

Abstract
Canines are excellent models for cancer studies due to their similar physiology and genomic sequence to humans, companion status and limited intra‐breed heterogeneity. Due to their affliction to mammary cancers, canines can serve as powerful genetic models of hereditary breast cancers. Variants within known human breast cancer susceptibility genes only explain a fraction of familial cases. Thus, further discovery is necessary but such efforts have been thwarted by genetic heterogeneity. Reducing heterogeneity is key, and studying isolated human populations have helped in the endeavour. An alternative is to study dog pedigrees, since artificial selection has resulted in extreme homogeneity. Identifying the genetic predisposition to canine mammary tumours can translate to human discoveries – a strategy currently underutilized. To explore this potential, we reviewed published canine mammary tumour genetic studies and proposed benefits of next generation sequencing canine cohorts to facilitate moving beyond incremental advances.
Keywords: comparative oncology, canines, animal model, hereditary breast cancer, comparative genomics, heterogeneity, canine mammary tumours, gene discovery, next‐generation sequencing
Background
As a component of the One Health Initiative (One Health Initiative, 2016), comparative oncology capitalizes on the fact that naturally occurring tumours in companion animals are excellent models of human cancer and can accelerate genetic, pathological and pharmaceutical discoveries (Davis & Ostrander 2014). Cancer is the leading cause of death in aged dogs, and is often spontaneous and similar to human cancers in its clinical presentation and pathophysiology (Davis & Ostrander 2014; Liu et al. 2014; Schiffman & Breen 2015). As loved members of human families, dogs are generally kept until old age, and are second only to humans in the level of health care that they receive (Rowell et al. 2011). Furthermore, canines age five to eight times faster than humans, providing an expedited model of disease onset and progression (Rowell et al. 2011). These facts, considered in conjunction with the large population of pet dogs in the United States (about 70 million) (Schiffman & Breen 2015), as well as the number of concerned breeders, establish dogs as the best‐known and most attainable mammalian model of human cancers (Rowell et al. 2011; Ostrander & Franklin 2012; Davis & Ostrander 2014; Liu et al. 2014; Schiffman & Breen 2015).
Although scientists are still deciphering the exact date and aetiology of canine domestication from wolves (Vonholdt et al. 2010), it is well known that the creation of modern dog breeds is a relatively recent phenomenon that occurred approximately 200 years ago and represents a significant evolutionary bottleneck (Sutter & Ostrander 2004; Rivera & von Euler 2011; Rowell et al. 2011; Ostrander & Franklin 2012). The artificial selection of dog breeds is largely based on human preferences and current trends, which is likely to continue. Through generations of linebreeding and influence of the popular sire effect (breeding one champion stud dog widely), most dog breeds were established from a small number of founders. The genetic characteristics of those founders are therefore currently overrepresented in the breed population (Patterson 2000). These give each breed its distinctive morphologic and behavioural traits, as well as predispositions to genetic diseases (Ostrander & Franklin 2012). In 2000, 370 canine genetic disorders were recognized and >50% were breed‐specific (Patterson 2000). This makes sense because strict pedigree barriers prevent interbreeding, suggesting that each breed represents an isolated population with potentially its own unique mutations and resultant diseases (Patterson 2000; Melin et al. 2016).
Breed or kennel‐based studies represent the human familial approach to disease gene discovery with the additional resource of well‐documented and large pedigrees, making them extremely attractive for genetic linkage studies. Generally, obtaining large, informative and properly ascertained human families is difficult, and to achieve the necessary statistical strength, families must often be grouped together for analysis (Chandler et al. 2016). This is not an ideal approach for genetically heterogeneous disorders, especially in outbred human populations. Thus, similar to studying humans from geographically isolated or founder populations, studying dog breeds provides a strategy to reduce genetic heterogeneity, since ancestral mutations occur at higher frequencies and contribute towards breed‐specific diseases (Ostrander & Franklin 2012). Highlighting the power of canine linkage analysis, in 2000, a single dog pedigree stemming from a German Shepherd sire that had litters with 6 different females was studied to identify the genetic locus of RCND (renal cystadenocarcinoma and nodular dermatofibrosis), a rare inherited cancer syndrome (Jonasdottir et al. 2000). A year later, the equivalent human cancer syndrome, Birt‐Hogg‐Dube syndrome, was mapped (Schmidt et al. 2001), and identification of the orthologous disease gene in both species immediately followed (Nickerson et al. 2002; Lingaas et al. 2003). Since that time, genetic mapping of canine disorders has contributed to major medical advancements. A short list of human disease genes that were first mapped in dogs includes those for narcolepsy, copper toxicosis, neuronal ceroid lipofuscinosis and ichthyosis (Ostrander & Franklin 2012).
In 2005, the first version of the canine genome was published (Lindblad‐Toh et al. 2005), which truly set the stage for comparative genomics. Since that time, both genome‐wide association studies (GWAS) and whole exome sequencing (WES) studies have been carried out to identify disease genes in canines that also explain the equivalent trait in humans, for example (Sloan et al. 2011; Grall et al. 2012). Compared to human GWAS studies, smaller dog cohorts can be studied (Davis & Ostrander 2014). It is possible that only 50 000 single nucleotide polymorphisms (SNPs) and 200 dogs would be needed to determine disease loci (Ostrander & Franklin 2012), compared to the thousands of cases and millions of SNPs needed in a human experiment (Michailidou et al. 2013). Not surprisingly, several medical research institutes have launched programs/projects promoting the use of purebred dogs as human cancer models, including two institutes of the National Institutes of Health (NIH), the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI), as well as the Broad Institute. The NCI developed the Comparative Oncology Program in 2003; the NHGRI has an internal research branch devoted to Cancer Genetics and Comparative Genomics; the Broad Institute initiated the Dog Disease Mapping Project. Furthermore, related extramural funding opportunities are available through the NIH as well as national breed registration organizations such as the American Kennel Club. With such current interest in this research area as well as the recent design and optimization of canine exome enrichment kits (Broeckx et al. 2015), even more translatable discoveries should unfold. This includes discoveries that enhance our knowledge of hereditary breast cancer (BC) genetics.
BC is prevalent in both humans and canines. It affects one in eight American women and, referred to as canine mammary tumours (CMTs) in the veterinary field, is the most common neoplasia in intact female dogs. In both species, identified risk factors include age, obesity, hormonal effects and genetics (Rivera & von Euler 2011; ACS, 2014; Melin et al. 2016). A true understanding of the genetic contributions to BC has yet to be fully grasped due to the heterogeneity of the disease and studied human cohorts, and its apparent polygenic inheritance (Chandler et al. 2016). The aims of this review are to argue for the use of the dog as a hereditary BC model and to summarize CMT genetic variant analyses reported to date. Regrettably, CMT genetic studies have been limited.
Canines as models of hereditary BC
Barriers to hereditary BC susceptibility gene discovery
Hereditary BC is characterized by a strong family history, early ages of onset (less than 45 years of age), bilateral presentations, affected males, as well as the appearance of other associated cancers, such as ovarian and prostate cancer, in the family (Berliner & Fay 2007). The genetic variant(s), or germline mutations, that segregate in hereditary BC families and increase risk of the disease, have an apparent autosomal dominant pattern of inheritance. Overall, genetic variants that increase risk of developing BC are divided into three broad groups of penetrance/risk, including high, moderate and low penetrant variants. Genes that harbour such variants are referred to as BC susceptibility genes (Chandler et al. 2016).
High and moderate risk variants are rare (normally defined as having a minor allele frequency (MAF) of less than 1%), and confer lifetime risks of over 50% and between 25 and 50%, respectively (Chandler et al. 2016). Over 35 hereditary BC susceptibility genes have been suggested to contain such risk variants, however, not all have sufficient statistical data confirming risk (Easton et al. 2015). A high or moderate penetrant variant that segregates in a family can be the major contributing allele that explains the increased familial risk, yet less than 30% of BC‐affected individuals with a personal or family history of the disease have such a variant in a currently reported BC susceptibility gene (Chandler et al. 2016). Two of these genes, BRCA1 (Miki et al. 1994) and BRCA2 (Wooster et al. 1995), were discovered in the 1990s and have been well‐documented as harbouring high and moderate risk variants. These are the most frequently mutated BC susceptibility genes to date, and the variants within them convey lifetime BC risks of 55–85% and 35–60%, respectively (Chandler et al. 2016). Together, BRCA1/2 mutations explain 15% of hereditary BC cases (Shiovitz & Korde 2015). Although extensively studied, in total, these findings leave over 70% of hereditary BC cases genetically unsolved.
Low penetrant BC variants are generally common (have a MAF >1%, which are referred to as SNPs) and, individually, only increase risk of disease by approximately 1.5‐fold compared to the average American women (Chandler et al. 2016). Over 70 BC‐associated SNPs have been reported, mainly identified through GWAS (Michailidou et al. 2013); and recently, researchers have tried to quantify the contributions of multiple low penetrant variants, theorizing that these could additively contribute to BC risk. Polygenic risk scores (PRS) were calculated and could explain up to a 3‐fold increased risk (for women in the highest percentile of PRS); however, women diagnosed with BC under the age of 40 years and/or with a family history of the disease were in the lowest PRS percentile (Mavaddat et al. 2015). This suggests that more rare and penetrant genetic variants likely explain the occurrence of hereditary BCs.
Since 2011, a number of WES studies have been carried out to identify additional rare variants that increase risk of hereditary BC (Chandler et al. 2016). WES is a next generation sequencing (NGS) approach (Shendure & Ji 2008) that targets the exome (all the coding regions in the genome) and was initially reported to aid in disease gene discovery efforts in 2010 (Ng et al. 2010a, 2010b). The hereditary BC WES studies that produced the most statistically significant data involved studying isolated/founder human populations (Chandler et al. 2016), suggesting that reducing heterogeneity is key to BC susceptibility gene discovery.
CMTs as a genetic model of human BCs
CMTs represent very practical models for human BCs since they share clinical, molecular, histological and epidemiological characteristics (Rivera & von Euler 2011; Visan et al. 2016). For instance in both species, females are primarily affected, but some male cases have been reported. Additionally, both species often develop mammary tumours as they age, with the average age of incidence hovering at about 10 years for most breeds (Rivera et al. 2009). Furthermore, hormonal influence is another commonality between BC and CMTs, and veterinarians and clinical researchers have extensively recorded the benefit of ovariohysterectomy (spaying) of female dogs to prevent CMT development (Egenvall et al. 2005; Jitpean & Hagman 2012; Liu et al. 2014). Ultimately, similar to human BCs, CMTs take a variety of histopathological forms, and about 50% of cases are malignant (Rivera et al. 2009; ACS, 2014; Melin et al. 2016). This, however, also highlights the importance of noting CMT and BC differences. For example, unlike in humans, it is common for CMTs to occur at multiple sites in an affected dog with varying histology within and between the different tumours. Plus, certain histological characteristics, such as myoepithelial cell proliferation, occur more often in CMTs than human BC (Visan et al. 2016). It is important to note that all animal models of disease present differences from the human condition, but, overall, the spontaneous and heterogeneous nature of CMTs best mimic human BC risk and development making the domestic dog a valuable genetic model of human BC.
Unlike for other forms of cancer, researchers do not agree upon which breeds have the greatest mammary cancer susceptibility or prevalence, as geographic and breed popularity contributions are confounding, and sources that report primary data are few (Sutter & Ostrander 2004; Egenvall et al. 2005; Borge et al. 2011, 2013; Jitpean & Hagman 2012; Davis & Ostrander 2014). One suspected “high‐risk” breed mentioned by multiple studies is the English Springer Spaniel (ESS) from Sweden. Average age of CMT onset in the ESS breed is approximately 7 years, mirroring the early‐onset observed in human familial BC cases (Rivera et al. 2009; Borge et al. 2013; Melin et al. 2016). While this breed may present an attractive model, it should be noted that spaying of dogs is uncommon in Sweden (Egenvall et al. 2005; Jitpean & Hagman 2012), so care must be taken to discern genetic from hormonal stimuli.
The genetics of CMTs is understudied, despite the fact that genetic analysis of CMTs began twenty years ago (Szabo et al. 1996; Van Leeuwen et al. 1996). It has long been documented that high‐risk BC susceptibility genes are well conserved between humans and dogs. In 1996, (Szabo et al. 1996) categorized known human BRCA1 missense mutations, and sequenced dog and mouse DNA (complementary and/or genomic) to investigate homology between species. Several of the mutations were located at shared amino acid residues across species, and a handful of those were also within conserved domains. Notably, BRCA1 sequence homology data was supportive of the use of a dog as a comparative genomics model: the researchers reported an 84% dog‐human nucleotide similarity versus 72% mouse‐human, and 73.8% dog‐human amino acid identity versus 55.9% mouse‐human identity (Szabo et al. 1996). The amino and carboxyl termini of the BRCA1 protein were highly conserved among all three species, especially including the zinc finger motif. The central portion of the BRCA1 protein is the most divergent among species, but closer in dog‐human (85% identity) than in mouse‐human (74%) (Szabo et al. 1996). Such conservation and similarity of these gene and protein segments is indicative of functional significance in both species.
CMT genetic studies – detecting germline risk variants
With the introduction of NGS, it is now well documented that tumours are tremendously genetically diverse regarding acquired (somatic) mutations (Russnes et al. 2011). Identifying such mutations is a hot area of research that began over two decades ago in both humans (Hollstein et al. 1994) and canines (Devilee et al. 1994). In fact, the initial focus of CMT genetic research involved identifying somatic mutations (Van Leeuwen et al. 1996), and a recent WES study has begun to advance our knowledge in this area (Liu et al. 2014). Regarding hereditary cancers, discovering the germline (inherited) genetic variants that drive cancer development before somatic mutations accumulate is an area that needs research focus. However, to date, just over a handful of publications have focused on identifying inherited genetic risk factors of CMT.
The first germline mutation associated with CMT was reported in the p53 tumour suppressor gene, which has long been classified as an important cancer catalyst in humans (Veldhoen et al. 1999). Matched normal and cancerous mammary tissues were selected from a cohort of 10 dogs, and a wealth of clinical information was available (age, breed, intact/spayed status, tumour histopathology and veterinarian's prognosis). One patient, a five‐year‐old Boxer, had two distinct germline mutations reported, each on a different parental chromosome. This included a large deletion of exons three through seven, and a P69L substitution in exon three – both predicted to be pathogenic (Table 1) (Veldhoen et al. 1999). These appear to be the only canine germline p53 mutations reported to date. It will be interesting to determine the true contribution of p53 mutations towards CMT genetics upon additional and larger sequencing studies.
Table 1.
Reported canine germline coding variants in orthologs of known or candidate BC susceptibility genes
| Gene | rs§ | Nomenclature | Reference | ||
|---|---|---|---|---|---|
| Genomic* | mRNA† | Protein‡ | |||
| BRCA1 | rs397510981 | chr9:g.19985060 | c.723A>G§ | p.K241K | (Enginler et al. 2014) |
| rs397512112 | chr9:g.19985075 | c.738T>A§ | p.T246T | (Enginler et al. 2014) | |
| rs397509570 | chr9:g.19988291 | c.3954G>A | p.S1318S | (Borge et al. 2011) | |
| BRCA2 | rs23250374 | chr25:g.7787056 | c.428A>G∥ | p.H143R | (Borge et al. 2011) |
| rs23244160 | chr25:g.7775050 | c.2401A>C | p.K801Q | (Borge et al. 2011) | |
| – | – | c.2414G>A*** | p.R805L | (Hsu et al. 2010) | |
| rs397511123 | chr25:g.7768691_7768693 | c.6918_6920delGTT | p.L2307del | (Borge et al. 2011; Enginler et al. 2014) | |
| rs23255542 | chr25:g.7768681 | c.6930C>T§ | p.F2310F | (Rowell et al. 2011; Enginler et al. 2014) | |
| rs397509895 | chr25:g.7747589 | c.9138A>G¶ | p.L3046L | (Enginler et al. 2014) | |
| – | chr25:g.7747332 | c.9308A>G | p.K3103R | (Borge et al. 2011; Enginler et al. 2014) | |
| rs397510884 | chr25:g.7735440 | c.9968G>A | p.S3323N | (Enginler et al. 2014) | |
| rs853007536 | chr25:g.7735654 | c.9995_9996insAAA** | p.M3332delinsIK | (Yoshikawa et al. 2005; Borge et al. 2011; Enginler et al. 2014) | |
| BRIP | rs397511741 | chr9:g.34983082 | c.3029G>A | p.R1010H | (Borge et al. 2011) |
| rs397512960 | chr9:g.34983223 | c.3170C>T | p.P1057L | (Borge et al. 2011) | |
| CDH1 | – | chr5:g.80784440_80784442 | c.387_389delCCA | p.H129del | (Borge et al. 2011) |
| rs397512866 | chr5:g.80776897 | c.945C>T | p.S315S | (Borge et al. 2011) | |
| EGFR | rs9206306 | chr18:g.5996046 | c.677G>A | p.R226Q | (Borge et al. 2011) |
| rs397513721 | chr18:g.5996076 | c.707C>T∥ | p.P236L | (Borge et al. 2011) | |
| HER2 (ERBB2) | rs397510212 | chr9:g.22773443 | c.1105A>G∥ | p.K369E | (Borge et al. 2011) |
| rs24616607 | chr9:g.22770524 | c.1575G>C | p.P525P | (Borge et al. 2011) | |
