

Abstract
The development of artificial nanomotor systems that are stimuli‐responsive is still posing many challenges. Herein, we demonstrate the self‐assembly of a redox‐responsive stomatocyte nanomotor system, which can be used for triggered drug release under biological reducing conditions. The redox sensitivity was introduced by incorporating a disulfide bridge between the hydrophilic poly(ethylene glycol) block and the hydrophobic polystyrene block. When incubated with the endogenous reducing agent glutathione at a concentration comparable to that within cells, the external PEG shells of these stimuli‐responsive nanomotors are cleaved. The specific bowl‐shaped stomatocytes aggregate after the treatment with glutathione, leading to the loss of motion and triggered drug release. These novel redox‐responsive nanomotors can not only be used for remote transport but also for drug delivery, which is promising for future biomedical applications.
Keywords: copolymers, drug delivery, nanomotors, redox sensitivity, self-assembly
In nature, organisms are capable of complex motion behavior in response to environmental changes to achieve different functions.1 To mimic this fantastic self‐migrating behavior from nature, artificial centimeter‐scale motors (hemicylindrical plates) were first reported by Whitesides and co‐workers with platinum on one side of the surface.2 These plates were propelled by oxygen bubbles that were generated by platinum‐catalyzed decomposition of a hydrogen peroxide (H2O2) solution. After a decade's effort, synthetic self‐propelled motors with miniature size were rapidly developed over the last few years.3 Owing to their small size, micro‐ and nanomotors show huge potential in different ranges of applications, such as environmental remediation, sensing, assisted fertilization, and drug delivery.4
Compared to normal drug delivery systems, the most important advantage of micro‐ and nanomotor systems is the impetus that is independent from the blood flow, which is necessary for tissue penetration and crossing cellular barriers.4d Therefore, to develop the next generation of drug delivery systems, both propulsion and in‐site drug release are important aspects for the design of active and directional delivery systems. Although some studies on artificial self‐propelled motors towards drug delivery applications have been reported, most of them either have micrometer dimensions or low drug loading efficiencies or cannot undergo controlled release of the loaded drug, which is not very practical for drug delivery.5 Therefore, nanomotor systems that can sense different stimuli produced by cells to control the properties and achieve transport behavior are a promising and also important research field. Recently, calcium carbonate Janus microparticles were reported that can move under extremely weakly acidic conditions generated by HeLa cells in situ.6 However, the development of nanomotors that are capable of controlled drug release is still challenging.
To achieve in‐site drug release, the specific environment and the differences between the desired and non‐desired sites of release should be taken into account. For example, the human body hosts many gradients of chemical signaling molecules, resulting in pH differences for inflamed, infected, or tumor tissue or even in different compartments of the cells. Temperature fluctuations can be observed at specific pathological sites, and redox potentials exist between extracellular and intracellular microenvironments.7 With regard to the latter, the intracellular compartments possess a much higher glutathione concentration (GSH, 2–10 mm) than the extracellular microenvironment (2–10 μm), which can be used as a trigger to achieve controlled release of encapsulated molecules from nanocarrier systems.8 Aside from the differences in the physiological oxidative conditions between extracellular and intracellular compartments, pathophysiological oxidative aspects in inflamed or tumor tissues can also be exploited to destabilize the drug delivery systems.
In our previous work, platinum nanoparticles (PtNPs) were loaded into the cavity of bowl‐shaped stomatocyte nanomotors that self‐assembled from the block copolymer poly(ethylene glycol)‐b‐polystyrene (PEG‐b‐PS).9 It was demonstrated that the motion3l of these polymeric nanomotors can be controlled and that they can also be guided3n and steered10 in desired directions in the presence of H2O2 and alternative biofuels.11 However, owing to the glassy and compact PS bilayer structure, the nanomotor cannot be degraded, and therefore, the loaded drugs were trapped either in the hydrophobic membrane or in the hydrophilic lumen, which is not suitable for further biomedical applications. Herein, we demonstrate the successful design of a first redox‐responsive nanomotor system through the incorporation of disulfide bonds between the hydrophobic PS block and the hydrophilic PEG block (PEG‐SS‐PS). After the introduction of disulfide bonds, the morphology of the bowl‐shaped structure and the encapsulation of model PtNP catalysts remained the same. The multifunctional nanomotor systems are able to probe the environment by sensing a change in the outside redox conditions, which leads to the cleavage of the hydrophilic PEG part and further disables the motion function. In human bodies, tumor tissues can produce H2O2 gradients owing to their high production rate of up to 5 nmol per 104 cells per hour, which is not too low when related to the size of a tumor.12 Our redox‐sensitive nanomotors can follow the resulting H2O2 gradient in the body and move towards to the diseased area.3n After accumulation, the redox‐responsive nanomotors can be taken up and release the loaded drug owing to the higher intracellular GSH concentration. In addition, the higher concentration of GSH also results in inhibition of the motion behavior owing to catalyst poisoning by the thiol group in GSH.13 These multifunctional bioreducible nanomotor systems can potentially lead to a new approach for a more efficient and controlled way to deliver drugs in the future.
The redox‐sensitive block copolymer PEG‐SS‐PS (Figure 1 a) and the standard polymer PEG‐b‐PS were synthesized by atom transfer radical polymerization (ATRP) of styrene by starting from PEG‐SS‐Br and PEG‐Br macroinitiators, respectively (see the Supporting Information, Scheme S1).14 After determination of the average molecular weight of PEG‐SS‐PS and PEG‐b‐PS by 1H NMR spectroscopy, redox‐sensitive polymersomes were then self‐assembled by a solvent switch method (Figure 1 b). During the self‐assembly, the hydrophilic anticancer drug doxorubicin (Dox) was loaded into the lumen of the polymersomes. In the presence of an organic solvent, the formed polymersomes with a flexible membrane underwent a morphology change into bowl‐shaped stomatocyte structures owing to an osmotic shock introduced by dialysis.15 During the shape transformation, model catalysts, namely PtNPs, were entrapped in the cavities of the stomatocytes to serve as engines for the nanomotors (Figure 1 b). After dialysis to remove the organic solvent, Dox‐loaded redox‐responsive nanomotors and redox‐insensitive nanomotors were obtained. According to dynamic light scattering (DLS) analysis, the self‐assembled structures have similar sizes and also a narrow polydispersity index (PDI; Table S1). Transmission electron microscopy (TEM) was further used to visualize the structure of the stomatocyte nanomotors. Well‐defined stomatocytes with a clear bilayer structure and also PtNP encapsulation were observed for both redox‐sensitive and ‐insensitive nanomotor samples (Figures 2 a and S1), indicating that the introduction of the disulfide bond does not affect the formation of the polymeric vesicles.
Figure 1.
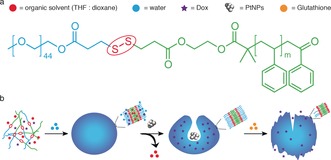
a) Chemical structure of the redox‐responsive block copolymer PEG‐SS‐PS used for the stomatocyte assembly. b) Self‐assembly and GSH‐triggered disassembly of the redox‐sensitive stomatocyte nanomotor.
Figure 2.
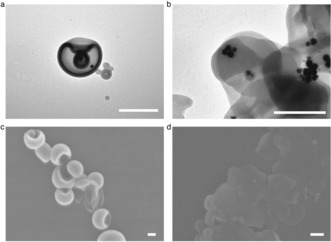
Characterization of the redox‐sensitive stomatocyte nanomotor before and after incubation with GSH. a) TEM image of a stomatocyte nanomotor. b) TEM image of a stomatocyte nanomotor after incubation with GSH. c) SEM image of a stomatocyte nanomotor. d) SEM image of a stomatocyte nanomotor after treatment with GSH. Scale bars: 200 nm.
The redox‐sensitive nature of the disulfide bond between the two blocks was exploited by addition of the endogenous reducing agent GSH. TEM and scanning electron microscopy (SEM) were used to observe the morphology changes of the nanomotor structures before and after incubation with GSH. Before addition of GSH, regular stomatocytes with small openings were observed in both TEM and SEM images (Figure 2 a, c) while aggregation without a clear bilayer membrane was observed after overnight treatment with GSH (Figure 2 b, d). The small GSH molecules can enter through the PEG shell of the nanomotor and then break down the redox‐responsive disulfide bonds that covalently link the PEG and PS blocks, resulting in cleavage of the outside PEG shell. The resulting structures are hydrophobic and unstable owing to the loss of the hydrophilic PEG layer, which therefore leads to the formation of aggregates, as observed in the TEM and SEM images.
To confirm the structural changes of redox‐responsive stomatocytes induced by GSH, DLS was also used to measure the size before and after incubation with the reducing agent for both sensitive and insensitive stomatocytes. For the insensitive stomatocytes, the sizes remained almost the same (diameter ca. 345 nm) after treatment with GSH, even when the concentration of the reducing agent was increased to 120 mm (Figure S3 a). The intensity DLS curve showed a narrow distribution, which was further confirmed by the low PDI (Supplementary Table 2). For stomatocytes composed of the stimuli‐responsive polymer PEG‐SS‐PS, the size distribution underwent a significant shift. The particle size changed from 349 nm to 1392 nm after treatment with 20 mm GSH (Figure S3 b and Table S2), indicating the formation of aggregates, which was also confirmed by TEM and SEM (Figure 2 b, d). Similar behavior was observed for GSH at concentrations of 40 mm and 120 mm. The size and count rate of the redox stomatocytes were also measured during the treatment with GSH (Figure S4). The size increased gradually with the GSH incubation time while the count rate decreased accordingly. Furthermore, the zeta potential of the redox stomatocytes before and after incubation with GSH was measured (Table S3). After treatment with GSH, the zeta potential of the redox‐sensitive stomatocytes had changed from −30.9 mV to −45.2 mV, again indicating PEG cleavage.
To confirm the successful PEG cleavage from the redox‐responsive stomatocytes, diffusion NMR spectroscopy was used to measure the diffusion coefficient of the cleaved PEG after incubation of the redox‐sensitive sample with GSH and to compare it with that of free PEG polymer (2 kDa, the same molecular weight as the PEG part of PEG‐SS‐PS). After GSH‐triggered PEG cleavage, the resulting sample was centrifuged to obtain the supernatant, which contains the cleaved PEG and GSH. The supernatant was then freeze‐dried and redissolved in deuterium oxide (D2O). GSH resonances were observed at δ=3.88 (2 H, NHCH 2COOH) and 3.73 ppm (1 H, CH2CHNH2COOH; Figure S5 a). Specific PEG signals around δ=3.63 ppm (broad singlet, PEG backbone) and 3.31 ppm (singlet, 3 H, CH 3OCH2) were observed for both cleaved PEG and free PEG (Figure S5 a, c). After fitting the decay curve of the NMR intensity, the diffusion coefficient of the cleaved PEG was determined to be 2.76±0.47×10−6 cm2 s−1 (Figure S5 b). For the free PEG sample, the diffusion coefficient in D2O was determined to be 2.55±0.05×10−6 cm2 s−1 (Figure S5 d). As the diffusion coefficient of the PEG cleaved from the redox‐responsive stomatocytes was almost the same as that of the free PEG control, this is an another clear indication that the external PEG shell was indeed cleaved by a GSH‐triggered chemical reaction.
As the cleavage of the PEG shells could disassemble the entire nanomotor system, stimuli‐triggered drug release might be possible. The Dox‐loaded redox‐sensitive nanomotors were incubated at different GSH concentrations at room temperature in vitro. Without GSH treatment, almost no Dox release was observed for the sensitive nanomotors (Figure S6). Incubation of the motor solution with 20 mm GSH, which is comparable to the intracellular concentration, resulted in a cumulative release of 68 % Dox from our bioreducible nanomotors. This release is significantly higher than the release in 1 mm GSH.
Having confirmed the redox responsiveness and potential in vitro drug release, nanoparticle tracking analysis (NTA) was used to investigate the motion behavior of these nanomotors in the presence of H2O2 solution. Nanosight NS500 was used to record the movement of the nanomotors in real time in a video. The movement of the nanomotors was recorded for 90 s with 30 frames per second. H2O2 solution was added to the nanomotors, and the resulting solutions were studied at 30 and 37 °C (final nanomotor concentration: 108 particles mL−1, final H2O2 concentration: 4.98 mm). The mean square displacement (MSD) was extracted from the simultaneous recording of the x,y coordinates of multiple particles (20 particles) by the NTA technique. Fitting of the MSD curves allowed for a calculation of the average velocity of the nanomotors by using the self‐diffusiophoretic model proposed by Golestanian and co‐workers.16 The MSDs (Figure 3 a) and velocities (Figure 3 b) of the redox‐responsive nanomotors at both 30 and 37 °C show directional autonomous movement as demonstrated in our previous studies.9 Higher MSDs and velocities were observed at 37 °C than at 30 °C owing to the higher catalytic efficiency of the PtNPs. However, almost Brownian motion was observed after treatment with GSH. The MSD curve had a typical shape (linear) and size for Brownian motion (Figure 3 a), which was similar to the behavior of the nanomotors in the absence of H2O2 fuel. In the presence of the GSH reducing agent, the PEG shell was cleaved, destabilizing the self‐assembled structure. In addition, the hydrophobicity of the resulting system led to further aggregation. Representative tracking trajectories of redox‐responsive nanomotors before and after the addition of GSH are presented in Figure 3 c. The trajectories of the sensitive nanomotors after degradation exhibited Brownian motion as expected while directional motion was observed before incubation with GSH, indicating active self‐propulsion.
Figure 3.
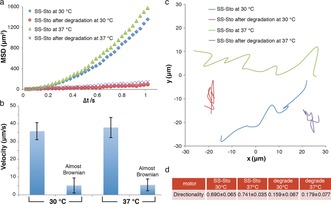
Motion evaluation of the redox‐sensitive stomatocyte nanomotor. a) MSD of redox‐sensitive nanomotors before and after degradation at different temperatures in the presence of H2O2. b) Velocity of redox‐sensitive nanomotors before and after degradation at different temperatures in the presence of H2O2. The respective velocities were calculated by fitting with MSD. The directional motion was fitted using the equation (4D)Δt+(v 2)(Δt 2), and the Brownian motion was fitted using the equation (4D)Δt. Error bars indicate the standard deviation of the velocity of 20 motors. c) Trajectories of redox‐sensitive nanomotors. d) Directionality of the movement of redox‐sensitive nanomotors as the average of 20 motors. SS‐Sto=redox‐sensitive nanomotor.
Directionality was also used for the characterization of the motion behavior of our multifunctional nanomotors.17 The directionality was calculated by comparing the Euclidian distance to the accumulated distance over a certain time interval. Here, the value of the directionality over a long time interval (around 1 s) was used to distinguish between non‐Brownian and Brownian motion (Figure 3 d). For the sensitive nanomotors in H2O2 at 30 °C, the directionality was calculated to be 0.690, indicating directional non‐Brownian motion. However, the directionality calculated at 30 °C was only 0.159 (demonstrated to be Brownian motion) when the sensitive nanomotors were dismantled by GSH, further confirming that our sensitive nanomotors lost their motility after cleavage of the PEG shell.
HeLa cells were used to demonstrate the cellular internalization and release behavior of the redox‐responsive nanomotors. The cells were incubated with Dox‐loaded redox‐sensitive or ‐insensitive nanomotors in the presence of H2O2. For the bioreducible nanomotors (Figure 4 b), the fluorescence signal from Dox diffused over the Hela cells, indicating the triggered release of Dox owing to the higher intracellular GSH concentration.18 However, for the insensitive nanomotors (Figure 4 a), dot‐like fluorescence signals was observed, which meant that Dox was still localized within the nanomotors even after they had been taken up by the cells.
Figure 4.
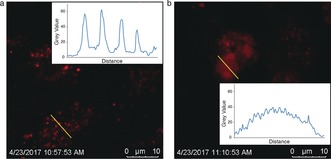
Cell uptake of Dox‐loaded redox‐sensitive and ‐insensitive nanomotors in the presence of H2O2 solution. a) Confocal image of Hela cells after exposure to Dox‐loaded nanomotors. b) Confocal image of cells after exposure to Dox‐loaded redox‐responsive nanomotors.
In summary, we have designed a multifunctional self‐propelled stomatocyte nanomotor with redox‐responsive behavior by introducing a disulfide bridge between the hydrophobic and hydrophilic blocks of the block copolymer constituents. Owing to the redox sensitivity of the disulfide bonds, the reducing agent GSH could be used to degrade the sensitive nanomotor system at physiological concentrations. Our nanomotor system is able to sense its local environment, in this case, the presence of reducing agents, which cleave the external PEG shell, thereby disabling the motion behavior and triggering drug release. As an endogenous reducing agent, GSH is commonly found at higher concentrations in intracellular compartments than in extracellular compartments, which makes our redox‐responsive nanomotor promising for drug delivery applications in the future.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the ERC under the European Union's Seventh Framework Programme (FP7/2007–2012)/ERC‐StG 307679 “StomaMotors”. We acknowledge support from the Ministry of Education, Culture and Science (Gravitation program 024.001.035). D.A.W. acknowledges financial support from the NWO Chemiche Wetenschappen VIDI grant 723.015.001. F.P. acknowledges funding from the China scholarship council.
Y. Tu, F. Peng, P. B. White, D. A. Wilson, Angew. Chem. Int. Ed. 2017, 56, 7620.
References
- 1.
- 1a. Urry D. W., Angew. Chem. Int. Ed. Engl. 1993, 32, 819; [Google Scholar]; Angew. Chem. 1993, 105, 859; [Google Scholar]
- 1b. Tu Y., Peng F., Adawy A., Men Y., Abdelmohsen L. K., Wilson D. A., Chem. Rev. 2016, 116, 2023; [DOI] [PubMed] [Google Scholar]
- 1c. Dickinson M. H., Farley C. T., Full R. J., Koehl M., Kram R., Lehman S., Science 2000, 288, 100. [DOI] [PubMed] [Google Scholar]
- 2. Ismagilov R. F., Schwartz A., Bowden N., Whitesides G. M., Angew. Chem. Int. Ed. 2002, 41, 652; [Google Scholar]; Angew. Chem. 2002, 114, 674. [Google Scholar]
- 3.
- 3a. Paxton W. F., Kistler K. C., Olmeda C. C., Sen A., Angelo S. K. St., Cao Y., Mallouk T. E., Lammert P. E., Crespi V. H., J. Am. Chem. Soc. 2004, 126, 13424; [DOI] [PubMed] [Google Scholar]
- 3b. Ma X., Wang X., Hahn K., Sánchez S., ACS Nano 2016, 10, 3597; [DOI] [PubMed] [Google Scholar]
- 3c. Wang H., Khezri B., Pumera M., Chem. 2016, 1, 473; [Google Scholar]
- 3d. Gao W., Dong R., Thamphiwatana S., Li J., Gao W., Zhang L., Wang J., ACS Nano 2015, 9, 117; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Wu Y., Wu Z., Lin X., He Q., Li J., ACS Nano 2012, 6, 10910; [DOI] [PubMed] [Google Scholar]
- 3f. Magdanz V., Sanchez S., Schmidt O. G., Adv. Mater. 2013, 25, 6581; [DOI] [PubMed] [Google Scholar]
- 3g. Mei Y., Huang G., Solovev A. A., Ureña E. B., Mönch I., Ding F., Reindl T., Fu R. K., Chu P. K., Schmidt O. G., Adv. Mater. 2008, 20, 4085; [Google Scholar]
- 3h. Chen C., Mou F., Xu L., Wang S., Guan J., Feng Z., Wang Q., Kong L., Li W., Wang J., Adv. Mater. 2017, 29, 1603374; [DOI] [PubMed] [Google Scholar]
- 3i. Schattling P., Thingholm B., Stadler B., Chem. Mater. 2015, 27, 7412; [Google Scholar]
- 3j. Dai B., Wang J., Xiong Z., Zhan X., Dai W., Li C.-C., Feng S.-P., Tang J., Nat. Nanotechnol. 2016, 11, 1087; [DOI] [PubMed] [Google Scholar]
- 3k. Wang W., Li S., Mair L., Ahmed S., Huang T. J., Mallouk T. E., Angew. Chem. Int. Ed. 2014, 53, 3201; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3265; [Google Scholar]
- 3l. Tu Y., Peng F., Sui X., Men Y., White P. B., van Hest J. C., Wilson D. A., Nat. Chem. 2017, 9, 480; [DOI] [PubMed] [Google Scholar]
- 3m. Jang B., Wang W., Wiget S., Petruska A. J., Chen X., Hu C., Hong A., Folio D., Ferreira A., Pané S., ACS Nano 2016, 10, 9983; [DOI] [PubMed] [Google Scholar]
- 3n. Peng F., Tu Y., van Hest J., Wilson D. A., Angew. Chem. Int. Ed. 2015, 54, 11662; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11828; [Google Scholar]
- 3o. Kim K., Liang Z., Liu M., Fan D., ACS Appl. Mater. Interfaces 2017, 9, 6144; [DOI] [PubMed] [Google Scholar]
- 3p. Sengupta S., Ibele M. E., Sen A., Angew. Chem. Int. Ed. 2012, 51, 8434; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8560; [Google Scholar]
- 3q. Wu Z., Si T., Gao W., Lin X., Wang J., He Q., Small 2016, 12, 577; [DOI] [PubMed] [Google Scholar]
- 3r. Lin X., Wu Z., Wu Y., Xuan M., He Q., Adv. Mater. 2016, 28, 1060; [DOI] [PubMed] [Google Scholar]
- 3s. Wang H., Pumera M., Chem. Rev. 2015, 115, 8704; [DOI] [PubMed] [Google Scholar]
- 3t. Moo J. G. S., Presolski S., Pumera M., ACS Nano 2016, 10, 3543. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Gao W., Wang J., ACS Nano 2014, 8, 3170; [DOI] [PubMed] [Google Scholar]
- 4b. Wang J., Biosens. Bioelectron. 2016, 76, 234; [DOI] [PubMed] [Google Scholar]
- 4c. Medina-Sánchez M., Schwarz L., Meyer A. K., Hebenstreit F., Schmidt O. G., Nano Lett. 2016, 16, 555; [DOI] [PubMed] [Google Scholar]
- 4d. Gao W., Wang J., Nanoscale 2014, 6, 10486; [DOI] [PubMed] [Google Scholar]
- 4e. Abdelmohsen L. K., Peng F., Tu Y., Wilson D. A., J. Mater. Chem. B 2014, 2, 2395; [DOI] [PubMed] [Google Scholar]
- 4f. Patra D., Sengupta S., Duan W., Zhang H., Pavlick R., Sen A., Nanoscale 2013, 5, 1273; [DOI] [PubMed] [Google Scholar]
- 4g. Wu Z., Wu Y., He W., Lin X., Sun J., He Q., Angew. Chem. Int. Ed. 2013, 52, 7000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7138; [Google Scholar]
- 4h. Duan W., Wang W., Das S., Yadav V., Mallouk T. E., Sen A., Annu. Rev. Anal. Chem. 2015, 8, 311. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Wu Z., Lin X., Zou X., Sun J., He Q., ACS Appl. Mater. Interfaces 2015, 7, 250; [DOI] [PubMed] [Google Scholar]
- 5b. Mou F., Chen C., Zhong Q., Yin Y., Ma H., Guan J., ACS Appl. Mater. Interfaces 2014, 6, 9897; [DOI] [PubMed] [Google Scholar]
- 5c. Srivastava S. K., Medina-Sánchez M., Koch B., Schmidt O. G., Adv. Mater. 2016, 28, 832; [DOI] [PubMed] [Google Scholar]
- 5d. Esteban-Fernández de Ávila B., Angell C., Soto F., Lopez-Ramirez M. A., Báez D. F., Xie S., Wang J., Chen Y., ACS Nano 2016, 10, 4997; [DOI] [PubMed] [Google Scholar]
- 5e. Wu Y., Lin X., Wu Z., Möhwald H., He Q., ACS Appl. Mater. Interfaces 2014, 6, 10476; [DOI] [PubMed] [Google Scholar]
- 5f. Garcia-Gradilla V., Sattayasamitsathit S., Soto F., Kuralay F., Yardımcı C., Wiitala D., Galarnyk M., Wang J., Small 2014, 10, 4154. [DOI] [PubMed] [Google Scholar]
- 6. Guix M., Meyer A. K., Koch B., Schmidt O. G., Sci. Rep. 2016, 6, 21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Ganta S., Devalapally H., Shahiwala A., Amiji M., J. Controlled Release 2008, 126, 187; [DOI] [PubMed] [Google Scholar]
- 7b. Mura S., Nicolas J., Couvreur P., Nat. Mater. 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- 8. Saito G., Swanson J. A., Lee K.-D., Adv. Drug Delivery Rev. 2003, 55, 199. [DOI] [PubMed] [Google Scholar]
- 9. Wilson D. A., Nolte R. J., Van Hest J. C., Nat. Chem. 2012, 4, 268. [DOI] [PubMed] [Google Scholar]
- 10. Peng F., Tu Y., Men Y., van Hest J., Wilson D. A., Adv. Mater. 2017, 29, 1604996. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Abdelmohsen L. K., Nijemeisland M., Pawar G. M., Janssen G.-J. A., Nolte R. J., van Hest J. C., Wilson D. A., ACS Nano 2016, 10, 2652; [DOI] [PubMed] [Google Scholar]
- 11b. Nijemeisland M., Abdelmohsen L. K., Huck W. T., Wilson D. A., van Hest J. C., ACS Cent. Sci. 2016, 2, 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Szatrowski T. P., Nathan C. F., Cancer Res. 1991, 51, 794. [PubMed] [Google Scholar]
- 13. Zhao G., Sanchez S., Schmidt O. G., Pumera M., Nanoscale 2013, 5, 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan W., Zou H., Guo W., Shen T., Ren J., Polym. Chem. 2013, 4, 2658. [Google Scholar]
- 15.
- 15a. Kim K. T., Zhu J., Meeuwissen S. A., Cornelissen J. J., Pochan D. J., Nolte R. J., van Hest J. C., J. Am. Chem. Soc. 2010, 132, 12522; [DOI] [PubMed] [Google Scholar]
- 15b. Meeuwissen S. A., Kim K. T., Chen Y., Pochan D. J., van Hest J., Angew. Chem. Int. Ed. 2011, 50, 7070; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7208. [Google Scholar]
- 16. Howse J. R., Jones R. A., Ryan A. J., Gough T., Vafabakhsh R., Golestanian R., Phys. Rev. Lett. 2007, 99, 048102. [DOI] [PubMed] [Google Scholar]
- 17. Kline T. R., Paxton W. F., Mallouk T. E., Sen A., Angew. Chem. Int. Ed. 2005, 44, 744; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 754. [Google Scholar]
- 18. Tu Y., Peng F., André A. A., Men Y., Srinivas M., Wilson D. A., ACS Nano 2017, 11, 1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary