

Abstract
Cobalt‐porphyrin‐catalysed intramolecular ring‐closing C−H bond amination enables direct synthesis of various N‐heterocycles from aliphatic azides. Pyrrolidines, oxazolidines, imidazolidines, isoindolines and tetrahydroisoquinoline can be obtained in good to excellent yields in a single reaction step with an air‐ and moisture‐stable catalyst. Kinetic studies of the reaction in combination with DFT calculations reveal a metallo‐radical‐type mechanism involving rate‐limiting azide activation to form the key cobalt(III)‐nitrene radical intermediate. A subsequent low barrier intramolecular hydrogen‐atom transfer from a benzylic C−H bond to the nitrene‐radical intermediate followed by a radical rebound step leads to formation of the desired N‐heterocyclic ring products. Kinetic isotope competition experiments are in agreement with a radical‐type C−H bond‐activation step (intramolecular KIE=7), which occurs after the rate‐limiting azide activation step. The use of di‐tert‐butyldicarbonate (Boc2O) significantly enhances the reaction rate by preventing competitive binding of the formed amine product. Under these conditions, the reaction shows clean first‐order kinetics in both the [catalyst] and the [azide substrate], and is zero‐order in [Boc2O]. Modest enantioselectivities (29–46 % ee in the temperature range of 100–80 °C) could be achieved in the ring closure of (4‐azidobutyl)benzene using a new chiral cobalt‐porphyrin catalyst equipped with four (1S)‐(−)‐camphanic‐ester groups.
Keywords: C−H activation, cobalt porphyrin, nitrogen heterocycles, radical reactions, ring-closing reactions
Introduction
Organic azides are interesting nitrene precursors for direct C−H amination reactions, producing linear or cyclic amines and only N2 as a harmless side product.1, 2 For example, Warren and co‐workers reported on the Cu‐catalyzed activation of adamantyl azide, followed by intermolecular nitrene insertion into several different C−H bonds.3 In general, however, most reported C−H amination examples involve activation of electronically pre‐activated and/or bulky, sterically protected tertiary azide substrates, such as tBuN3, adamantly azide, aryl azides (ArN3), phosphoryl azides [(RO)2(O)PN3], sulfonyl azides (RSO2N3) or carbonazidates (RO(CO)N3) as the nitrene‐precursors.1, 2, 3, 4, 5, 6, 7, 8 Intramolecular C−H amination reactions involving organic azides constitute an interesting protocol for the preparation of N‐heterocyclic compounds, but to date, most of the reported protocols are limited to the use of electronically pre‐activated azides.9, 10, 11, 12 More recently, and most relevant to the investigations described in this paper, some interesting examples involving intramolecular amination of C−H bonds using primary aliphatic organic azides (RCH2N3) as the “nitrene“ source were reported (see Figure 1).13, 14, 15
Figure 1.

Comparison of previously reported catalysts and those used in the present study for intramolecular ring‐closing C−H bond amination taking (4‐azidobutyl)benzene as a benchmark.
Although these examples provide interesting lead‐reactivity for the synthesis of saturated heterocycles (in particular the system reported by Betley and co‐workers13a), there are still a number of hurdles to overcome; (1) most of the previously reported catalysts for this reaction are highly sensitive to air and water and require relatively high catalyst loadings, thus resulting in low catalytic turnover numbers (TONs);13, 14, 15 (2) several of the reported catalysts produce significant amounts of (Boc‐protected) linear amines as undesired side products;13, 14, 15 (3) asymmetric versions of intramolecular ring‐closing C−H bond amination reactions of unactivated aliphatic azides are unknown; and (4) relatively limited mechanistic information for these types of reactions is available to guide scientists in finding solutions to the above challenges, thus hampering the design and development of more efficient catalysts. Cobalt porphyrin systems have been reported for related C−H functionalization protocols. The reported mechanistic proposals for these types of reactions are quite interesting. Based on spectroscopic studies and supporting DFT calculations, activation of electronically pre‐activated organic azides [i.e., CH3C6H4SO2N3 (TsN3), NO2C6H4SO2N3 (NsN3), CCl3CH2O(C=O)N3 (TrocN3)] by cobalt(II)‐porphyrin catalysts is believed to produce unique cobalt(III)‐nitrene‐radical intermediates (CoIII‐N‐Y), which subsequently react with C−H bonds in a hydrogen‐atom transfer (HAT) radical rebound process.16 Related nitrene‐radical intermediates (albeit with more complicated electronic structures) have been proposed as intermediates in the Fe‐ and Pd‐catalysed formation of N‐heterocycles from (4‐azidobutyl)benzene.13, 14, 15 However, kinetic and/or spectroscopic evidence to unambiguously support these mechanistic proposals is thus far lacking. Furthermore, the mechanism of activation of primary aliphatic azides might differ from the catalytic pathways followed by more activated organic azides such as NsN3 and TrocN3 (also for cobalt‐based catalysts).
Alternative resting states and rate‐limiting steps may be operational, and the C−H activation steps could either involve an HAT‐rebound mechanism, direct nitrene insertion into the C−H bond or even both at the same time.13a Moreover, as the activation of aliphatic azides typically requires elevated reaction temperatures, even the involvement of free nitrene intermediates in the C−H bond‐amination steps cannot be excluded.17 Hence, the mechanistic features previously disclosed for reactions involving activated azides cannot be directly translated to those involving much less reactive aliphatic azides. As such, we became interested in the activation of primary aliphatic azides by low‐spin, metallo‐radical cobalt(II) catalysts, which have never been reported, to the best of our knowledge. Here, we disclose and discuss the cobalt(II)‐porphyrin‐catalysed ring‐closing C−H bond amination of primary aliphatic azides, focusing in particular on the mechanism, scope and prospect of these reactions (see Figure 1).
The catalysts reported here are air and water stable, and cleanly produce cyclic (Boc‐protected) amines in good yields and with higher TONs than reported for other catalysts. Additionally, detailed mechanistic information was gathered from reaction progress kinetics, kinetic isotope competition experiments and supporting DFT studies, all of which strongly support the involvement of aliphatic CoIII‐N.‐R species as key intermediates in these reactions. Furthermore, as a proof of principle, we present the first example of an enantioselective C−H amination reaction of the type shown in Figure 1, strongly suggesting that the C−H bond activation and C−N bond formation steps of the overall catalytic ring‐closing reaction occur in the coordination sphere of cobalt. No free nitrene intermediates seem to be involved in these reactions.
Results and Discussion
We started our evaluation of the catalytic activity of cobalt(II)‐porphyrin complexes under various conditions by monitoring the conversion of azide 5 a into pyrrolidine 5 b (Table 1) with the commercially available cobalt(II)‐tetraphenylporphyrin complex 1 ([Co(TPP)], Figure 2). In the absence of an amine trapping agent, complete recovery of the starting material was observed after 16 hours at 100 °C using 1 mol % of the catalyst (Table 1, entry 1). Some conversion could, however, be observed when high catalyst loadings were used (see the Supporting Information). In the presence of di‐tert‐butyldicarbonate (Boc2O), we observed the desired Boc‐protected cyclic amine product 5 b (tert‐butyl 2‐phenylpyrrolidine‐1‐carboxylate) by 1H NMR spectroscopy. The product was isolated in 17 % yield (Table 1, entry 2, TON=17) after 16 hours of reaction. We were pleased to see that under these conditions the TONs obtained with [Co(TPP)] catalyst 1 in the conversion of substrate 5 a to product 5 b are already higher than those of most of the recently reported homogeneous catalysts for this reaction.13, 14, 15
Table 1.
Catalyst evaluation in intramolecular ring‐closing C−H bond amination of azide 5 a.[a]

| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Loading [mol %] | Conversion [%] | Yield [%] | TON |
| 1[d] | [Co(TPP)] | 1 | –[b] | –[b] | n.d.[c] |
| 2 | [Co(TPP)] | 1 | 24 | 17 | 17 |
| 3 | [Co(TPF20P)] | 1 | <5 | n.d.[c] | n.d.[c] |
| 4 | [Co(TMP)] | 1 | 38 | 36 | 35 |
| 5[e] | [Co(TMP)] | 1 | 39 | 32 | 30 |
| 6[f] | [Co(TMP)] | 1 | 30 | n.d.[c] | 30 |
| 7[g] | [Co(Salophen)] | 1 | –[b] | –[b] | n.d.[c] |
| 8 | AIBN | 4 | –[b] | –[b] | n.d.[c] |
| 9 | [Co(TMP)] | 2 | 57 | 54 | 27 |
| 10 | [Co(TMP)] | 4 | >95 | 89 | 21 |
[a] Conditions: Substrate 5 (0.3 mmol), catalyst (1–4 mol %), Boc2O (0.36 mmol) and solvent (3.0 mL) were mixed and reacted for 16 h at 100 °C. After the reaction only starting material and expected products were observed. Conversions are based on 1H NMR analysis of the ratio between starting materials and products. Yields are of isolated material. [b] No product was detected by 1H NMR. [c] Not determined. [d] No Boc2O was used in the reaction. [e] Benzene was used as the solvent. [f] 3 % v/v water was present in the solvent. [g] Salophen=N,N′′‐bis‐(3,5‐di‐tert‐butyl‐salicylidene)‐1,2‐phenylenediamine).
Figure 2.

Achiral cobalt(II) complexes used in this study: cobalt(II) tetraphenylporphyrin ([Co(TPP)], 1), cobalt(II) pentafluorophenylporphyrin ([Co(TPF20P)], 2), cobalt(II) tetramesitylporphyrin ([Co(TMP)], 3) and cobalt(II) salophen ([Co(salophen], 4).
We continued our optimisation by changing the catalyst; with cobalt(II)‐tetra(pentafluorophenyl)‐porphyrin ([Co(TPF20P)] complex 2, Figure 2) as the catalyst, only trace amounts of the desired product 5 b (<5 %) were obtained (Table 1, entry 3). [Co(salophen)] complex 4 (Figure 2)18 proved to be completely inactive (entry 7). In contrast, marked improvements both in yield and TON were observed using the electron‐rich tetra(2,4,6‐trimethylphenyl)‐porphyrin complex 3 ([Co(TMP)], Figure 2) as the catalyst. The desired product 5 b was obtained in 36 % isolated yield using 1 mol % of catalyst (TON=35; Table 1, entry 4) at 38 % conversion of the substrate. No significant change in the isolated product yield was observed upon changing the solvent to benzene (entry 5). To our surprise, the presence of up to 3 % v/v of water in the solvent mixture did not result in a significant decrease in activity (entry 6), which is quite remarkable as previously reported catalysts are all very sensitive to the presence of even trace amounts of water.
To verify whether the cobalt‐porphyrin complex is an active component in the catalysis or merely initiating a metal‐free radical reaction, we replaced cobalt(II) by azobis(isobutyronitrile) (AIBN, entry 8). However, use of this well‐known radical initiator did not lead to any product formation. At the low cobalt catalyst loadings used so far, incomplete conversion of the azide 5 a was observed after 16 hours, which nicely allows for proper comparison of the TONs of the different catalysts (Table 1, entries 1–7). To obtain synthetically useful yields of the desired product 5 b within the same reaction time, however, higher catalyst loadings were used. With 2 and 4 mol % of [Co(TMP)] catalyst 3, the product yield increased to 54 % and 89 %, respectively (Table 1, entries 9 and 10). Importantly, none of these reactions produced any significant amounts of undesired linear (Boc‐protected) amine products previously observed for related systems.13, 15
Next, we explored the substrate scope by examining the synthesis of oxazolidines and imidazolidines using the above [Co(por)]‐catalyzed ring‐closing C−H bond‐amination protocol. By introducing an oxygen atom in the aliphatic chain at the alpha‐position to the benzylic C−H (Table 2, entry 1, 6 a), the oxazolidine product was obtained in high isolated yield (6 b, 93 %; TON=23). Introduction of two methyl groups on the aliphatic chain was expected to further improve the efficiency of the cyclisation reaction (by virtue of the Thorpe–Ingold effect19). However, this modification led to a significant decrease in both the yield and TON for formation of oxazolidine product 7 b (Table 2, entry 2). Steric hindrance introduced by the two methyl groups apparently has a larger effect on the reactivity than pre‐organisation for cyclisation when using substrate 7 a. The reaction did however proceed cleanly, as only product 7 b and starting material 7 a were observed upon analysis of the crude reaction mixture. Substitution of oxygen for an NH‐functionality in the aliphatic chain resulted in a small decrease in the product yield using [Co(TMP)] complex 3 as the catalyst (69 % yield, TON=17; entry 3). In this reaction, [Co(TPP)] complex 1 produced imidazolidine product 8 b in a comparable yield (67 %, TON=17). In all reactions involving a secondary NH‐group in the substrate (entries 3–10), we used 2.4 equiv of Boc2O to protect both nitrogen atoms.
Table 2.
Catalytic formation of oxazolidines and imidazolidines.[a]
| Entry | Substrate | Product | Yield [%] | TON |
|---|---|---|---|---|
| 1 |

|

|
93 | 23 |
| 6 a | 6 b | |||
| 2 |

|

|
32 | 8 |
| 7 a | 7 b | |||
| 3 |

|

|
69[b] 67[c] | 17[b] 17[c] |
| 8 a | 8 b | |||
| 4 |
|
|
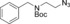
69[b] | 16[b] |
| 8 c | 8 b | |||

|

|
|||
| 5[b,c] 6[b,c] 7[b,c] 8[b,c] 9[b,c] 10[b,c] | R=Me, 9 a R=Ph, 10 a R=F, 11 a R=Cl, 12 a R=Br, 13 a R=OMe, 14 a | R=Me, 9 b R=Ph, 10 b R=F, 11 b R=Cl, 12 b R=Br, 13 b R=OMe, 14 b | 96 91 84 87 95 82 | 24 22 21 22 24 20 |
[a] Conditions: Substrate (0.3 mmol), Boc2O (1.2 equivalent), [Co(TMP)] (4 mol %), and toluene (3.0 mL) were added and reacted for 16 h at 100 °C. At the end of the reaction only starting material and products were observed. [b] 2.4 equiv of Boc2O were added. Yields are of isolated material. In entries 3 and 5–10, full conversion of starting material was always observed. The only observed side product was the corresponding azide with a Boc‐protected secondary nitrogen (compound 8 c and compounds S6‐S11, see the Supporting Information). [c] Commercially available [Co(TPP)] 1 was used as the catalyst.
The decrease in yield could be due to the increased bulk of the substrate after Boc‐protection of the secondary amine. This also explains why the difference in activity between the two catalysts 1 and 3 is small for this substrate. When substrate 8 a was treated with Boc2O to yield substrate 8 c prior to the cyclisation reaction, no change in the product yield was observed as compared to the one‐pot procedure (Table 2, entry 4). We further decided to investigate the tolerance of the reaction for substitution on the phenyl ring of the amine‐containing substrates (Table 2, entries 5–10). Because of the small difference in activity between [Co(TPP)] (1) and [Co(TMP)] (3) for substrate 8 a, the commercially available [Co(TPP)] catalyst 1 was used for substrates 9 a–14 a. In these cases, we also used 2.4 equiv of Boc2O in the reaction (Table 2, entries 5–10) to prevent product inhibition. Using 4 mol % catalyst loading, high yields (up to 96 %) were obtained for all substrates containing a substituent in the para‐position of the phenyl ring. The effect of electron‐donating and electron‐withdrawing substituents is marginal, which is in agreement with the kinetic and computational studies described below, showing that azide activation is rate‐limiting. At the end of the reaction, we always observed full conversion of the starting material for substrates 8 a–14 a. The only observed side product in these reactions was the corresponding Boc‐protected azide (compound 8 c and compounds S6–S11, see the Supporting Information), again without formation of any significant amounts of undesired linear (Boc‐protected) amines from the azide substrates.13, 15
To further explore the scope of the cobalt‐porphyrin catalyst for the preparation of N‐saturated heterocycles from alkyl‐azide fragments, we examined the azide‐containing disubstituted arenes 15 a–17 a to obtain bicyclic isoindoline and tetrahydroisoquinoline derivatives (Scheme 1). The presence of the phenyl ring resulted in a decrease in the selectivity because isoindoline product 15 b was obtained in only 27 % yield. However, full conversion of the substrate was observed. The protected linear amine product 15 c (35 %) and some ill‐defined oligomeric/polymeric side products were observed (see the Supporting Information). It seems that if the desired ring‐closing step requires breaking of a stronger C−H bond [higher bond dissociation energy (BDE)], the reaction is partially driven to intermolecular HAT reactions (most likely from the alpha position of another azide substrate), producing undesired side products. This is consistent with the formation of oligomeric/polymeric products, linear Boc‐protected amines as well as nitriles from substrates containing even stronger C−H bonds (see the Supporting Information).
Scheme 1.

Application of the [Co(por)]‐mediated intramolecular C−H bond‐amination ring‐closing protocol for the synthesis of alternative heterocycles 15 b–18 b.
It is worth mentioning that substrate 16 a also reacts selectively to form the N‐heterocylic ring product 16 b, which supports a correlation between the BDE of the reacting C−H bond and the ease and selectivity of the ring‐closing step. Note that replacing the methyl group in 15 a by an ethyl group (substrate 16 a) lowers the BDE of the reacting C−H bond. Full conversion was observed after 16 h using 2 mol % catalyst loading to yield the desired product 16 b in 93 % isolated yield (TON=45). Apparently, the increased stabilisation of the benzylic radical is sufficient to avoid the competitive oligomerisation/polymerisation reactions observed for substrate 15 a. Finally, a six‐membered heterocycle 17 b was found to be accessible using substrate 17 a, albeit in only 28 % yield (TON=7). As observed for substrate 15 a, but in contrast to all other reactions described above, 37 % of the corresponding linear Boc‐protected amine product 17 c and some ill‐defined oligomers/polymers were obtained as side products in this case. In addition, activation of the allylic C−H bond of substrate 18 a yields the six‐membered heterocycle product 18 b (38 %, TON=10). Other compounds detected in the reaction mixture were the starting material 18 a (37 %), linear product 18 c (12 %) and small amounts of unidentified products. Additional substrates used in the C−H amination protocol, varying the BDE of the reacting C−H bond, are described in the Supporting Information (Table S1). The results obtained with these substrates indicate that intermolecular HAT starts to compete with intramolecular HAT when the barrier for C−H bond activation at the targeted position for ring‐closing amination becomes too strong (i.e., higher BDEs of the targeted C−H bond and/or formation of six‐membered rings instead of five‐membered rings). Substrates with weaker benzylic C−H bonds at the delta position react selectively to form five‐membered N‐heterocylic rings without any indication for competing intermolecular HAT reactions (see Tables 1 and 2).
Mechanistic investigations
Kinetic studies
The mechanism of the intramolecular ring‐closing C−H bond‐amination reaction was explored experimentally by kinetic analysis of the reaction progress monitoring the conversion of (4‐azidobutyl)benzene substrate 5 a into tert‐butyl 2‐phenylpyrrolidine‐1‐carboxylate product 5 b, using [Co(TMP)] catalyst 3 in the presence of Boc2O. Following the reaction in time (see Supporting Information) revealed clean first‐order kinetics in both the [substrate] and the [catalyst], but zero‐order kinetics in [Boc2O]. No substrate saturation effects were detected over a broad concentration range of azide 5 a. This translates into the following empirical rate equation, with second‐order rate constant k=12 m −1 h−1 [Eq. (1)]:
| (1) |
To exclude the alternative possibility of a rate‐limiting C−H bond‐activation step, we performed an inter‐ and intramolecular kinetic isotope competition experiment using the bis‐deuterated (d 2‐5 a) and mono‐deuterated (d 1‐5 a) analogues of (4‐azidobutyl)benzene substrate 5 a (see Scheme 2). In the intermolecular competition experiment (Scheme 2, top), no kinetic isotope effect (KIE=1) was observed, thus showing that the C−H bond‐activation step occurs after the rate‐limiting azide activation step.20 The intramolecular kinetic isotope competition experiment (Scheme 2, bottom) does reveal a substantial kinetic isotope effect (KIE=7), showing that the C−H activation step is in itself not barrierless (see also Scheme 3). The rather large intramolecular KIE is perhaps suggestive of some tunnelling contribution to the C−H activation step (see computational mechanistic studies), as can be expected for a radical‐type hydrogen‐atom transfer (HAT) process.21, 22 Hence, the kinetic data point to a pre‐equilibrium involving weak and reversible binding of azide 5 a to [Co(TMP)] catalyst 3, followed by rate‐limiting substrate activation involving dinitrogen loss from the coordinated azide. Subsequent C−H activation leading to C−N bond formation and ring closure is faster, but not barrierless (see also the proposed mechanism in Scheme 3).
Scheme 2.

Intermolecular kinetic isotope competition experiment between non‐deuterated (4‐azidobutyl)benzene substrate 5 a and its bis‐deuterated analogue d 2 ‐ 5 a (top) and intramolecular kinetic isotope competition experiment using mono‐deuterated substrate d 1‐5 a (bottom).
Scheme 3.

Proposed mechanism for [Co(por)]‐catalysed intramolecular C−H bond‐amination ring‐closing reaction of 5 a to 6 b and the corresponding DFT‐computed energies (BP86, def2‐TZVP, disp3).[a] Free energies (ΔG°373K in kcal mol−1), all energies (also TS1, TS2 and TS3) relative to A. [b] L=□=vacant site (5‐coordinate pathway). [c] L=5 c (6‐coordinate pathway).
The reaction rate was further evaluated at various temperatures (80–115 °C) to obtain the activation parameters from the Arrhenius and Eyring equations (see Supporting Information for details). This led to the following experimental values: E a=+18.7±2.3 kcal mol−1, ΔG ≠=+26.2±4.6 kcal mol−1, ΔH ≠=+18.0±2.3 kcal mol−1 and ΔS ≠=−22.0±6.2 cal mol−1 K−1. The rather large negative activation entropy (−22.0±6.2 e.u.) points to an ordered, associative transition state, suggestive of an uphill substrate binding event preceding rate‐limiting azide activation (see proposed mechanism in Scheme 3). When the reaction was performed in presence of TEMPO as a radical trap, a large decrease in the turnover number of catalyst 2 was observed (with 5 equiv TEMPO, TON=9; with 25 equiv TEMPO, TON=3), in good agreement with the proposed radical mechanism (Scheme 3).
Because product inhibition was observed in the reaction without Boc2O, we determined the association constant for 2‐phenylpyrrolidine (5 c) binding to the [Co(TMP)] catalyst 3. The equilibrium constant for the formation of five‐coordinate complex [Co(TMP)(5 c)] from [Co(TMP)] and 5 c at room temperature was found to be K 1=1300, whereas binding of a second molecule 5 c to [Co(TMP)(5 c)] to form six‐coordinate complex [Co(TMP)(5 c)2] has a very low equilibrium constant of K 2=80 (see Supporting Information). We were unable to determine the binding constant for (4‐azidobutyl)benzene coordination to 3 because the equilibrium is shifted towards the starting materials. This has previously also been observed for aromatic azide substrates.22 Assuming that substrate binding and activation requires the free four‐coordinate [Co(TMP)] catalyst, and thus competes with product binding, the considerable binding constant K 1 associated with formation of the product adduct [Co(TMP)(5 c)] should indeed result in a significant additional energy penalty to the rate‐limiting step (also at 373 K) when the reaction is performed in absence of Boc2O (see Scheme 3).
Computational mechanistic studies
The mechanism was further explored computationally (DFT, BP86, def2‐TZVP, disp3), using a simplified model of the catalyst without substituents at the porphyrin ring (Scheme 3). Based on the kinetic studies described above, we anticipated that the initial steps of the mechanism involve coordination of the aliphatic azide 5 a to cobalt(II), followed by dinitrogen loss to produce a nitrene‐radical intermediate. Rate‐limiting azide activation has also been proposed in the mechanisms for [Co(por)]‐catalysed C−H bond‐amination reactions using activated azides (carbonazidates [ROC(O)N3] or sulfonyl azides [RSO2N3]).5, 11, 16 This indeed is a plausible pathway according to the DFT calculations (Scheme 3; for more details see the Supporting Information). Azide coordination is slightly endergonic (+3.2 kcal mol−1) and subsequent loss of dinitrogen from intermediate B via TS1 to produce the key nitrene‐radical species C is also the rate‐ limiting step in the computed mechanism, which is in good agreement with the experimental kinetic studies (vide supra).
The computations further show a lower barrier for the rate‐limiting azide activation step along the five‐coordinate pathway (ΔG ≠=+24.5 kcal mol−1; L=□ in Scheme 3) than along the six‐coordinate pathway with an additional molecule of product 5 c bound to cobalt as an axial ligand (ΔG ≠=+28.5 kcal mol−1; L=5 c in Scheme 3). This is in agreement with the experimentally observed inhibiting effect of the unprotected amine product 5 c on the reaction rates of catalytic reactions performed in absence of Boc2O. The computed activation parameters for TS1 (ΔG ≠=+24.5 kcal mol−1, ΔH ≠=+16.3 kcal mol−1, ΔS ≠=−21.9 e.u.) are in excellent agreement with the experimental values determined from the Arrhenius and Eyring equations (E a=+18.7±2.3 kcal mol−1, ΔG ≠=+26.2±4.6 kcal mol−1, ΔH ≠=+18.0±2.3 kcal mol−1 and ΔS ≠=−22.0±6.2 e.u.). A spin density plot of five‐coordinate species C (Figure 3) shows 88 % spin population at the nitrene moiety and only 6 % at Co, thus confirming the nitrene‐radical nature of this key intermediate (see also Scheme 3).16 HAT from the benzylic position of the activated substrate to the nitrene‐radical moiety (TS2) to produce amido‐benzyl radical intermediate D and the subsequent radical rebound step (TS3) both have remarkably low barriers. From the relative barriers of hydrogen and deuterium abstraction in the HAT step, a KIE=3.4 was calculated. Such calculations neglect any tunnelling effects, as they take only differences in zero point energy (ZPE) into account, resulting from isotope exchange in the vibrational analysis.
Figure 3.

Spin‐density plot of intermediate C present in Scheme 3.
Although the predicted KIE calculated as such is in good qualitative agreement with the experiments, the experimental value is larger (KIE=7). This is suggestive of some tunnelling contribution to the C−H bond‐splitting process, as can be expected for a radical‐type HAT process.21, 22 In good agreement with the experimental data, dissociation of amine product 5 c from cobalt(II) species E is endergonic, thus resulting in product inhibition (i.e., slower reactions) in the absence of Boc2O (see the Supporting Information for more DFT details). Attack of Boc2O on radical intermediate D prior to radical rebound cannot be fully excluded on the basis of the available experimental data. However, in view of the very low computed barrier for the radical rebound step (<2 kcal mol−1), this pathway seems unlikely.
Additional catalytic experiments based on the mechanistic insights gathered
The mechanistic information gathered above called for some additional experimental studies. First of all, binding of unprotected product 5 c to the catalyst is substantial, but the magnitude of the experimentally determined binding constant (K 1=1300) suggests that the activation barrier of the rate‐limiting azide activation step in absence of Boc2O is raised only to a limited amount, and thus it should be possible to overcome this increased barrier at higher temperatures. As such, we argued that it might be possible to obtain the unprotected cyclic amine 5 c by conversion of substrate 5 a without Boc2O at elevated temperatures. Indeed, full conversion of substrate 5 a was observed when performing the catalytic reaction with 4 mol % of [Co(TMP)] catalyst 3 at 140 °C in chlorobenzene. As expected, the desired 2‐phenylpyrrolidine 5 c was indeed formed, albeit in a low yield (19 %, TON=5). Several undesired and unidentified side products were formed under these non‐optimised reaction conditions (see Supporting Information).
In marked contrast to the reaction performed at 140 °C in absence of Boc2O, the catalytic reactions (and kinetic studies) performed in the presence of Boc2O allowed for clean conversion of azide 5 a to the Boc‐protected cyclic amine 5 b at lower reaction temperatures (80–115 °C). Combined with clean first‐order kinetics in both the [catalyst] and the [azide substrate], the data suggest that catalyst deactivation processes are minor and slow. As such, we wondered if higher TONs would be attainable when performing the reaction at lower catalyst loading but, at the same time, at higher absolute concentration of both the substrate and the catalyst. Such conditions are expected to shift the pre‐equilibrium for substrate binding somewhat further in the direction of the coordinated complex B (Scheme 3), and simultaneously, may reduce any detrimental effects of impurities in the solvent. This indeed leads to higher TONs. Performing the reaction at a high substrate concentration of 0.44 m, using 1 mol % of [Co(TMP)] catalyst 3, led to formation of product 5 b in 58 % yield (TON=59) after 16 h, and the yield further increased to 73 % (TON=76) after 72 h of reaction. These are the highest TONs for the cyclisation of substrate 5 a reported to date.
Enantioselective intramolecular ring‐closing C−H bond amination
In the proposed reaction mechanism shown in Scheme 3, the substrate remains coordinated to the cobalt centre throughout the catalytic cycle. If correct, this should allow for enantioselective reactions when using a chiral cobalt porphyrin catalyst. We therefore performed the ring‐closing C−H bond‐amination reaction of substrate 5 a with a new chiral cobalt porphyrin catalyst equipped with four (1S)‐(−)‐camphanic‐ester substituents (19, Figure 4).
Figure 4.

Structure of (1S)‐(−)‐camphanic acid based chiral catalyst 19 used in the enantioselective ring‐closing C−H bond‐amination reaction of substrate 5 a with 4 mol% catalyst loading.
Synthesis and characterisation of the new catalyst 19 is described in the Supporting Information. The chiral catalyst 19 produces product 5 b in lower yields (33 % yield, TON=8, at 4 mol % catalyst loading) compared to [Co(TMP)] catalyst 3, but indeed allowed enantioselective product formation in 29 % enantiomeric excess (ee) at 100 °C (enantiomeric ratio 35:65). The ee value increased to 46 % (enantiomeric ratio 27:73) by lowering the reaction temperature to 80 °C, although at a somewhat reduced product yield (22 %, TON=6). Chirality transfer at these rather high reaction temperatures is quite remarkable, and the results demonstrate for the first time the feasibility of enantioselective radical‐type ring closure reactions from aliphatic azides using metallo‐radical catalysis (Figure 4).23
This clearly calls for additional screening of chiral catalysts for these types of reactions in follow‐up studies. Perhaps most importantly, the result strongly suggests that the reaction does not proceed through formation of free nitrenes in a metal‐free C−H bond‐activation pathway. This is an important observation, because in some Fe‐ and Ru‐catalysed nitrene‐transfer reactions, formation of free nitrenes (reacting outside the coordination sphere of the metal in an uncontrolled manner) has been observed.17 The observed chirality transfer thus holds important additional mechanistic information, and shows unambiguously that the C−H bond‐activation and C−N bond‐formation steps in the overall catalytic ring‐closing reaction occur within the coordination sphere of cobalt.
Conclusion
A cobalt‐catalysed ring‐closing C−H amination protocol was developed for the synthesis of a variety of saturated N‐heterocycles. The applied air‐ and moisture‐stable [Co(por)] catalysts give significantly higher turnover numbers (up to 76) than other reported homogeneous catalyst systems based on Fe and Pd for these type of reactions. The use of di‐tert‐butyldicarbonate (Boc2O) prevents competitive binding of the formed amine product, thereby significantly enhancing the reaction rate. Detailed kinetic studies, kinetic isotope competition experiments and supporting DFT calculations reveal a metallo‐radical‐type mechanism involving rate‐limiting azide activation to form the key cobalt(III)‐nitrene‐radical intermediate. Subsequent low barrier intramolecular HAT from a benzylic C−H bond to the nitrene‐radical intermediate followed by a radical rebound step leads to formation of the desired N‐heterocyclic ring products. Kinetic isotope competition experiments are in agreement with a radical‐type C−H bond‐activation step (intramolecular KIE=7), which occurs after rate‐limiting azide activation (intermolecular KIE=1). Enantioselective ring‐closing amination proved possible when using a new chiral cobalt‐porphyrin catalyst equipped with four (1S)‐(−)‐camphanic‐ester substituents in the second coordination sphere. Modest enantioselectivities (up to 46 % ee) were achieved in the ring closure of (4‐azidobutyl)benzene, despite the high reaction temperature used (80 °C). This demonstrates for the first time the feasibility of enantioselective radical‐type ring closure of aliphatic azides using metallo‐radical catalysis,23 and strongly suggests that the C−H bond‐activation and C−N bond‐formation steps of the overall catalytic ring‐closing reaction occur in the coordination sphere of cobalt. The involvement of free nitrenes in these reactions seems unlikely. The ring‐closing amination reactions described in this paper are rare examples of selective catalytic transformations proceeding through one‐electron substrate‐activation steps producing discrete metal‐bound nitrogen‐centred radical intermediates.24 We hope that the prospects and mechanistic information provided in this paper will guide others and stimulate further research in this field.
Experimental Section
General intramolecular C−H bond‐amination procedure: To a dry Schlenk flask was added catalyst (12 μmol), substrate (0.30 mmol) and di‐tert‐butyldicarbonate (0.36 mmol) in dry toluene (3.0 mL). The stirred reaction mixture was heated to 100 °C for 16 h. After cooling to room temperature, the crude mixture was purified by flash chromatography (SiO2, DCM/hexanes/TEA 50:50:1).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Ed Zuidinga is acknowledged for mass analysis of the obtained compounds. Financial support from NWO‐CW (VICI project 016.122.613), the ERC (Starting Grants 202886 & 279097) and the University of Amsterdam (RPA Sustainable Chemistry) is gratefully acknowledged.
P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek, B. de Bruin, Chem. Eur. J. 2017, 23, 7945.
References
- 1. Intrieri D., Zardi P., Casellia A., Gallo E., Chem. Commun. 2014, 50, 11440–11453. [DOI] [PubMed] [Google Scholar]
- 2. Lu H.-J., Zhang X. P., Chem. Soc. Rev. 2011, 40, 1899–1909. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Gephart R. T., Warren T. H., Organometallics 2012, 31, 7728–7752; [Google Scholar]
- 3b. Badiei Y. M., Dinescu A., Dai X., Palomino R. M., Heinemann F. W., Cundari T. R., Warren T. H., Angew. Chem. Int. Ed. 2008, 47, 9961–9964; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 10109–10112. [Google Scholar]
- 4.
- 4a. Fantauzzi S., Gallo E., Caselli A., Ragaini F., Casati N., Macchi P., Cenini S., Chem. Commun. 2009, 3952–3954; [DOI] [PubMed] [Google Scholar]
- 4b. Cenini S., Gallo E., Penoni A., Ragaini F., Tollari S., Chem. Commun. 2000, 2265–2266. [Google Scholar]
- 5.
- 5a. Jin L.-M., Lu H.-J., Cui Y., Lizardi C. L., Arzua T. N., Wojtas L., Cui X., Zhang X. P., Chem. Sci. 2014, 5, 2422–2427; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Lu H.-J., Subbarayan V., Tao J., Zhang X. P., Organometallics 2010, 29, 389–393. [Google Scholar]
- 6. Suárez J. R., Chiara J. L., Chem. Commun. 2013, 49, 9194–9196. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Shin K., Baek Y., Chang S., Angew. Chem. Int. Ed. 2013, 52, 8031–8036; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8189–8194; [Google Scholar]
- 7b. Park S. H., Kwak J., Shin K., Ryu J., Park Y., Chang S., J. Am. Chem. Soc. 2014, 136, 2492–2502; [DOI] [PubMed] [Google Scholar]
- 7c. Kim H. J., Ajitha M. J., Lee Y., Ryu J., Kim J., Lee Y., Jung Y., Chang S., J. Am. Chem. Soc. 2014, 136, 1132–1140. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Nishioka Y., Uchida T., Katsuki T., Angew. Chem. Int. Ed. 2013, 52, 1739–1742; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1783–1786; [Google Scholar]
- 8b. Omura K., Murakami M., Uchida T., Irie R., Katsuki T., Chem. Lett. 2003, 32, 354–355; [Google Scholar]
- 8c. Uchida T., Katsuki T., Chem. Rec. 2014, 14, 117–129. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Stokes B. J., Dong H., Leslie B. E., Pumphrey A. L., Driver T. G., J. Am. Chem. Soc. 2007, 129, 7500–7501; [DOI] [PubMed] [Google Scholar]
- 9b. Jones C., Nguyen Q., Driver T. G., Angew. Chem. Int. Ed. 2014, 53, 785–788; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 804–807; [Google Scholar]
- 9c. Nguyen Q., Nguyen T., Driver T. G., J. Am. Chem. Soc. 2013, 135, 620–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Intrieri D., Mariani M., Caselli A., Ragaini F., Gallo E., Chem. Eur. J. 2012, 18, 10487–10490. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Lu H., Li C., Jiang H., Lizardi C. L., Zhang X. P., Angew. Chem. Int. Ed. 2014, 53, 7028–7032; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7148–7152; [Google Scholar]
- 11b. Lu H.-J., Jiang H.-L., Hu Y., Wojtas L., Zhang X. P., Org. Lett. 2012, 14, 5158–5161; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Lu H.-J., Jiang H.-L., Hu Y., Wojtas L., Zhang X. P., Chem. Sci. 2011, 2, 2361–2366. [Google Scholar]
- 12. Singh R., Kolev J. N., Sutera P. A., Fasan R., ACS Catal. 2015, 5, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Hennessy E. T., Betley T. A., Science 2013, 340, 591–595; [DOI] [PubMed] [Google Scholar]
- 13b. Hennessy E. T., Liu R. Y., Iovan D. A., Duncan R. A., Betley T. A., Chem. Sci. 2014, 5, 1526–1532; [Google Scholar]
- 13c. Iovan D. A., Betley T. A., J. Am. Chem. Soc. 2016, 138, 1983–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Thacker N. C., Lin Z., Zhang T., Gilhula J. C., Abney C. W., Lin W., J. Am. Chem. Soc. 2016, 138, 3501–3509; [DOI] [PubMed] [Google Scholar]
- 14b. Villanueva O., Mace Weldy N., Blakey S. B., MacBeth C. E., Chem. Sci. 2015, 6, 6672–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Broere D. L. J., de Bruin B., Reek J. N. H., Lutz M., Dechert S., van der Vlugt J. I., J. Am. Chem. Soc. 2014, 136, 11574–11577; [DOI] [PubMed] [Google Scholar]
- 15b. Broere D. L. J., van Leest N. P., de Bruin B., Siegler M. A., van der Vlugt J. I., Inorg. Chem. 2016, 55, 8603–8611; [DOI] [PubMed] [Google Scholar]
- 15c.After this paper was accepted, we also disclosed a less sensitive Fe catalyst: Bagh B., Broere D. L. J., Sinha V., Kuijpers P. F., van Leest N. P., de Bruin B., Demeshko S., Siegler M. A., van der Vlugt J. I., J. Am. Chem. Soc. 2017, 139, 5117–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Goswami M., Lyaskovskyy V., Domingos S. R., Buma W. J., Woutersen S., Troeppner O., Ivanović-Burmazović I., Lu H., Cui X., Zhang X. P., Reijerse E. F., DeBeer S., van Schooneveld M. M., Pfaff F. F., Ray K., de Bruin B., J. Am. Chem. Soc. 2015, 137, 5468–5479; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Olivos Suarez A. I., Jiang H., Zhang X. P., de Bruin B., Dalton Trans. 2011, 40, 5697–5705; [DOI] [PubMed] [Google Scholar]
- 16c. Lyaskovskyy V., Olivos Suarez A. I., Lu H., Jiang H., Zhang X. P., de Bruin B., J. Am. Chem. Soc. 2011, 133, 12264–12273. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Mankad N. P., Müller P., Peters J. C., J. Am. Chem. Soc. 2010, 132, 4083–4085; [DOI] [PubMed] [Google Scholar]
- 17b. Takaoka A., Moret M.-E., Peters J. C., J. Am. Chem. Soc. 2012, 134, 6695–6706. [DOI] [PubMed] [Google Scholar]
- 18. Wöltinger J., Bäckvall J.-E., Zsigmond Á., Chem. Eur. J. 1999, 5, 1460–1467. [Google Scholar]
- 19. Beesley R. M., Ingold C. K., Thorpe J. F., J. Chem. Soc. Trans. 1915, 107, 1080–1106. [Google Scholar]
- 20. Simmons E. M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 3066–3072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3120–3126. [Google Scholar]
- 21.
- 21a. Wu A., Mayer J. M., J. Am. Chem. Soc. 2008, 130, 14745–14754; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21b. Gómez-Emeterio B. P., Urbano J., Mar Díaz-Requejo M., Pérez P. J., Organometallics 2008, 27, 4126–4130. [Google Scholar]
- 22. Ragaini F., Penoni A., Gallo E., Tollari S., Gotti C. L., Lapadula M., Mangioni E., Cenini S., Chem. Eur. J. 2003, 9, 249–259. [DOI] [PubMed] [Google Scholar]
- 23.Reviews on metallo-radical catalysis:
- 23a. Lyaskovskyy V., de Bruin B., ACS Catal. 2012, 2, 270–279; [Google Scholar]
- 23b. Jahn U., Top. Curr. Chem. 2011, 320, 191–322; [DOI] [PubMed] [Google Scholar]
- 23c. Clark A., Eur. J. Org. Chem. 2016, 2231–2243; [Google Scholar]
- 23d. Lu H.-J., Zhang X. P., Chem. Soc. Rev. 2011, 40, 1899–1909; see also ref. [2]. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Olivos Suarez A. I., Lyaskovskyy V., Reek J. N. H., van der Vlugt J. I., de Bruin B., Angew. Chem. Int. Ed. 2013, 52, 12510–12529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12740–12760; [Google Scholar]
- 24b. Xiong T., Zhang Q., Chem. Soc. Rev. 2016, 45, 3069–3087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary