

Abstract
Alkenyl‐to‐allyl 1,4‐rhodium(I) migration enables the generation of nucleophilic allylrhodium(I) species by remote C−H activation. This new mode of reactivity was employed in the diastereoselective reaction of arylboron reagents with substrates containing a 1,3‐enyne tethered to a ketone, to give products containing three contiguous stereocenters. The products can be obtained in high enantioselectivities using a chiral sulfur‐alkene ligand.
Keywords: allylic compounds, cyclization, isomerization, reaction mechanisms, rhodium
Catalytic C−H functionalizations have revolutionized chemical synthesis by providing powerful new tools for bond construction.1 However, a critical objective for the advancement of this field is its application to a more diverse range of transformations. Nucleophilic allylations2 are important reactions that could benefit from C−H functionalization principles. Most typically, these processes have employed allylmetal(loid) reagents such as allyltin, allylboron, or allylsilicon compounds.2 The generation of nucleophilic allylmetal species by the activation of allylic C−H bonds would bypass the need to prepare such reagents and potentially increase efficiency by streamlining synthetic sequences. This strategy would also be a valuable complement to nucleophilic allylations involving migratory insertions of allenes,3, 4 the use of simple π‐unsaturated compounds in hydrogenative or redox‐triggered additions,5, 6 hetero‐ene reactions,7 and Prins reactions.8
Although generating electrophilic allylmetal species by allylic C−H activation is well‐known,9, 10 there is, to our knowledge, limited precedent for corresponding processes that provide nucleophilic allylmetals.11 Very recently, the groups of Schneider,11a Kanai,11b and Mita and Sato11c described the formation and trapping of nucleophilic allylmetal species from simple hydrocarbons. In view of the nucleophilic character of allylrhodium(I) species,4a, 12 we envisaged that activation of a remote C−H bond by 1,4‐rhodium(I) migration12d, 13, 14 could also achieve this goal. Specifically, rhodium(I)‐catalyzed reaction of an arylboron reagent with the alkyne of a 1,3‐enyne would provide the alkenylrhodium species A (Scheme 1). This intermediate could then undergo a 1,4‐rhodium(I) shift to the cis‐allylic substituent to give the allylrhodium(I) species B, which could be trapped by an electrophile. This approach was expected to be challenging, given that there is only very limited precedent for rhodium(I) to migrate to C(sp3) centers.12d, 14k,14m Nevertheless, the generation of electrophilic allylrhodium(III) species by a similar strategy in our rhodium(III)‐catalyzed oxidative annulations of 1,3‐enynes provided some encouragement.10 Herein, we describe the implementation of this strategy in arylative intramolecular allylations of ketones to give stereochemically complex fused bicycles with high diastereoselectivities. Preliminary results of enantioselective reactions are also provided.
Scheme 1.
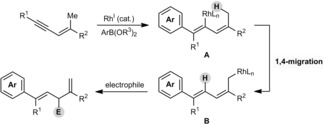
Proposed alkenyl‐to‐allyl 1,4‐rhodium(I) migration.
This study began with the reaction of the enynone 1 a with 3,5‐dimethylphenyl pinacol boronate (1.3 equiv), [{Rh(cod)Cl}2] (1.5 mol %), and K3PO4 (0.3 equiv) at 65 °C for 16 hours in various solvents (Table 1). A 3,5‐disubstituted arylboron reagent was used to minimize 1,4‐rhodium(I) migration onto the aryl group as described previously,15 as it is well‐known that migration onto an aryl ring ortho to a substituent is unfavorable.14a,14i, 15a Pinacol boronates were used because 3,5‐disubstituted variants are easily accessed through iridium‐catalyzed C−H borylation.16 The reaction conducted in THF/MeOH (10:1) gave diastereomeric bicycles 2 aa 17 and 2 ab 18 in a 13:87 ratio (entry 1). After purification, 2 aa and 2 ab were isolated in 11 and 46 % yield, respectively. Traces of the diketone 3 a were also formed, and resulted from arylrhodation of the alkyne of 1 a with the regioselectivity opposite to that seen in the formation of 2 aa/2 ab, followed by a cyclization‐fragmentation pathway.15a, 19 Notably, switching the solvent to MeCN/MeOH (10:1) reversed the sense of diastereoselectivity and gave 2 aa and 2 ab in 66 and 5 % yield, respectively (entry 2). Using TBME/tBuCN/MeOH (10:1.2:1) gave a further increase in diastereoselectivity (entry 3).
Table 1.
Evaluation of solvents.[a]
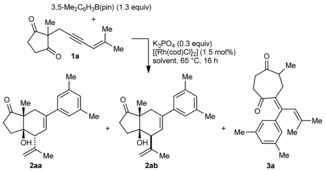
| Entry | Solvent | d.r. | Yield [%][c] | ||
|---|---|---|---|---|---|
| 2 aa/2 ab [b] | 2 aa | 2 ab | 3 a | ||
| 1 | THF/MeOH (10:1) | 17:83 | 11 | 46 | n.d.[d] |
| 2 | MeCN/MeOH (10:1) | 91:9 | 66 | 5 | 9 |
| 3 | TBME/tBuCN/MeOH (10:1.2:1) | 94:6 | 73 | 4 | 14 |
[a] Reactions employed 0.50 mmol of 1 a. [b] Determined by 1H NMR analysis of the crude reaction mixtures. [c] Yield of the isolated product. [d] n.d.=not determined. cod=1,5‐cyclooctadiene, TBME=tert‐butyl methyl ether, THF=tetrahydrofuran.
In the proposed catalytic cycle (Scheme 2), transmetalation of the arylboronate with the rhodium methoxide 4 provides the arylrhodium species 5, which undergoes migratory insertion with the alkyne of 1 a to give alkenylrhodium species 6. 1,4‐Rhodium migration gives the allylrhodium species (Z)‐7, which cyclizes onto a ketone to provide the rhodium alkoxide 8. Methanolysis of 8 liberates the product 2 aa or 2 ab and regenerates 4.
Scheme 2.

Proposed catalytic cycle.
Scheme 3 presents the reactions of 1 a with various arylboronic acid pinacol esters. Products analogous to 3 a were generally formed in up to 20 % yield (by 1H NMR analysis of the crude reaction mixtures) but were not isolated. The reaction is tolerant of halide (2 ba, 2 ea, and 2 ha), methoxy (2 ca and 2 fa), trifluoromethyl (2 da), and carbomethoxy groups (2 ea) on the arylboronate. In addition, 3,5‐disubstituted (2 aa–2 ea), 3,4,5‐trisubstituted (2 fa), and 2,5‐disubstituted arylboronates (2 ga and 2 ha) are tolerated. 2,5‐Disubstituted arylboronates gave lower yields (2 ga and 2 ha), which is presumably a consequence of the steric hindrance of the ortho‐substituent. Finally, a heteroarylboronate is also tolerated (2 ia).
Scheme 3.
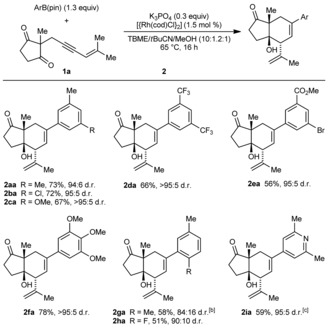
[a] Reaction of 1 a with various arylboronates. Reactions employed 0.50 mmol of 1 a. Diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixtures. Yields are of isolated, diastereomerically pure products. [b] Reaction performed with 2.5 mol % [{Rh(cod)Cl}2]. [c] Reaction employed 0.45 mmol of 1 a.
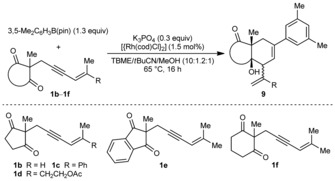
Next, variation of the enynone was explored, and the substrates 1 b–f, containing methyl groups cis to the alkyne, all reacted successfully with 3,5‐dimethylphenyl pinacol boronate (Table 2). Substrates containing hydrogen, phenyl, or alkyl groups trans to the alkyne are tolerated (entries 1–3). With the phenyl‐containing substrate 1 c, however, application of the standard reaction conditions gave no diastereoselectivity (1:1 d.r.).20 Fortunately, switching the solvent to 2‐MeTHF/MeOH (10:1) gave the syn,syn‐diastereomer 9 cb in greater than 95:5 d.r. and 62 % yield (entry 2).17 In contrast to our findings using rhodium(III) catalysis,10 substrates containing methylene groups (as opposed to methyl groups) cis to the alkyne are unreactive. Variation of the 1,3‐diketone is also possible. For example, the indane‐1,3‐dione 1 e gave 9 ea in 74 % yield and >95:5 d.r. (entry 4).17 Under the standard reaction conditions, the six‐membered cyclic 1,3‐diketone 1 f underwent decomposition in competition with arylative allylation. However, by changing the arylboronate to the more reactive neopentyl glycol ester, and using K2CO3 and tAmOH in place of K3PO4 and MeOH, respectively, 9 fa was formed in 67 % yield (entry 5).17 The process is not limited to cyclic 1,3‐diketones as the β‐ketoester 10 reacted smoothly using 2.5 mol % of [{Rh(cod)Cl}2] to give 11 in 62 % yield and 95:5 d.r. [Eq. 1].
Table 2.
Arylative allylation of various enynones.[a]
| Entry | Enynone | Product (Ar=3,5‐Me2C6H3) | d.r.[b] | Yield [%][c] | |
|---|---|---|---|---|---|
| 1[d] | 1 b |
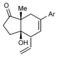
|
9 ba | n.d.[e] | 50 (+7)[f] |
| 2[g] | 1 c |
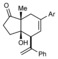
|
9 cb | >95:5 | 62 |
| 3 | 1 d |
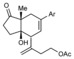
|
9 da | 84:16 | 52 |
| 4 | 1 e |

|
9 ea | >95:5 | 74 |
| 5[h] | 1 f |
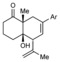
|
9 fa | 84:16 | 67 |
[a] Reactions employed 0.50 mmol of 1 b–f. [b] Determined by 1H NMR analysis of the crude reaction mixtures. [c] Yield of isolated, diastereomerically pure products. [d] Using 2.5 mol % of [{Rh(cod)Cl}2]. [e] The d.r. value could not be determined by 1H NMR analysis. [f] Yield of the isolated minor syn‐syn diastereomer 9 bb. [g] Using 2‐MeTHF/MeOH (10:1) in place of TBME/tBuCN/MeOH (10:1.2:1). [h] Using 3,5‐Me2C6H3B(neo) (1.3 equiv), K2CO3 (1.3 equiv), and tAmOH (1.5 equiv) as the reagents in TBME/tBuCN (8.3:1). neo=neopentyl glycol.

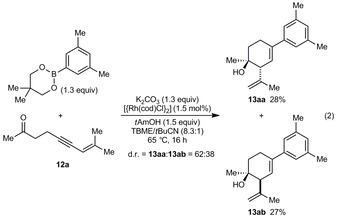
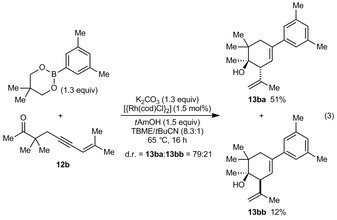
Furthermore, the fully acyclic substrates 12 a and 12 b also underwent successful arylative intramolecular allylation [Eq. 2 and 3], although the diastereoselectivities were lower compared with substrates containing cyclic ketones. For acceptable yields, it was important to use neopentyl glycol boronate, K2CO3, and tAmOH. Under these reaction conditions, 12 a reacted with 3,5‐dimethylphenyl neopentyl glycol boronate to give the diastereomeric products 13 aa and 13 ab in 28 and 27 % yield, respectively [Eq. (2)].21 Improved results were obtained with 12 b, which contains a geminal dimethyl group in the tether, and 13 ba and 13 bb were obtained in 51 and 12 % yield, respectively [Eq. (3)].21 The same reactions conducted in 2‐MeTHF instead of TBME/tBuCN gave 13 ab and 13 bb as the major products, but were lower yielding.
The substrate 14, which contains an E‐1,3‐enyne, did not undergo the reaction, and only starting materials were recovered [Eq. 4]. This result confirms the requirement for cis‐allylic hydrogen atoms to be present in the enyne to allow 1,4‐rhodium(I) migration to occur (compare with Table 2, entry 1 using the Z‐isomer 1 b). In addition, reaction of hexadeuterated enynone [D]6‐1 a with 3,5‐dimethylphenylboronic acid pinacol ester gave [D]6‐2 aa with greater than 95 % deuterium transfer to the alkene of the cyclohexene [Eq. 5]. This outcome is consistent with 1,4‐rhodium(I) migration occurring by a C−H oxidative addition/reductive elimination through a rhodium(III) hydride intermediate as hypothesized previously for alkenyl‐to‐aryl 1,4‐rhodium(I) migration.14j


Up until this point, all of the arylboronates evaluated possess substitution patterns that disfavor 1,4‐rhodium(I) migration of intermediates such as 6 onto the aryl group. To assess whether alkenyl‐to‐allyl 1,4‐rhodium(I) migration would still be favored when a sterically more accessible site is available, 1 a was reacted with phenylboronic acid (Scheme 4). The reaction in TBME/tBuCN (8:1) in the presence of t‐amyl alcohol (1.5 equiv) gave a 95:5 mixture of inseparable products, 15 and 2 ja. The product 15 results from 1,4‐rhodium(I) migration onto the phenyl group followed by intramolecular ketone arylation,15 whereas 2 ja is the arylative allylation product. When the solvent was changed to 2‐MeTHF, the allylation product 2 jb was formed preferentially (36:64 ratio of 15/2 jb) in 89:11 d.r., and was isolated as a single diastereomer in 45 % yield. The reasons for this switch in chemoselectivity are not currently known.
Scheme 4.

Reaction of 1 a with PhB(OH)2.
Consistent with models proposed in prior rhodium‐catalyzed nucleophilic allylations,4a, 12b–12e we suggest that allylation occurs through cyclic six‐membered transition states (Scheme 5). In the absence of a nitrile in the reaction medium (Table 1, entry 1), we assume that (Z)‐7, formed from 1,4‐rhodium(I) migration of 6, cyclizes through a chairlike arrangement (TS1) to give 2 aa (Scheme 5). The boatlike structure TS2 should be disfavored. However, when a coordinating nitrile is present (Table 1, entries 2 and 3), the rate of cyclization could be decreased, allowing isomerization of (Z)‐7 into (E)‐7.22 Thereafter, we assume that cyclization of (E)‐7 occurs through the chairlike conformation TS5 to give 2 ab (Scheme 5). The alternative conformation TS3 is likely to be disfavored because of 1,3‐diaxial interactions and allylic 1,3‐strain. The boatlike structure TS4 is also likely to be unfavorable. However, we do not exclude the possibility that when a nitrile is present, 2 aa is formed by cyclization of (E)‐7 through an open transition state because of preferential coordination of rhodium to the nitrile rather than the ketone.
Scheme 5.
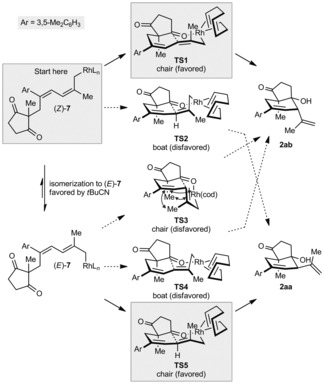
Possible stereochemical models.
Similar chairlike transition states can be used to explain the outcomes of the reactions 12 a and 12 b [Eqs. (2) and (3)], and the diastereomeric ratios observed may be a consequence of their more flexible nature (see the Supporting Information).
Finally, preliminary efforts at developing enantioselective reactions were conducted (Table 3).23 Only modest results were obtained with chiral diene ligands24 (see Supporting Information), while no reaction occurred when chiral bisphosphines were used. However, the reaction of 1 a with 3,5‐dimethylphenylboronic acid (1.3 equiv) in the presence of [{Rh(C2H4)2Cl}2] (2.5 mol %), the sulfur‐alkene L1 25 (5.0 mol %), and KF (1.5 equiv) in TBME/tBuCN/MeOH (40:5:1) gave (+)‐2 aa 17 in 61 % yield and 91 % ee (entry 1). The diastereomeric product (+)‐2 ab was obtained in 11 % yield and 88 % ee. Similar results were obtained with 3‐chloro‐5‐methylphenylboronic acid (entry 2).
Table 3.
Enantioselective reactions.[a]
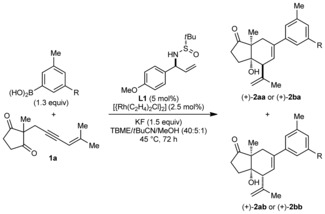
| Entry | R | d.r.[b] | Major isomer[c] | Minor isomer[c] |
|---|---|---|---|---|
| 1 | Me | 80:20 | (+)‐2 aa 61 %, 91 % ee | (+)‐2 ab 11 %, 88 % ee |
| 2 | Cl | 78:22 | (+)‐2 ba 47 %, 90 % ee | (+)‐2 bb 10 %, 90 % ee |
[a] Reactions employed 0.25 mmol of 1 a. [b] Determined by 1H NMR analysis of the crude reaction mixture. [c] Yields are of isolated, diastereomerically pure products. Enantiomeric excesses were determined HPLC analysis on a chiral stationary phase.
In summary, we have reported the rhodium‐catalyzed arylative allylation of enynones with arylboron reagents. The key step of the reaction is the alkenyl‐to‐allyl 1,4‐rhodium(I) migration, a new mode of reactivity which enables the generation of nucleophilic allylrhodium(I) species without prefunctionalization of the allylic position. Cyclization of the allylrhodium species onto a pendant ketone leads to bicyclic products containing three contiguous stereocenters with high diastereoselectivities. The products can be obtained in high enantioselectivities using a chiral sulfur‐alkene ligand. Further applications of this promising platform for generating allylmetal species are in progress.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the ERC (Starting Grant No. 258580), the EPSRC (Leadership Fellowship to H.W.L.; grants EP/I004769/1 and EP/I004769/2), the University of Nottingham, and GlaxoSmithKline for financial support.
B. M. Partridge, M. Callingham, W. Lewis, H. W. Lam, Angew. Chem. Int. Ed. 2017, 56, 7227.
Contributor Information
Dr. Benjamin M. Partridge, http://www.nottingham.ac.uk/∼pczhl/.
Prof. Hon Wai Lam, Email: hon.lam@nottingham.ac.uk.
References
- 1.Selected reviews:
- 1a. Chen Z., Wang B., Zhang J., Yu W., Liu Z., Zhang Y., Org. Chem. Front. 2015, 2, 1107–1295; [Google Scholar]
- 1b. Engle K. M., Mei T.-S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788–802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Yamaguchi J., Yamaguchi A. D., Itami K., Angew. Chem. Int. Ed. 2012, 51, 8960–9009; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9092–9142; [Google Scholar]
- 1d. Yeung C. S., Dong V. M., Chem. Rev. 2011, 111, 1215–1292; [DOI] [PubMed] [Google Scholar]
- 1e. Davies H. M. L., Morton D., Chem. Soc. Rev. 2011, 40, 1857–1869. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Huo H.-X., Duvall J. R., Huang M.-Y., Hong R., Org. Chem. Front. 2014, 1, 303–320; [Google Scholar]
- 2b. Yus M., González-Gómez J. C., Foubelo F., Chem. Rev. 2013, 113, 5595–5698; [DOI] [PubMed] [Google Scholar]
- 2c. Yus M., González-Gómez J., Foubelo F., Chem. Rev. 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]
- 3. Jeganmohan M., Cheng C.-H., Chem. Commun. 2008, 3101–3117. [DOI] [PubMed] [Google Scholar]
- 4.For selected, recent examples, see:
- 4a. Tran D. N., Cramer N., Angew. Chem. Int. Ed. 2010, 49, 8181–8184; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8357–8360; [Google Scholar]
- 4b. Yoshida Y., Murakami K., Yorimitsu H., Oshima K., J. Am. Chem. Soc. 2010, 132, 8878–8879; [DOI] [PubMed] [Google Scholar]
- 4c. Meng F., Jang H., Jung B., Hoveyda A. H., Angew. Chem. Int. Ed. 2013, 52, 5046–5051; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5150–5155; [Google Scholar]
- 4d. Meng F., McGrath K. P., Hoveyda A. H., Nature 2014, 513, 367–374; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Yeung K., Ruscoe R. E., Rae J., Pulis A. P., Procter D. J., Angew. Chem. Int. Ed. 2016, 55, 11912–11916; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12091–12095. [Google Scholar]
- 5.For reviews, see:
- 5a. Feng J., Kasun Z. A., Krische M. J., J. Am. Chem. Soc. 2016, 138, 5467–5478; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Ketcham J. M., Shin I., Montgomery T. P., Krische M. J., Angew. Chem. Int. Ed. 2014, 53, 9142–9150; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9294–9302; [Google Scholar]
- 5c. Dechert-Schmitt A.-M. R., Schmitt D. C., Gao X., Itoh T., Krische M. J., Nat. Prod. Rep. 2014, 31, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected, recent examples, see:
- 6a. Chen T.-Y., Tsutsumi R., Montgomery T. P., Volchkov I., Krische M. J., J. Am. Chem. Soc. 2015, 137, 1798–1801; [DOI] [PubMed] [Google Scholar]
- 6b. Liang T., Nguyen K. D., Zhang W., Krische M. J., J. Am. Chem. Soc. 2015, 137, 3161–3164; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Liang T., Zhang W., Chen T.-Y., Nguyen K. D., Krische M. J., J. Am. Chem. Soc. 2015, 137, 13066–13071; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. Liang T., Zhang W., Krische M. J., J. Am. Chem. Soc. 2015, 137, 16024–16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu X., Zheng K., Feng X., Synthesis 2014, 46, 2241–2257. [Google Scholar]
- 8.
- 8a. Olier C., Kaafarani M., Gastaldi S., Bertrand M. P., Tetrahedron 2010, 66, 413–445; [Google Scholar]
- 8b. Pastor I. M., Yus M., Curr. Org. Chem. 2007, 11, 925–957. [Google Scholar]
- 9.Selected examples:
- 9a. Hansson S., Heumann A., Rein T., Aakermark B., J. Org. Chem. 1990, 55, 975–984; [Google Scholar]
- 9b. Covell D. J., White M. C., Angew. Chem. Int. Ed. 2008, 47, 6448–6451; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6548–6551; [Google Scholar]
- 9c. Vermeulen N. A., Delcamp J. H., White M. C., J. Am. Chem. Soc. 2010, 132, 11323–11328; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9d. Trost B. M., Thaisrivongs D. A., Donckele E. J., Angew. Chem. Int. Ed. 2013, 52, 1523–1526; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1563–1566; [Google Scholar]
- 9e. Osberger T. J., White M. C., J. Am. Chem. Soc. 2014, 136, 11176–11181; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9f. Trost B. M., Donckele E. J., Thaisrivongs D. A., Osipov M., Masters J. T., J. Am. Chem. Soc. 2015, 137, 2776–2784; [DOI] [PubMed] [Google Scholar]
- 9g. Tao Z.-L., Li X.-H., Han Z.-Y., Gong L.-Z., J. Am. Chem. Soc. 2015, 137, 4054–4057; [DOI] [PubMed] [Google Scholar]
- 9h. Wang P.-S., Liu P., Zhai Y.-J., Lin H.-C., Han Z.-Y., Gong L.-Z., J. Am. Chem. Soc. 2015, 137, 12732–12735. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Burns D. J., Lam H. W., Angew. Chem. Int. Ed. 2014, 53, 9931–9935; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10089–10093; [Google Scholar]
- 10b. Burns D. J., Best D., Wieczysty M. D., Lam H. W., Angew. Chem. Int. Ed. 2015, 54, 9958–9962; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10096–10100. [Google Scholar]
- 11.
- 11a. Bao W., Kossen H., Schneider U., J. Am. Chem. Soc. 2017, 139, 4362–4365; [DOI] [PubMed] [Google Scholar]
- 11b. Wei X.-F., Xie X.-W., Shimizu Y., Kanai M., J. Am. Chem. Soc. 2017, 139, 4647–4650; [DOI] [PubMed] [Google Scholar]
- 11c. Michigami K., Mita T., Sato Y., J. Am. Chem. Soc. 2017, 139, 6094–6097. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Takada Y., Hayashi S., Hirano K., Yorimitsu H., Oshima K., Org. Lett. 2006, 8, 2515–2517; [DOI] [PubMed] [Google Scholar]
- 12b. Kimura M., Nojiri D., Fukushima M., Oi S., Sonoda Y., Inoue Y., Org. Lett. 2009, 11, 3794–3797; [DOI] [PubMed] [Google Scholar]
- 12c. Luo Y., Hepburn H. B., Chotsaeng N., Lam H. W., Angew. Chem. Int. Ed. 2012, 51, 8309–8313; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 8434–8438; [Google Scholar]
- 12d. Hepburn H. B., Lam H. W., Angew. Chem. Int. Ed. 2014, 53, 11605–11610; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11789–11794; [Google Scholar]
- 12e. Martínez J. I., Smith J. J., Hepburn H. B., Lam H. W., Angew. Chem. Int. Ed. 2016, 55, 1108–1112; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1120–1124. [Google Scholar]
- 13.For reviews of 1,4-metal migration, see:
- 13a. Ma S., Gu Z., Angew. Chem. Int. Ed. 2005, 44, 7512–7517; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7680–7685; [Google Scholar]
- 13b. Shi F., Larock R. C., Top. Curr. Chem. 2009, 292, 123–164. [DOI] [PubMed] [Google Scholar]
- 14.Selected examples:
- 14a. Oguma K., Miura M., Satoh T., Nomura M., J. Am. Chem. Soc. 2000, 122, 10464–10465; [Google Scholar]
- 14b. Hayashi T., Inoue K., Taniguchi N., Ogasawara M., J. Am. Chem. Soc. 2001, 123, 9918–9919; [DOI] [PubMed] [Google Scholar]
- 14c. Miura T., Sasaki T., Nakazawa H., Murakami M., J. Am. Chem. Soc. 2005, 127, 1390–1391; [DOI] [PubMed] [Google Scholar]
- 14d. Yamabe H., Mizuno A., Kusama H., Iwasawa N., J. Am. Chem. Soc. 2005, 127, 3248–3249; [DOI] [PubMed] [Google Scholar]
- 14e. Shintani R., Takatsu K., Hayashi T., Angew. Chem. Int. Ed. 2007, 46, 3735–3737; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3809–3811; [Google Scholar]
- 14f. Matsuda T., Shigeno M., Murakami M., J. Am. Chem. Soc. 2007, 129, 12086–12087; [DOI] [PubMed] [Google Scholar]
- 14g. Seiser T., Roth O. A., Cramer N., Angew. Chem. Int. Ed. 2009, 48, 6320–6323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6438–6441; [Google Scholar]
- 14h. Shigeno M., Yamamoto T., Murakami M., Chem. Eur. J. 2009, 15, 12929–12931; [DOI] [PubMed] [Google Scholar]
- 14i. Shintani R., Isobe S., Takeda M., Hayashi T., Angew. Chem. Int. Ed. 2010, 49, 3795–3798; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3883–3886; [Google Scholar]
- 14j. Sasaki K., Nishimura T., Shintani R., Kantchev E. A. B., Hayashi T., Chem. Sci. 2012, 3, 1278–1283; [Google Scholar]
- 14k. Zhang J., Liu J.-F., Ugrinov A., Pillai A. F. X., Sun Z.-M., Zhao P., J. Am. Chem. Soc. 2013, 135, 17270–17273; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14l. Shintani R., Iino R., Nozaki K., J. Am. Chem. Soc. 2014, 136, 7849–7852; [DOI] [PubMed] [Google Scholar]
- 14m. Masarwa A., Weber M., Sarpong R., J. Am. Chem. Soc. 2015, 137, 6327–6334. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Partridge B. M., Solana González J., Lam H. W., Angew. Chem. Int. Ed. 2014, 53, 6523–6527; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6641–6645; [Google Scholar]
- 15b. Johnson T., Choo K.-L., Lautens M., Chem. Eur. J. 2014, 20, 14194–14197. [DOI] [PubMed] [Google Scholar]
- 16. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- 17.The relative configurations of the products 2 aa, 9 cb, 9 ea, and 9 fa, and the absolute configuration of (+)-2 aa were determined by X-ray crystallography. CCDC 1493830–1493833 and 1535509 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre..
- 18.The relative configurations of the majority of the products described herein were determined by NOESY experiments. The relative configurations of 2 ab and 2 jb were assigned tentatively by analogy with 9 cb, the relative configuration of which was determined by X-ray crystallography (see Ref. [17]).
- 19. Miura T., Shimada M., Murakami M., Angew. Chem. Int. Ed. 2005, 44, 7598–7600; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7770–7772. [Google Scholar]
- 20.This experiment gave 9 ca in 22 % yield (see the Supporting Information).
- 21.The relative configurations of 13 aa and 13 ab could not be determined unambiguously and were assigned tentatively by analogy with the reaction producing 13 ba and 13 bb, on the assumption that the major products in each case possess the same relative configuration. The relative configurations of 13 ba and 13 bb were determined by NOESY experiments. The product 13 bb contained small quantities of an inseparable impurity.
- 22. Z/E Isomerization could occur through a sequence involving 1,3-rhodium transposition, bond rotation, and a second 1,3-transposition.
- 23.For enantioselective rhodium-catalyzed desymmetrizating arylative cyclizations of enone-diones, see: Bocknack B. M., Wang L. C., Krische M. J., Proc. Natl. Acad. Sci. USA 2004, 101, 5421–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For reviews, see:
- 24a. Feng C.-G., Xu M.-H., Lin G.-Q., Synlett 2011, 1345–1356; [Google Scholar]
- 24b. Shintani R., Hayashi T., Aldrichimica Acta 2009, 42, 31–38; [Google Scholar]
- 24c. Johnson J. B., Rovis T., Angew. Chem. Int. Ed. 2008, 47, 840–871; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 852–884; [Google Scholar]
- 24d. Defieber C., Grutzmacher H., Carreira E. M., Angew. Chem. Int. Ed. 2008, 47, 4482–4502; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 4558–4579. [Google Scholar]
- 25. Feng X., Wang Y., Wei B., Yang J., Du H., Org. Lett. 2011, 13, 3300–3303. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary