X | X | X | X | |||||
| Biomarker substudyi | X | X | X | X | X | |||||||||
| Concomitant medicationsj | X | X | X | X | X | X | X | X | X | X | X | X | X | X |
| Vital status and clinical outcome assessments | X | X | X | X | X | X | X | X | X | X | X | X | ||
| Assessment of readmission | X | X | X | X | ||||||||||
| Adverse and serious adverse eventsk | X | X | X | X | X | X | X | X | ||||||
BP, blood pressure; ECG, electrocardiogram; HF, heart failure; HR, heart rate; PSW, premature patient withdrawal; X, assessment.
ECGs will be performed and interpreted locally at screening and at day 5 or discharge, whichever occurs first.
ECGs will be collected in a subset of randomized patients participating in the ECG substudy and sent to a central ECG vendor for evaluation. ECGs will be collected at baseline and at the end of study drug infusion.
The echocardiogram should be performed as soon as possible post‐randomization, but prior to discharge. If an echocardiogram is performed during the screening period (i.e. within the 16 h window) and the patient is subsequently randomized, the screening echocardiogram will qualify as the index hospitalization echocardiogram and a repeat echocardiogram post‐randomization will not be necessary.
A complete physical examination will be performed at screening; an abbreviated physical examination will be performed at all other specified time points.
BP and HR measurements are to be performed at 30 and 60 min and then every hour for the first 6 h of study drug infusion, and then every 3 h during study drug infusion, including night‐time hours. Post‐infusion, BP and HR are to be measured every 3 h until 12 h following end of infusion, then every 6 h for 48 h and then every 24 h until the earlier of day 5 or discharge. BP and HR are to be measured with the patient in the same position and with the same equipment using the same arm, throughout study drug infusion. These measurements may be made and recorded by trained health care personnel as part of their routine clinical duties, as well as study personnel.
At hours 24, 48, 72, 96, and 120, physician assessment of HF signs and symptoms will include an assessment of the occurrence of worsening HF in the interval preceding the visit.
Blood will be locally collected and analysed daily during hospitalization. If discharge occurs prior to day 5, local blood collection will not be required at the day 5 hospital/clinic visit.
Blood will be collected in a subset of randomized patients participating in the laboratory substudy for measurement of biochemistry, haematology, and plasma glycated haemoglobin by the central laboratory. Urine dipstick will be measured locally at screening to rule out any conditions requiring further diagnostic evaluation or treatment.
Blood will be collected in a subset of randomized patients participating in the biomarker substudy to be performed by the central laboratory.
Major cardiovascular and non‐cardiovascular classes of medication taken by a subject ∼30 days prior to study drug initiation and on a daily basis while hospitalized through day 5, at discharge, and at days 14, 60, 120, and 180 will be recorded. Only those medications currently being taken or that were taken within 24 h prior to the visit will be collected.
Non‐serious and serious adverse events will be reported from the signing of the informed consent form through days 5 and 14, respectively.
The occurrence of WHF is reported by the investigator through day 5 post‐randomization, and is defined as worsening signs and/or symptoms of HF that require an intensification of i.v. therapy or mechanical ventilatory, renal or circulatory support for HF. Such treatment can include the institution or up‐titration of i.v. diuretic, i.v. nitrates, or any other i.v. medication for HF, or institution of mechanical support such as ventilation, ultrafiltration, haemodialysis, intra‐aortic balloon pump, or ventricular assist device. Worsening HF can occur within the 5 day post‐randomization period either during the index admission, or after discharge as an unplanned rehospitalization or unscheduled physician office/emergency department visit due to a primary diagnosis of HF. This endpoint also includes patients who die in this 5 day period from any cause.
ECGs are performed and interpreted locally for all patients at screening and at day 5 or discharge, whichever occurs first. In addition, ECGs are performed locally and interpreted by a central core lab at baseline and end of study infusion in a substudy of at least 500 patients in selected centres to evaluate the impact of serelaxin on ECG variables. Samples through day 14 are collected for central haematology, blood chemistry, and biomarker assays in ∼1600 patients at selected centres.
Statistical considerations
Primary and secondary efficacy outcomes, except all‐cause mortality, will be compared between treatment groups on an intent‐to‐treat basis using a sequentially rejective multiple testing procedure22 controlling the overall two‐sided α at 5%. Treatment groups will be compared regarding the time to CV death through day 180 with a log‐rank test at an initial significance level of (4/5)α, and regarding time to WHF through day 5 at an initial significance level of (1/5)α using a Gehan's generalized Wilcoxon test. All‐cause mortality will be tested independently at the two‐sided 5% significance level, if the test of either or both of the primary endpoints is significant. The significance level for the final test will be adjusted to account for the interim efficacy analysis, planned to occur after ∼60% (i.e. 329) of confirmed CV deaths have accrued. A Lan–DeMets spending function approximating an O'Brien–Fleming stopping boundary will be employed to control the overall one‐sided statistical testing of the CV mortality endpoint at the 2% level and the interim analysis will only be performed on the CV mortality endpoint. An independent Data Monitoring Committee (Appendix), supported by an independent, unblinded statistical centre, regularly reviews safety data as well as the interim efficacy analysis.
Accounting for the one interim efficacy analysis, 547 confirmed CV deaths are needed for 80% power to detect a 22% relative risk reduction. Assuming the 180 day CV death rate is 9.0% in the placebo group, which is ∼80% of 11.3% of all‐cause death observed in the placebo group in RELAX‐AHF,19 ∼6800 patients will need to be enrolled. The observed overall rate of the primary CV mortality endpoint will be assessed on a blinded basis, and adjustments to patient enrolment made in order to achieve the required number of events. Using the proposed multiple testing procedure, the power for WHF is at least 80% with the sample size of 6800 assuming at least a 20% relative risk reduction with 12.2% placebo event rate based on RELAX‐AHF data.
Discussion
Over the last two decades, morbidity and mortality from chronic HFrEF has decreased dramatically with the adoption of ACE inhibitors/angiotensin receptor blockers, beta‐blockers, and mineralocorticoid receptor antagonists. These therapies comprise the cornerstone of pharmacological chronic HF treatment. More recently, further progress has been made with the addition of an angiotensin receptor/neprilysin inhibitor (ARNI) as an alternative to ACE inhibitors, and ivabradine as an adjunct to maximally tolerated beta‐blocker therapy in patients in sinus rhythm.1, 23 However, these patients remain at high risk for acute decompensation, an event associated with a marked increase in mortality and HF recurrence either in the form of in‐hospital or post‐discharge WHF. Despite the urgent need to improve the outcomes of patients with AHF, only three therapies have gained regulatory approval in the last two decades: i.v. milrinone and nesiritide in the US and levosimendan in Europe.1, 2 Despite their approval, none of these drugs has demonstrated favourable effects on outcomes. Current treatment of AHF is based on drugs that have limited evidence of efficacy based on formal randomized controlled data. While diuretics have a Class I recommendation with a C level of evidence for the treatment of patients with congestion for symptom relief, other therapies such as nitrates have Class IIa recommendations with a B level of evidence.1 None of these therapies has an indication for improvement in clinical outcomes.
On the other hand, trials of novel therapies for AHF have failed to significantly improve symptoms or outcomes.24 Given the number of failures, variables other than the drug itself may have influenced these results. The heterogeneity of AHF patients, the inclusion of patients with non‐cardiac causes of symptoms, and the failure to align the drug's mechanism of action with the optimal patient population most likely to benefit from the study treatment are also potentially contributing causes.
To address these limitations, the RELAX‐AHF trials have enrolled patients admitted for AHF with persistent symptoms well defined by objective clinical diagnostics, including congestion on chest radiograph and elevated natriuretic peptide plasma levels. The patients are required to remain symptomatic despite initial treatment with i.v. diuretics, and in RELAX‐AHF‐2 only patients with severe enough AHF anticipated to require 48 h of i.v. therapy are enrolled. The inclusion of at least mild renal dysfunction among the entry criteria allows selection of a higher‐risk patient population, where serelaxin's potential renoprotective properties could provide benefit.20, 25 Enrolling patients with normal‐to‐elevated blood pressure (SBP ≥125 mmHg) selects patients most likely to benefit from the vasodilatory properties of serelaxin and those less likely to suffer from the untoward effects of hypotension. Drug‐induced hypotension has been a major cause of failure in previous AHF trials.9, 26 Whether serelaxin could provide benefit to patients with lower blood pressures or initial evidence of compromised perfusion can be addressed in future clinical studies and clinical practice. Enrolling patients early enough to maximize the potential end‐organ protective and haemodynamic benefits of serelaxin, yet late enough to be confident in the diagnosis of AHF, is central to the design of the RELAX‐AHF trials. Other studies that focused on dyspnoea relief suggested that earlier initiation of therapy provides greater symptom relief.27 However, in one study, only two‐thirds of the enrolled patients with a suspected diagnosis of AHF within 1 h of presentation were confirmed to have AHF at 6 h after presentation, suggesting that too rapid enrolment might result in patients without AHF being included in the trial.28 In both Pre‐RELAX‐AHF21 and RELAX‐AHF19 patients were randomized within 16 h of presentation at a median of 8–9 h; this time frame is the goal for RELAX‐AHF‐2 as well.
In contrast with studies in patients with chronic HF where a reduced ejection fraction has been a useful criterion for patient selection, the serelaxin trials enrolled patients with both HFrEF and HFpEF.19, 21 This is consistent with previous trials testing drugs acting on peripheral vessels and/or renal function in patients with AHF.29, 30 The prognosis of the patients with AHF was not found to be related to left ventricular ejection fraction31 and a similar benefit of serelaxin on clinical outcomes was observed in both patients with preserved and reduced ejection fraction.32 However, to provide additional information on the potential beneficial effects of serelaxin in these groups of patients, echocardiograms are obtained during the index hospitalization in RELAX‐AHF‐2. The timing of the echocardiograms was not specified, since a prior study suggested that left ventricular function does not change significantly in these types of AHF patients33 and it would be undesirable to have the echocardiogram interfere with randomization and initiation of study drug.
There is no limitation on concomitant oral therapies during study drug administration, but i.v. vasoactive therapies are either excluded or limited. Given the entry criterion of SBP ≥125 mmHg, there is no indication for vasopressors or inotropic agents, and while relatively safe, nesiritide has demonstrated no clinical benefits and has limited regulatory approval globally. Intravenous nitrates are the only i.v. vasoactive drugs allowed in addition to diuretics at the time of enrolment. Despite lack of evidence about their efficacy in the treatment of AHF34 and a recent guideline specifically stating, ‘Do not routinely offer nitrates to people with AHF’,35 many authorities continue to support their use. This may be especially true in patients with increased blood pressure at the time of randomization and hence, administration of i.v. nitrates was allowed only in this subgroup of patients (i.e. SBP >150 mmHg at the time of screening). The dose of nitrates allowed in RELAX‐AHF‐2 is clinically relevant and not particularly restrictive; an 80 kg person could be receiving up to 133 µg/min nitroglycerin or three times the dose achieved in VMAC,36 and over 6–10 times the recommended starting dose of the ESC guidelines.1
While few would challenge the clinical importance of reducing CV mortality, some have questioned the biological plausibility of a 48 h infusion of any drug having a significant effect on 180 day mortality in patients with AHF. While the ability of a brief infusion of a thrombolytic can clearly improve survival in patients with acute myocardial infarction, our understanding of AHF has not yet revealed a similarly specific ‘clot’ to target. However, an emerging concept of AHF as a combination of a haemodynamic, neurohormonal, inflammatory, and cytokine storm that results in small, but clinically significant end‐organ damage suggests that early and effective interventions could have long‐term, beneficial effects. Results from RELAX‐AHF support this hypothesis,19, 20 where evidence of myocardial, renal, liver, and other organ protection by serelaxin was associated with a 37% reduction in both CV and all‐cause mortality. In addition, there is evidence to support the converse, where a 48 h infusion of milrinone decreased long‐term survival compared to placebo with the survival curves continuing to diverge beyond the infusion.37 While the Pre‐RELAX‐AHF and RELAX‐AHF studies suggest a survival advantage of serelaxin in patients with AHF, mortality was not a primary endpoint in either study and the HF literature is replete with programmes that have failed to confirm early signals of improved survival. Consequently, RELAX‐AHF‐2 is appropriately powered to detect a clinically meaningful 22% reduction in risk of CV mortality.
While reducing CV mortality is an undisputedly important goal, WHF has only more recently emerged as a clinically meaningful endpoint in itself with increasing recognition also by regulators. In‐hospital WHF is generally defined as WHF symptoms and signs requiring an intensification of therapy,8, 10, 38, 39, 40, 41 and occurs in a variable proportion of patients admitted for AHF, ranging from 5 to 42%.41 Worsening HF is associated with a prolonged length of hospitalization, increased release of biomarkers related to myocardial damage and renal dysfunction and, more importantly, with a poorer long‐term outcome both with respect to rehospitalizations and mortality. The clinical importance of WHF has been demonstrated in retrospective analyses of patient databases and intervention trials, as well as in a recent pooled analysis of 3691 patients from AHF trials.42 The occurrence of WHF is also sensitive to treatment as it may be reduced by drugs active on symptoms in the patients with AHF,9 and serelaxin treatment in RELAX‐AHF was associated with a 30% decrease in WHF within 14 days. Due to these encouraging results and the importance of this event to patients, RELAX‐AHF‐2 was designed from its initiation to include robust report forms and detailed documentation to appropriately collect and characterize WHF events, an endpoint elevated from exploratory in RELAX‐AHF to key secondary in RELAX‐AHF‐2 since the trial initiation. Moreover, after a strategic reconsideration following the reviews of marketing authorization applications submitted based on RELAX‐AHF results to health authorities worldwide and after further consultations with regulators, the Sponsor and the Executive Committee decided to further elevate WHF through day 5 to a second primary endpoint in RELAX‐AHF‐2, in addition to CV mortality through day 180. Thus, RELAX‐AHF‐2 is designed to definitively assess the effect of serelaxin on WHF as an additional primary endpoint of the trial.
In conclusion, RELAX‐AHF‐2 is expected to answer a key question in AHF therapeutics: can a 48 h infusion of serelaxin reduce 180 day CV mortality of patients admitted for AHF or reduce the occurrence of WHF? If this question is answered in agreement with previous evidence from Pre‐RELAX‐AHF and RELAX‐AHF, it will revolutionize therapy for patients with AHF and for the first time provide an addition to the AHF therapeutic armamentarium with definitive outcome data.
Funding
The RELAX‐AHF‐2 study is funded by Novartis Pharma AG.
Conflict of interest: J.R.T. reports grants, personal fees, and non‐financial support from Novartis, during the conduct of the study; grants, personal fees, and non‐financial support from Amgen, Bayer, Bristol‐Myers Squibb, and Novartis; personal fees and non‐financial support from Cytokinetics, and Trevena; personal fees from Relypsa, and ZS Pharma, outside the submitted work. M.M. reports personal fees from Novartis, during the conduct of the study; personal fees from Bayer, Servier, Relypsa, and Stealth Therapeutics, outside the submitted work. A.A.V. reports grants and personal fees from Novartis, during the conduct of the study; personal fees from Boehringer, personal fees from GSK, grants and personal fees from Vifor, grants and personal fees from Stealth, grants and personal fees from Bayer, personal fees from Trevena, outside the submitted work. P.P. reports personal fees from Novartis, during the conduct of the study; grants and personal fees from Vifor Pharma, personal fees from Amgen, personal fees from Servier, personal fees from Bayer, grants and personal fees from Coridea, personal fees from Celladon, personal fees from Cardiorentis, grants from Singulex, outside the submitted work. P.S.P. reports personal fees and non‐financial support from Novartis, during the conduct of the study; personal fees and non‐financial support from Bristol‐Myers Squib, Relypsa, scPharmaceuticals, personal fees from Medtronic and Trevena, personal fees and other from Roche Diagnostics, and grants from PCORI, outside the submitted work. B.H.G. reports personal fees and non‐financial support from Novartis, during the conduct of the study. G.F. reports Committee fees from Novartis, during the conduct of the study; he is Committee member of trials sponsored by Novartis, Bayer, Cardiorentis, Vifor, Servier, outside the submitted work. G.M.F. reports grants and personal fees from Novartis, during the conduct of the study; grants and personal fees from Amgen, grants from Trevena, personal fees from GSK, personal fees from Bristol‐Myers Squibb, personal fees from Myokardia, grants from Otsuka, outside the submitted work. B.A.D. and G.C. report grants and personal fees from Novartis, during the conduct of the study; grants from Celyad, Trevena, Inc., grants from Sorbent Therapeutics, ChanRx, Laguna Pharmaceuticals, Singulex, outside the submitted work. B.A.D. and G.C. are employees of Momentum Research, Inc. L.B.M., C.G., M.W., T.A.H., and T.S. report personal fees from Novartis, during the conduct of the study; personal fees and other from Novartis, outside the submitted work, and are employees of Novartis.
Supporting information
Appendix S1. RELAX‐AHF‐2 Amendments.
Appendix 1.
Executive Committee: M. Metra (Co‐Chair), J.R. Teerlink (Co‐Chair), G.M. Felker, G. Filippatos, B.H. Greenberg, P.S. Pang, P. Ponikowski, A.A. Voors, G. Cotter, B.A. Davison, C. Gimpelewicz, T. Severin.
Data Monitoring Committee: M. Konstam (Chair), K. Dickstein, S. Goldstein, M. Komajda; Independent Statistician: S. Emerson.
Endpoint Adjudication Committee: G.M. Felker (Co‐Chair), J. Butler (Co‐Chair), L. Allen, P. Carson, Z. Eapen, A. Hernandez, J. Januzzi, D. Lanfear, A. Miller, I. Piña.
National Leaders: K. Adams (USA), S. Anker (EU), A. Arias Mendoza (Mexico), P. Avendaño (Chile), F. Bacal (Brazil), M. Böhm (Germany), G. Bortman (Argentina), J. Cleland (UK), A. Cohen‐Solal (France), M. Crespo (Spain), R. Ferrari (Italy), M. Dorobantu (Romania), L. Echeverría (Columbia), G. Filippatos (Greece), E. Goncalvesova (Slovakia), A. Goudev (Bulgaria), H. Haddad (Canada), A. Katz/S. Goland (Israel), L. Køber (Denmark), H. Krum (Australia), J. Lema Osores (Peru), P. Levy (USA), P. Manga (South Africa), K. McDonald (Ireland), B. Merkely (Hungary), C. Müller (Switzerland), B. Pieske/D. von Lewinski (Austria), M. Ruda (Russia), J. Silva‐Cardoso (Portugal), J. Spinar (Czech Republic), I. Squire (UK), J. Stepinska (Poland), W. Van Mieghem (Belgium), A. Voors (The Netherlands), G. Wikström (Sweden), M. Yilmaz (Turkey).
Trial registration. NCT01870778.
References
- 1. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 2. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;62:e147–239. [DOI] [PubMed] [Google Scholar]
- 3. Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, Nichol G, Pham M, Pina IL, Trogdon JG. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail 2013;6:606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Damasceno A, Mayosi BM, Sani M, Ogah OS, Mondo C, Ojji D, Dzudie A, Kouam CK, Suliman A, Schrueder N, Yonga G, Ba SA, Maru F, Alemayehu B, Edwards C, Davison BA, Cotter G, Sliwa K. The causes, treatment, and outcome of acute heart failure in 1006 Africans from 9 countries. Arch Intern Med 2012;172:1386–1394. [DOI] [PubMed] [Google Scholar]
- 5. Shimokawa H, Miura M, Nochioka K, Sakata Y. Heart failure as a general pandemic in Asia. Eur J Heart Fail 2015;17:884–892. [DOI] [PubMed] [Google Scholar]
- 6. Weatherley BD, Milo‐Cotter O, Felker GM, Uriel N, Kaluski E, Vered Z, O'Connor CM, Adams KF, Cotter G. Early worsening heart failure in patients admitted with acute heart failure—a new outcome measure associated with long‐term prognosis? Fundam Clin Pharmacol 2009;23:633–639. [DOI] [PubMed] [Google Scholar]
- 7. Torre‐Amione G, Milo‐Cotter O, Kaluski E, Perchenet L, Kobrin I, Frey A, Rund MM, Weatherley BD, Cotter G. Early worsening heart failure in patients admitted for acute heart failure: time course, hemodynamic predictors, and outcome. J Card Fail 2009;15:639–644. [DOI] [PubMed] [Google Scholar]
- 8. Metra M, Teerlink JR, Felker GM, Greenberg BH, Filippatos G, Ponikowski P, Teichman SL, Unemori E, Voors AA, Weatherley BD, Cotter G. Dyspnoea and worsening heart failure in patients with acute heart failure: results from the Pre‐RELAX‐AHF study. Eur J Heart Fail 2010;12:1130–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Packer M, Colucci WS, Fisher L, Massie BM, Teerlink JR, Young JB, Padley RJ, Thakkar R, Delgado‐Herrera L, Salon J, Garratt C, Huang B, Sarapohja T; REVIVE Heart Failure Study Group . Effect of levosimendan on the short‐term clinical course of patients with acutely decompensated heart failure. JACC Heart Fail 2013;1:103–111. [DOI] [PubMed] [Google Scholar]
- 10. Mentz RJ, Metra M, Cotter G, Milo O, McKendry C, Chiswell K, Davison BA, Cleland JG, Bloomfield DM, Dittrich HC, Fiuzat M, Ponikowski P, Givertz MM, Voors AA, Teerlink JR, O'Connor CM. Early vs. late worsening heart failure during acute heart failure hospitalization: insights from the PROTECT trial. Eur J Heart Fail 2015;17:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gheorghiade M, Vaduganathan M, Fonarow GC, Bonow RO. Rehospitalization for heart failure: problems and perspectives. J Am Coll Cardiol 2013;61:391–403. [DOI] [PubMed] [Google Scholar]
- 12. Kristensen SL, Jhund PS, Kober L, Preiss D, Kjekshus J, McKelvie RS, Zile MR, Anand IS, Wikstrand J, Wedel H, Komajda M, Carson PE, Cleland JG, McMurray JJ. Comparison of outcomes after hospitalization for worsening heart failure, myocardial infarction, and stroke in patients with heart failure and reduced and preserved ejection fraction. Eur J Heart Fail 2015;17:169–176. [DOI] [PubMed] [Google Scholar]
- 13. Solomon SD, Dobson J, Pocock S, Skali H, McMurray JJ, Granger CB, Yusuf S, Swedberg K, Young JB, Michelson EL, Pfeffer MA. Influence of nonfatal hospitalization for heart failure on subsequent mortality in patients with chronic heart failure. Circulation 2007;116:1482–1487. [DOI] [PubMed] [Google Scholar]
- 14. Ahmed A, Allman RM, Fonarow GC, Love TE, Zannad F, Dell'italia LJ, White M, Gheorghiade M. Incident heart failure hospitalization and subsequent mortality in chronic heart failure: a propensity‐matched study. J Card Fail 2008;14:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abrahamsson P, Swedberg K, Borer JS, Bohm M, Kober L, Komajda M, Lloyd SM, Metra M, Tavazzi L, Ford I. Risk following hospitalization in stable chronic systolic heart failure. Eur J Heart Fail 2013;15:885–891. [DOI] [PubMed] [Google Scholar]
- 16. XJ Du, Bathgate RA, Samuel CS, Dart AM, Summers RJ. Cardiovascular effects of relaxin: from basic science to clinical therapy. Nat Rev Cardiol 2010;7:48–58. [DOI] [PubMed] [Google Scholar]
- 17. Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev 2013;93:405–480. [DOI] [PubMed] [Google Scholar]
- 18. Tietjens J, Teerlink JR. Serelaxin and acute heart failure. Heart 2016;102:95–99. [DOI] [PubMed] [Google Scholar]
- 19. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld LR, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin TM, Metra M; RELAXin in Acute Heart Failure (RELAX‐AHF) Investigators. Serelaxin, recombinant human relaxin‐2, for treatment of acute heart failure (RELAX‐AHF): a randomised, placebo‐controlled trial. Lancet 2013;381:29–39. [DOI] [PubMed] [Google Scholar]
- 20. Metra M, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, Ponikowski P, Unemori E, Voors AA, Adams KF Jr, Dorobantu MI, Grinfeld L, Jondeau G, Marmor A, Masip J, Pang PS, Werdan K, Prescott MF, Edwards C, Teichman SL, Trapani A, Bush CA, Saini R, Schumacher C, Severin T, Teerlink JR; Investigators RELAX‐AHF. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX‐AHF) development program: correlation with outcomes. J Am Coll Cardiol 2013;61:196–206. [DOI] [PubMed] [Google Scholar]
- 21. Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, Marmor A, Katz A, Grzybowski J, Unemori E, Teichman SL, Cotter G. Relaxin for the treatment of patients with acute heart failure (Pre‐RELAX‐AHF): a multicentre, randomised, placebo‐controlled, parallel‐group, dose‐finding phase IIb study. Lancet 2009;373:1429–1439. [DOI] [PubMed] [Google Scholar]
- 22. Bretz F, Maurer W, Brannath W, Posch M. A graphical approach to sequentially rejective multiple test procedures. Stat Med 2009;28:586–604. [DOI] [PubMed] [Google Scholar]
- 23. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol 2016;68:1476–1488. [DOI] [PubMed] [Google Scholar]
- 24. Mebazaa A, Longrois D, Metra M, Mueller C, Richards AM, Roessig L, Seronde MF, Sato N, Stockbridge NL, Gattis Stough W, Alonso A, Cody RJ, Cook Bruns N, Gheorghiade M, Holzmeister J, Laribi S, Zannad F. Agents with vasodilator properties in acute heart failure: how to design successful trials. Eur J Heart Fail 2015;17:652–664. [DOI] [PubMed] [Google Scholar]
- 25. Voors AA, Dahlke M, Meyer S, Stepinska J, Gottlieb SS, Jones A, Zhang Y, Laurent D, Slart RH, Navis GJ. Renal hemodynamic effects of serelaxin in patients with chronic heart failure: a randomized, placebo‐controlled study. Circ Heart Fail 2014;7:994–1002. [DOI] [PubMed] [Google Scholar]
- 26. Erdmann E, Semigran MJ, Nieminen MS, Gheorghiade M, Agrawal R, Mitrovic V, Mebazaa A. Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur Heart J 2013;34:57–67. [DOI] [PubMed] [Google Scholar]
- 27. Peacock WF 4th, Fonarow GC, Emerman CL, Mills RM, Wynne J; ADHERE Scientific Advisory Committee and Investigators ; ADHERE Study Group. Impact of early initiation of intravenous therapy for acute decompensated heart failure on outcomes in ADHERE. Cardiology 2007;107:44–51. [DOI] [PubMed] [Google Scholar]
- 28. Mebazaa A, Pang PS, Tavares M, Collins SP, Storrow AB, Laribi S, Andre S, Mark Courtney D, Hasa J, Spinar J, Masip J, Frank Peacock W, Sliwa K, Gayat E, Filippatos G, Cleland JG, Gheorghiade M. The impact of early standard therapy on dyspnoea in patients with acute heart failure: the URGENT‐dyspnoea study. Eur Heart J 2010;31:832–841. [DOI] [PubMed] [Google Scholar]
- 29. McMurray JJ, Teerlink JR, Cotter G, Bourge RC, Cleland JG, Jondeau G, Krum H, Metra M, O'Connor CM, Parker JD, Torre‐Amione G, van Veldhuisen DJ, Lewsey J, Frey A, Rainisio M, Kobrin I. Effects of tezosentan on symptoms and clinical outcomes in patients with acute heart failure: the VERITAS randomized controlled trials. JAMA 2007;298:2009–2019. [DOI] [PubMed] [Google Scholar]
- 30. Massie BM, O'Connor CM, Metra M, Ponikowski P, Teerlink JR, Cotter G, Weatherley BD, Cleland JG, Givertz MM, Voors A, DeLucca P, Mansoor GA, Salerno CM, Bloomfield DM, Dittrich HC. Rolofylline, an adenosine A1‐receptor antagonist, in acute heart failure. N Engl J Med 2010;363:1419–1428. [DOI] [PubMed] [Google Scholar]
- 31. Bello NA, Claggett B, Desai AS, McMurray JJ, Granger CB, Yusuf S, Swedberg K, Pfeffer MA, Solomon SD. Influence of previous heart failure hospitalization on cardiovascular events in patients with reduced and preserved ejection fraction. Circ Heart Fail 2014;7:590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Filippatos G, Teerlink JR, Farmakis D, Cotter G, Davison BA, Felker GM, Greenberg BH, Hua T, Ponikowski P, Severin T, Unemori E, Voors AA, Metra M. Serelaxin in acute heart failure patients with preserved left ventricular ejection fraction: results from the RELAX‐AHF trial. Eur Heart J 2014;35:1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gandhi SK, Powers JC, Nomeir AM, Fowle K, Kitzman DW, Rankin KM, Little WC. The pathogenesis of acute pulmonary edema associated with hypertension. N Engl J Med 2001;344:17–22. [DOI] [PubMed] [Google Scholar]
- 34. Wakai A, McCabe A, Kidney R, Brooks SC, Seupaul RA, Diercks DB, Salter N, Fermann GJ, Pospisil C. Nitrates for acute heart failure syndromes. Cochrane Database Syst Rev 2013;8:CD005151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. National Clinical Guideline Centre . Acute Heart Failure: Diagnosing and Managing Acute Heart Failure in Adults. NICE Clinical Guideline 187, Methods, evidence and recommendations. London: National Institute for Health and Care Excellence; 2014. p1–329. [PubMed] [Google Scholar]
- 36. Investigators VMAC. Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002;287:1531–1540. [DOI] [PubMed] [Google Scholar]
- 37. Felker GM, Benza RL, Chandler AB, Leimberger JD, Cuffe MS, Califf RM, Gheorghiade M, O'Connor CM; Investigators OPTIME‐CHF. Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME‐CHF study. J Am Coll Cardiol 2003;41:997–1003. [DOI] [PubMed] [Google Scholar]
- 38. Cotter G, Metra M, Weatherley BD, Dittrich HC, Massie BM, Ponikowski P, Bloomfield DM, O'Connor CM. Physician‐determined worsening heart failure: a novel definition for early worsening heart failure in patients hospitalized for acute heart failure—association with signs and symptoms, hospitalization duration, and 60‐day outcomes. Cardiology 2010;115:29–36. [DOI] [PubMed] [Google Scholar]
- 39. Cotter G, Metra M, Davison BA, Senger S, Bourge RC, Cleland JG, Jondeau G, Krum H, O'Connor CM, Parker JD, Torre‐Amione G, van Veldhuisen DJ, Milo O, Kobrin I, Rainisio M, McMurray JJ, Teerlink JR; VERITAS Investigators . Worsening heart failure, a critical event during hospital admission for acute heart failure: results from the VERITAS study. Eur J Heart Fail 2014;16:1362–1371. [DOI] [PubMed] [Google Scholar]
- 40. Kelly JP, Mentz RJ, Hasselblad V, Ezekowitz JA, Armstrong PW, Zannad F, Felker GM, Califf RM, O'Connor CM, Hernandez AF. Worsening heart failure during hospitalization for acute heart failure: Insights from the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND‐HF). Am Heart J 2015;170:298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Butler J, Gheorghiade M, Kelkar A, Fonarow GC, Anker S, Greene SJ, Papadimitriou L, Collins S, Ruschitzka F, Yancy CW, Teerlink JR, Adams K, Cotter G, Ponikowski P, Felker GM, Metra M, Filippatos G. In‐hospital worsening heart failure. Eur J Heart Fail 2015;17:1104–1113. [DOI] [PubMed] [Google Scholar]
- 42. Davison BA, Metra M, Cotter G, Massie BM, Cleland JG, Dittrich HC, Edwards C, Filippatos G, Givertz MM, Greenberg B, Ponikowski P, Voors AA, O'Connor CM, Teerlink JR; PROTECT and RELAX‐AHF Executive Committees. Worsening heart failure following admission for acute heart failure: a pooled analysis of the PROTECT and RELAX‐AHF studies. JACC Heart Fail 2015;3:395–403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. RELAX‐AHF‐2 Amendments.