| rs24537329 | chr9:g.22770473 | c.1626A>G | p.E542E | (Borge et al. 2011) | |
| rs24537331 | chr9:g.22770288 | c.1728C>T | p.C576C | (Borge et al. 2011) | |
| rs397510076 | chr9:g.22766833 | c.1905G>A | p.A635A | (Borge et al. 2011) | |
| rs397512599 | chr9:g.22763063 | c.2769T>C | p.Y923Y | (Borge et al. 2011) | |
| rs397512289 | chr9:g.22761328 | c.3486G>A | p.P1162P | (Borge et al. 2011) | |
| rs397510013 | chr9:g.22761055 | c.3759C>T | p.Y1253Y | (Borge et al. 2011) | |
| ESR1 | rs21960513 | chr1:g.42131190 | c.627T>C | p.F209F | (Borge et al. 2011) |
| rs397512038 | chr1:g.42208686 | c.979A>G | p.I327V | (Borge et al. 2011) | |
| rs397512133 | chr1:g.42364093 | c.1578G>A | p.L526L | (Borge et al. 2011) | |
| PTEN | – | chr26:g.37910150 | c.975C>T | p.L325L | (Borge et al. 2011) |
| TP53 | – | chr5:g.32564669 | c.206C>T∥ | p.P69L | (Veldhoen et al. 1999) |
| – | chr5:g.32564760_32562912 | Germline deletion of exons 3‐7∥ | (Veldhoen et al. 1999) | ||
*Genome build: Broad CanFam3.1/canFam3 (Dog Assembly. Sept. 2011). † Nucleotide accession numbers: BRCA1: NM_001013416.1, BRCA2: NM_001006653.4, BRIP: XM_847556.4, CDH1: NM_001287125.1, EGFR: ENSCAFT00000005575.3, HER2: NM_001003217.2, ESR1: NM_001286958.1, PTEN: NM_001003192, TP53: NM_000546.5. ‡ Protein accession numbers: BRCA1: NP_001013434, BRCA2: NP_001006654, BRIP: ENSCAFT00000045493.2, CDH1: NP_001274054.1, EGFR: ENSCAFT00000005575.3, HER2: NP_001003217, ESR1: NP_001273887.1, PTEN: NP_001003192, TP53: NP_001003210. §variants found only in CMT‐affected dogs. ¶claimed to be associated with CMT. ∥predicted to be pathogenic in respective papers. **variant was initially suspected pathogenic but is now considered neutral. ***variant is named as reported in Hsu et al.; due to lack of information presented, locating this variant within NM_001006653 and Broad CanFam3.1/canFam3 was not possible.
In the mid‐late 1990s and early 2000s, the canine sequences of BRCA1 (Szabo et al. 1996) and BRCA2 (Bignell et al. 1997; Ochiai et al. 2001) were generated to obtain a reference sequence that would aid in the analysis of CMT susceptibility. A number of variations were reported, but their identification in normal canine mammary or other non‐tumoral tissues(Szabo et al. 1996; Bignell et al. 1997; Ochiai et al. 2001) left no connection to disease. Benign and pathogenic variants exist (Richards et al. 2015), and well‐designed studies are required to associate variants with a disease. The first presumed canine BRCA2 cancer‐risk variant was reported in 2005 by Yoshikawa et al. (2005) (p.M3332IK; Table 1 and Fig. 1), the same group that previously published the complete canine BRCA2 sequence (Ochiai et al. 2001; Yoshikawa et al. 2005). However, this study involved the analysis of blood‐extracted DNA samples from 21 tumour‐free dogs with no clinical or breed data made available. Furthermore, the insertion was detected in 17 of the 21 dogs; 10 were homozygous and seven were heterozygous (Yoshikawa et al. 2005). The authors claimed that the variant altered a predicted nuclear localization segment within the C‐terminus of the canine BRCA2 protein, and noted that the same sequence in the human BRCA2 protein harboured two missense variants (p.I3312V and p.I3312M; Fig. 1). Noteworthy, these two human variants are reported to be of unknown significance in the Breast Cancer Information Core (BIC) database. Since BRCA2 was already known to interact with Rad51 at the extreme C‐terminus and predicted to play a role in the handover of Rad51 to DNA substrates, hybridization assays were carried out to note interaction differences with and without the variant. Ultimately, the presence of p.M3332IK reported a slightly stronger BRCA2/Rad51 interaction, which could disturb the transfer of Rad51 to its substrate, leading to the authors' pathogenicity prediction (Yoshikawa et al. 2005). Despite the fact that this variant has been reported in additional sequencing studies that aimed to identify germline CMT‐risk variants (Table 1), no associations could be claimed (Borge et al. 2011; Enginler et al. 2014); in fact, a subsequent paper by Yoshikawa et al. (2012) reported the variant to be neutral.
Figure 1.
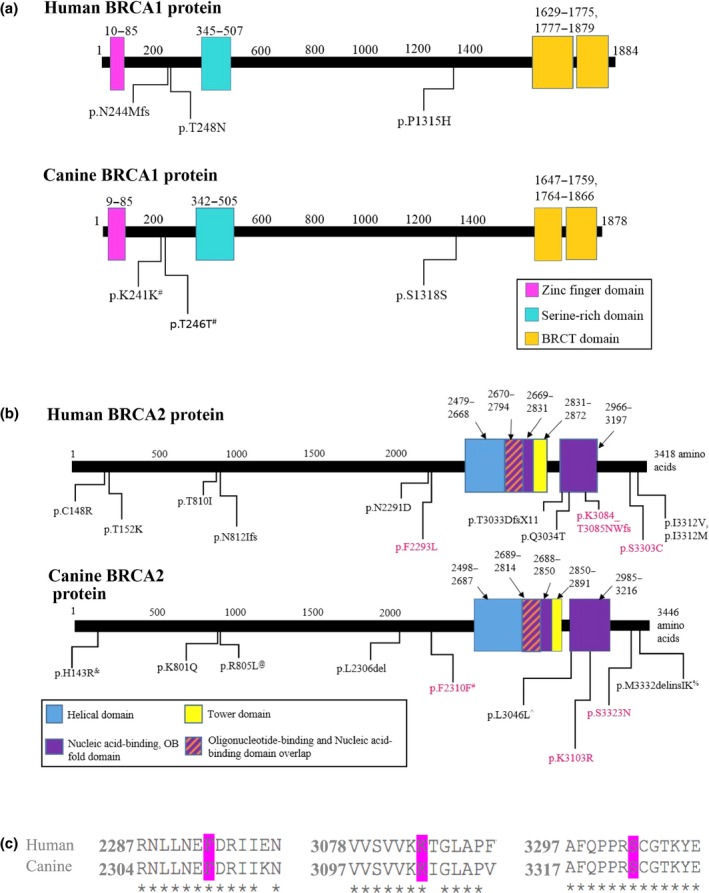
Illustrations of human and canine BRCA1 and BRCA2 proteins. All known canine coding variants in BRCA1 (Panel A) and BRCA2 (Panel B) proteins are noted on the diagram. Known human variants reported in Breast Cancer Information Core (BIC) within 2 amino acids of the conserved position of a canine variant were noted as well; the conserved human residues and locations were determined through a protein alignment. Hot pink text indicates conserved canine and human amino acid residues with variants; see Panel C for amino acid alignment. #variants found only in CMT‐affected dogs;^claimed to be associated with CMT; &predicted to be pathogenic in respective papers; and %initially suspected pathogenic but is now considered neutral, and @named as reported in Hsu et al. due to lack of information presented, which limited locating variant within NM_001006653 and Broad CanFam3.1/canFam3.
Human protein accession numbers: BRCA1: NP_009231, BRCA2: NP_000050; Canine protein accession numbers: BRCA1: NP_001013434, BRCA2: NP_001006654
Rivera et al. (2009) were the first to report an association between CMT and variants in BRCA1 and BRCA2. They studied ESSs from Sweden, where 36% of the population is affected, by carrying out a case–control study involving 212 CMT cases and 143 CMT‐unaffected controls. Ten canine orthologs of genes either known or predicted to increase risk to human BC were selected for study, and four to nine common SNPs were selected per gene, totalizing 63 genotyped SNPs. Overall, statistically significant associations were detected for one variant in BRCA2 and two in BRCA1. Interestingly, odds ratios of ~4 were calculated for both genes. Thus, the authors suggested that a common CMT‐predisposing allele exists in both BRCA1 and BRCA2 in this ESS population (Rivera et al. 2009). The associated SNPs were intronic or appeared non‐functional, so the quest to identify the exact risk variants remains open.
Several Sanger sequencing studies that aimed to detect germline CMT‐risk variants have been carried out. Firstly, it should be noted that in 2010 a study by Hsu et al. (2010) sequenced exon 11 of canine BRCA2 using DNA extracted from 11 CMTs and four randomly collected normal mammary tissues. The main objective was to identify genetic variants in the CMT samples associated with different tumour histological type and prognosis. Since matched normal mammary tissues were not sequenced in parallel, this approach prevented the true classification of the CMT reported variants as somatic or germline. However, two assumed somatic variants were identified as hot spots that may have prognostic potential since they were detected in the majority of the studied CMTs, and specifically, in all dogs with stage V mammary carcinosarcomas and rapid disease progression. Interestingly, one of those two hot spot alleles, an arginine at amino acid position 805 (p.R805, corresponding to a guanine at mRNA position 2414), is reported in two canine BRCA2 reference sequences (AB043895 and Z75664). The authors did report p.R805L (c.2414 G>A) in the normal mammary tissues that were sequenced as a reference in their study, which differed from the previously reported reference sequences, presumably classifying it as a germline BRCA2 variation (Table 1). Determining whether a germline allele at this position is associated with CMT requires further investigation.
In 2011, Borge et al. sequenced eleven genes (BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EGFR, ESR1, HER2, PTEN, STK11, and TP53) in 32 dogs from eight separate breeds (Borge et al. 2011). The breeds were evenly divided into “high risk” (Boxer, Cocker Spaniel, English Springer Spaniel, and Standard Poodle) and “low risk” (Bernese Mountain Dog, Cavalier King Charles Spaniel, Shetland Sheepdog and St. Bernard) groups to simulate a case–control study. Blood for DNA extraction was obtained from four randomly selected dogs from each breed and the authors did not have knowledge of clinical CMT status. Twenty‐five coding variants were reported; this included nine non‐synonymous, 13 synonymous, two deletions and one insertion (Table 1) (Borge et al. 2011). No statistically significant allele frequency differences were recorded between high and low risk breeds. However, three of the detected non‐synonymous variants were predicted to be damaging, BRCA2 p.H143R, EGFR p.P236L and HER2 p.K369E (Table 1; Fig. 1). The deletions (one each in BRCA2 and CDH1) and insertion (in BRCA2) affected 3 bps each (Table 1; Fig. 1); thus, no frameshifting mutations were identified. The BRCA2 insertion was the same as reported by Yoshikawa and colleagues (Table 1; Fig. 1) (Yoshikawa et al. 2005; Borge et al. 2011). Overall, the authors provided the first comprehensive list of coding variants in cancer‐associated genes and highlighted potentially pathogenic variants that they suggested are likely associated with CMT. Subsequently, the same group carried out a follow‐up case–control genotyping study that aimed to further investigate those probable associations (Borge et al. 2013). Common SNPs within all the genes listed above and sequenced in the previous study (Borge et al. 2011), minus TP53, which was previously investigated by Rivera et al. (2009) in a similar study, were genotyped in a case–control cohort of ESS and in a second group of dogs that were either at high or low risk of CMT. Ultimately, several SNPs of significance within the ESR1 gene were identified (Borge et al. 2013). Although broad in scope, these efforts are marred by the assumption that breeds have conferred differing propensities for CMTs, a yet‐to‐be‐proven assertion.
Confirmed CMT susceptibility genes BRCA1 and BRCA2 (Fig. 1) had thus far returned goading variant profiles. Thus, the next case–control study surveyed female dogs (25 with and 10 without a CMT diagnosis) from a variety of breeds for genetic differences specifically in BRCA1 and BRCA2 (Enginler et al. 2014). Clinical information such as age, age at spaying or intact status, tumour histopathology, body weight and breed were all noted. Using DNA extracted from the blood of affected and unaffected cohorts, selected regions of BRCA1 and BRCA2 were sequenced, and BRCA2 p.L3046L was shown to be significantly associated with CMTs (Table 1; Fig. 1). Several other variants were found only in the CMT‐affected dogs and not in controls, but were not statistically significant. These included BRCA1 p.K241K and p.T246T, and BRCA2 p.F2310F (Table 1; Fig. 1) (Enginler et al. 2014); the latter was also identified in a previous study (Borge et al. 2011). Additional investigation of these variants and application of a similar but larger experimental design could bolster future efforts.
The most recent CMT genetics publication that aimed to identify inherited CMT‐risk factors described the first CMT GWAS (Melin et al. 2016). The study cohort was comprised of only ESS dogs, but interestingly, dogs from Swedish as well as Norwegian and British populations were examined. Blood and buccal swab samples were acquired from client‐owned dogs at veterinary clinics over several years, along with pertinent clinical data. A total of 332 ESSs (188 cases and 144 controls) were genotyped for over 130 000 SNPs. Ultimately, genome wide significance was obtained for one SNP on chromosome 11; seven other SNPs on chromosomes 11 and 27 were nominally associated, identifying three potential CMT‐risk loci (one on chromosome 11 and two on chromosome 27) (Melin et al. 2016). Further analysis identified an associated haplotype on chromosome 11 that encompassed CDK5RAP2, which encodes a cyclin‐dependent kinase involved in cell cycle regulation. Moreover, LACRT and SLC38A4, which encode a glycoprotein involved in tear secretion and an amino acid transporter, respectively, were suggested candidate genes for the two chromosome 27 loci.
Overall, pitfalls of the current CMT genetic studies include sequencing small segments of already‐known high‐penetrant BC genes, and using cohorts where CMT‐affection status of the sequenced individuals and their predecessors remains unknown. Furthermore, when on a quest to identify breed‐specific risk variants, focusing on only one specific breed (ESS) or, on the complete opposite end of the spectrum, designing studies with small cohorts of multiple breeds, is not efficient. The currently published studies have specifically associated variants in BRCA1, BRCA2 (Rivera et al. 2009; Enginler et al. 2014), and ESR1 (Borge et al. 2013) with CMT risk. Drawing from known BC susceptibility and general DNA repair/maintenance genes, a host of potential CMT genes‐ BRIP, CDH1, ERBB2 (also called HER2), PTEN, STK11 and TP53‐ are suspected (Borge et al. 2011), but confirmatory case–control experiments have not been conducted. This assumption provides a springboard for CMT‐variant discovery, but should not limit future and more unbiased BC/CMT gene discovery investigation (Chandler et al. 2016).
Future directions
With little known about the complete list of genes responsible for both BC and CMTs, applications of NGS to canine cohorts seems like a logical step to facilitate novel susceptibility gene discovery. Although breed predispositions are currently unclear, a simple experimental design utilizing cases within a single breed and maximally unrelated controls from the same breed could be implemented to ascertain probable predispositions. An in depth pedigree analysis would aid in the success of this approach and identify a probable CMT inheritance pattern. Overall, this method would reduce sample size to a manageable, yet statistically significant, number (Davis & Ostrander 2014; Broeckx et al. 2016). Applying NGS technologies towards cohorts such as these would both confirm suggested associations of previously identified coding variants and unearth novel variants involved in the disease, including variants in regulatory elements. While WES is currently the most attractive and attainable NGS method due to cost and other constraints, analysing the entire canine genome through whole genome sequencing (WGS) is worthwhile since disease‐predisposing variants are not necessarily solely located in the coding DNA (Davis & Ostrander 2014; Melin et al. 2016). During these studies, ascertainment of clinical data including age, sex and tumour type are invaluable and could be individually tested for association with newly identified CMT‐risk variants.
Conclusions
Heterogeneity of the human genome has allowed our species to fight and prevail against a suite of illnesses throughout the ages, but it poses an unshakable obstacle in our advances against cancer. Embracing the use of dogs and their simplified genetic structure will likely help us overcome this barrier. Whether the end goal is to improve the health of the human or of the canine patient, in this new era of personalized medicine and comparative genomics, it is remiss to ignore the resources provided by the history of dog breeding, the sequencing of the canine genome, and latest WES and WGS technologies.
Declarations
Consent for publication
Not applicable.
Availability of data and material
Breast Cancer Information Core (https://research.nhgri.nih.gov/bic/)
dbSNP (http://www.ncbi.nlm.nih.gov/snp)
Dog Disease Mapping Project (http://www.broadinstitute.org/what-is-broad/areas-focus/project-spotlight/dog-disease-mapping-project-dogdna-studying-domesticated-do)
ExPASy Bioinformatics Resource Portal SIM Protein Alignment Tool (http://web.expasy.org/sim/).
Sources of funding
2016 Veterinary Summer Scholars Program at Auburn University, College of Veterinary Medicine (fellowship awarded to KG); American Association of Colleges of Pharmacy (AACP) New Investigator Award (2016), AURIC Seed Grant (2015–2016), AURIC Seed Grant (2016–2017), Auburn University Competitive Outreach Scholarship Grant (2016), Auburn University Innovative Research grant through the Internal Grant Program (2016–2018) (to NDM).
Conflict of interest
The authors declare that they have no conflicts of interests.
Ethics statement
No ethical approval was required as it is a review article.
Authors' contributions
NDM provided background information and was key in directing literature review efforts. NDM also contributed to the manuscript outline and revisions. KG compiled the variant data and wrote the first draft of the manuscript. Figures and tables were products of a collaborative effort from both NDM and KG. All authors read and approved the final manuscript.
Acknowledgements
The authors wish to acknowledge Erin Bigili and Stephanie Spina for their editing contributions. KG would specifically like to thank the Auburn University College of Veterinary Medicine, Merial, and other supporters of the 2016 Veterinary Summer Scholars Program for funding her research experience, during which time she contributed to this manuscript.
References
- ACS . (2014). American Cancer Society: Breast Cancer Facts and Figures 2013‐2014.
- Berliner J.L., Fay A.M. (2007) Risk assessment and genetic counseling for hereditary breast and ovarian cancer: recommendations of the National Society of Genetic Counselors. Journal of Genetic Counseling 16, 241–260. Epub 2007/05/18. Doi: 10.1007/s10897‐007‐9090‐7. [DOI] [PubMed] [Google Scholar]
- Bignell G., Micklem G., Stratton M.R., Ashworth A. & Wooster R. (1997) The BRC repeats are conserved in mammalian BRCA2 proteins. Human Molecular Genetics 6, 53–58. [DOI] [PubMed] [Google Scholar]
- Borge K.S., Borresen‐Dale A.L. & Lingaas F. (2011) Identification of genetic variation in 11 candidate genes of canine mammary tumour. Veterinary and Comparative Oncology 9, 241–250. Doi: 10.1111/j.1476‐5829.2010.00250.x. [DOI] [PubMed] [Google Scholar]
- Borge K.S., Melin M., Rivera P., Thoresen S.I., Webster M.T., von Euler H. et al (2013) The ESR1 gene is associated with risk for canine mammary tumours. BMC Veterinary Research 9, 69 Doi: 10.1186/1746-6148-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeckx B.J., Hitte C., Coopman F., Verhoeven G.E., De Keulenaer S., De Meester E. et al (2015) Improved canine exome designs, featuring ncRNAs and increased coverage of protein coding genes. Scientific Reports 5, 12810 Doi:10.1038/srep12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeckx B.J., Coopman F., Verhoeven G.E., De Keulenaer S., De Meester E., Bavegems V. et al (2016) Toward the most ideal case‐control design with related and unrelated dogs in whole‐exome sequencing studies. Animal Genetics 47, 200–207. Doi: 10.1111/age.12400. [DOI] [PubMed] [Google Scholar]
- Chandler M.R., Bilgili E.P. & Merner N.D. (2016) A Review of Whole Exome Sequencing Efforts Toward Hereditary Breast Cancer Susceptibility Gene Discovery. Human Mutation. Doi: 10.1002/humu.23017. [DOI] [PubMed] [Google Scholar]
- Davis B.W. & Ostrander E.A. (2014) Domestic dogs and cancer research: a breed‐based genomics approach. ILAR Journal 55, 59–68. Doi: 10.1093/ilar/ilu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilee P., Van Leeuwen I.S., Voesten A., Rutteman G.R., Vos J.H. & Cornelisse C.J. (1994) The canine p53 gene is subject to somatic mutations in thypoid carcinoma. Anticancer Research 14, 2039–2046. [PubMed] [Google Scholar]
- Easton D.F., Pharoah P.D., Antoniou A.C., Tischkowitz M., Tavtigian S.V., Nathanson K.L. et al (2015) Gene‐panel sequencing and the prediction of breast‐cancer risk. New England Journal of Medicine 372, 2243–2257. Doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egenvall A., Bonnett B.N., Ohagen P., Olson P., Hedhammar A. & von Euler H. (2005) Incidence of and survival after mammary tumors in a population of over 80,000 insured female dogs in Sweden from 1995 to 2002. Preventive Veterinary Medicine 69, 109–127. Doi: 10.1016/j.prevetmed.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Enginler S.O., Akis I., Toydemir T.S., Oztabak K., Haktanir D., Gunduz M.C. et al (2014) Genetic variations of BRCA1 and BRCA2 genes in dogs with mammary tumours. Veterinary Research Communications 38, 21–27. Doi: 10.1007/s11259‐013‐9577‐7. [DOI] [PubMed] [Google Scholar]
- Grall A., Guaguere E., Planchais S., Grond S., Bourrat E., Hausser I. et al (2012) PNPLA1 mutations cause autosomal recessive congenital ichthyosis in golden retriever dogs and humans. Nature Genetics 44, 140–147. Doi: 10.1038/ng.1056. [DOI] [PubMed] [Google Scholar]
- Hollstein M., Rice K., Greenblatt M.S., Soussi T., Fuchs R., Sorlie T. et al (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Research 22, 3551–3555. [PMC free article] [PubMed] [Google Scholar]
- Hsu W.L., Huang Y.H., Chang T.J., Wong M.L. & Chang S.C. (2010) Single nucleotide variation in exon 11 of canine BRCA2 in healthy and cancerous mammary tissue. The Veterinary Journal 184, 351–356. Doi: 10.1016/j.tvjl.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Jitpean S. & Hagman R. (2012) Strom Holst B, Hoglund OV, Pettersson A, Egenvall A. Breed variations in the incidence of pyometra and mammary tumours in Swedish dogs. Reproduction in Domestic Animals 47(Suppl 6), 347–350. Doi: 10.1111/rda.12103. [DOI] [PubMed] [Google Scholar]
- Jonasdottir T.J., Mellersh C.S., Moe L., Heggebo R., Gamlem H., Ostrander E.A. et al (2000). Genetic mapping of a naturally occurring hereditary renal cancer syndrome in dogs. Proceedings of the National Academy of Sciences USA 97, 4132–4137. Doi: 10.1073/pnas.070053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad‐Toh K., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M. et al (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438, 803–819. Doi:10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- Lingaas F., Comstock K.E., Kirkness E.F., Sorensen A., Aarskaug T., Hitte C. et al (2003) A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German Shepherd dog. Human Molecular Genetics 12, 3043–3053. Doi: 10.1093/hmg/ddg336. [DOI] [PubMed] [Google Scholar]
- Liu D., Xiong H., Ellis A.E., Northrup N.C., Rodriguez C.O. Jr, O'Regan R.M. et al (2014) Molecular homology and difference between spontaneous canine mammary cancer and human breast cancer. Cancer Research 74, 5045–5056. Doi: 10.1158/0008-5472.CAN-14-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavaddat N., Pharoah P.D., Michailidou K., Tyrer J., Brook M.N., Bolla M.K. et al (2015) Prediction of breast cancer risk based on profiling with common genetic variants. Journal of the National Cancer Institute 107, pii: djv036 Doi: 10.1093/jnci/djv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin M., Rivera P., Arendt M., Elvers I., Muren E., Gustafson U. et al (2016) Genome‐Wide Analysis Identifies Germ‐Line Risk Factors Associated with Canine Mammary Tumours. PLoS Genetics 12, e1006029 Doi: 10.1371/journal.pgen.1006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou K., Hall P., Gonzalez‐Neira A., Ghoussaini M., Dennis J., Milne R.L. et al (2013) Large‐scale genotyping identifies 41 new loci associated with breast cancer risk. Nature Genetics 45, 353–361, 61e1–2. Doi: 10.1038/ng.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y., Swensen J., Shattuck‐Eidens D., Futreal P.A., Harshman K., Tavtigian S. et al (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266, 66–71. [DOI] [PubMed] [Google Scholar]
- Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M. et al (2010a) Exome sequencing identifies the cause of a mendelian disorder. Nature Genetics 42, 30–35. Doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I. et al (2010b) Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nature Genetics 42, 790–793. Doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson M.L., Warren M.B., Toro J.R., Matrosova V., Glenn G., Turner M.L. et al (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt‐Hogg‐Dube syndrome. Cancer Cell 2, 157–164. [DOI] [PubMed] [Google Scholar]
- Ochiai K., Morimatsu M., Tomizawa N. & Syuto B. (2001) Cloning and sequencing full length of canine Brca2 and Rad51 cDNA. Journal of Veterinary Medical Science 63, 1103–1108. [DOI] [PubMed] [Google Scholar]
- One Health Initiative . (2016). Available at: http://www.onehealthinitiative.com/
- Ostrander E.A. & Franklin H. (2012) Epstein Lecture. Both ends of the leash–the human links to good dogs with bad genes. New England Journal of Medicine 367, 636–646. Doi: 10.1056/NEJMra1204453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson D.F. (2000) Companion animal medicine in the age of medical genetics. Journal of Veterinary Internal Medicine 14, 1–9. [PubMed] [Google Scholar]
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier‐Foster J. et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 17, 405–424. Doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera P. & von Euler H. (2011) Molecular biological aspects on canine and human mammary tumors. Veterinary Pathology 48, 132–146. Doi: 10.1177/0300985810387939. [DOI] [PubMed] [Google Scholar]
- Rivera P., Melin M., Biagi T., Fall T., Haggstrom J., Lindblad‐Toh K. et al (2009) Mammary tumor development in dogs is associated with BRCA1 and BRCA2. Cancer Research 69, 8770–8774. Doi: 10.1158/0008‐5472.CAN‐09‐1725. [DOI] [PubMed] [Google Scholar]
- Rowell J.L., McCarthy D.O. & Alvarez C.E. (2011) Dog models of naturally occurring cancer. Trends in Molecular Medicine 17, 380–388. Doi: 10.1016/j.molmed.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russnes H.G., Navin N., Hicks J. & Borresen‐Dale A.L. (2011) Insight into the heterogeneity of breast cancer through next‐generation sequencing. The Journal of Clinical Investigation 121, 3810–3818. Doi: 10.1172/JCI57088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman J.D., Breen M. (2015) Comparative oncology: what dogs and other species can teach us about humans with cancer. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences 370, pii: 20140231 Doi: 10.1098/rstb.2014.0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt L.S., Warren M.B., Nickerson M.L., Weirich G., Matrosova V., Toro J.R. et al (2001) Birt‐Hogg‐Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. American Journal of Human Genetics 69, 876–882. Doi: 10.1086/323744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J., Ji H. (2008) Next‐generation DNA sequencing. Nature Biotechnology 26, 1135–1145. Doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Shiovitz S. & Korde L.A. (2015) Genetics of breast cancer: a topic in evolution. Annals of Oncology. Doi: 10.1093/annonc/mdv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan J.L., Johnston J.J., Manoli I., Chandler R.J., Krause C., Carrillo‐Carrasco N. et al (2011) Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nature Genetics 43, 883–886. Doi: 10.1038/ng.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutter N.B. & Ostrander E.A. (2004) Dog star rising: the canine genetic system. Nature Reviews Genetics 5, 900–910. Doi: 10.1038/nrg1492. [DOI] [PubMed] [Google Scholar]
- Szabo C.I., Wagner L.A., Francisco L.V., Roach J.C., Argonza R., King M.C. et al (1996) Human, canine and murine BRCA1 genes: sequence comparison among species. Human Molecular Genetics 5, 1289–1298. [DOI] [PubMed] [Google Scholar]
- Van Leeuwen I.S., Hellmen E., Cornelisse C.J., Van den Burgh B. & Rutteman G.R. (1996) P53 mutations in mammary tumor cell lines and corresponding tumor tissues in the dog. Anticancer Research 16, 3737–3744. [PubMed] [Google Scholar]
- Veldhoen N., Watterson J., Brash M., Milner J. (1999) Identification of tumour‐associated and germ line p53 mutations in canine mammary cancer. British Journal of Cancer 81, 409–415. Doi: 10.1038/sj.bjc.6690709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visan S., Balacescu O., Berindan‐Neagoe I. & Catoi C. (2016) In vitro comparative models for canine and human breast cancers. Clujul Medical 89, 38–49. Doi: 10.15386/cjmed-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonholdt B.M., Pollinger J.P., Lohmueller K.E., Han E., Parker H.G., Quignon P. et al (2010) Genome‐wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464, 898–902. Doi: 10.1038/nature08837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooster R., Bignell G., Lancaster J., Swift S., Seal S., Mangion J. et al (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378, 789–792. Doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- Yoshikawa Y., Morimatsu M., Ochiai K., Nagano M., Yamane Y., Tomizawa N. et al (2005) Insertion/deletion polymorphism in the BRCA2 nuclear localization signal. Biomedical Research 26, 109–116. [DOI] [PubMed] [Google Scholar]
- Yoshikawa Y., Morimatsu M., Ochiai K., Okuda K., Taoda T., Chikazawa S. et al (2012) Establishment of a PCR analysis method for canine BRCA2. BMC Research Notes 5, 173 Doi: 10.1186/1756-0500-5-173. [DOI] [PMC free article] [PubMed] [Google Scholar]