

Abstract
Described herein is an effective and practical modular flow design for the meta‐selective C−H arylation of anilines. The design consists of four continuous‐flow modules (i.e., diaryliodonium salt synthesis, meta‐selective C−H arylation, inline copper extraction, and aniline deprotection) which can be operated either individually or consecutively to provide direct access to meta‐arylated anilines. With a total residence time of 1 hour, the desired product could be obtained in high yield and excellent purity without the need for column chromatography, and the residual copper content meets the standards for parenterally administered pharmaceutical substances.
Keywords: arylation, C−H activation, copper, hypervalent compounds, flow chemistry
Site‐selective C−H bond functionalization strategies are of paramount importance in modern organic synthesis.1 However, because of the ubiquitous presence of C−H bonds in organic molecules, the regioselective assembly of substituted arenes remains a major challenge. Traditionally, the ortho and, to some extent, the para substitution of arenes have been thoroughly explored with the use of Friedel–Crafts chemistry. More recently, much work has been carried out on the transition metal catalyzed ortho‐functionalization of arenes, and it proceeds by a cyclometalation strategy.2 In contrast, the development of meta‐selective transformations has required much more scientific investigation. Besides traditional approaches which rely on tuning steric and electronic properties of aromatic substrates, novel robust catalytic strategies have emerged.3 For example, the incorporation of directing‐group templates4 or the use of transient ligand mediators5 (e.g., Pd/norbornene) have been exploited to carefully navigate transition metals to the meta‐position.
Among the reported meta‐selective C−H functionalization strategies, the meta‐arylation of electron‐rich arenes is of high interest to access novel biaryl motifs, which represent a common moiety within medicines, agrochemicals, and functional materials.6 In particular, Gaunt and co‐workers first reported the meta‐selective C−H arylation of protected anilines.7 This transformation was believed to proceed via a highly electrophilic CuIII/aryl intermediate, obtained from CuI, and in presence of diaryliodonium salts as both oxidant and arylating agent.8 However, despite being shelf‐stable, nontoxic, and synthetically useful, diaryliodonium salts have limited availability and are expensive.9 The main cause is associated with its cumbersome preparation. Hereto, stoichiometric amounts of hazardous reagents (e.g., mCBPA and TfOH) are necessary to oxidize iodine to its hypervalent state (i.e., I+III) and the greatly exothermic nature of the reaction makes hot‐spot formation highly probable, thus resulting in reduced selectivities and safety issues on a large scale.
A central theme of our research is to develop continuous‐flow methods, thus delivering a set of new tools to facilitate challenging synthetic transformations and provide additional advantages over batch in terms of, for example, safety,10 scalability,11 time‐reduction,12 or selectivity.13 Given the importance of meta‐arylated anilines and its limited scalability potential in batch, we felt that a continuous‐flow strategy to access such compounds would represent an important advance. To prepare these compounds, we identified four key steps in its synthesis, including the synthesis of the diaryliodonium salt, the meta‐selective C−H arylation, and the removal of both the copper catalyst and the directing group (Scheme 1). While all these modules have great potential on its own, combining them would allow straightforward access to the meta‐arylated anilines within a reasonable time scale and effort.
Scheme 1.
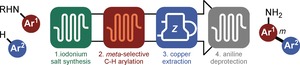
Modular flow design for the direct access to meta‐arylated anilines.
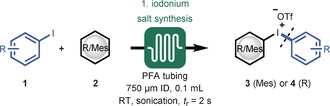
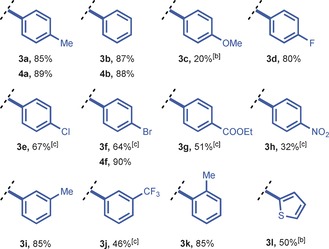
In taking on this challenge, we anticipated that diaryliodonium salt synthesis as the first module could highly benefit from continuous‐flow processing to alleviate the current safety limitations (i.e., highly exothermic nature and hazardous reagents handling).14 The one‐pot synthesis developed by the group of Olofsson was identified as the most convenient strategy to produce a diverse set of diaryliodonium salts.14b A 0.1 mL PFA reactor coil (750 μm I.D.) was constructed and reagents were introduced by three separate feed streams (i.e., reagent feed, oxidant feed, and acid feed; see Figure S4 in the Supporting Information). To prevent microreactor clogging and to ensure excellent heat dissipation, the reactor assembly was submerged in an ultrasonic bath kept at room temperature. After initial optimization (see Table S1), it was found that the target di‐p‐tolyliodonium triflate (4 a) could be obtained, after only a two‐seconds residence time, in excellent yield (89 %) after crystallization (Table 1). Notably, our flow protocol was highly reproducible and the yields were typically higher than those obtained with conventional batch labware (52–67 %).14b, 15 The procedure can be readily scaled and, as an example, we obtained 2.04 grams of 4 a (5 mmol scale, 89 %). Next, a small library of symmetrical and unsymmetrical diaryliodonium salts was established in flow (Table 1). A wide variety of unsymmetrical diaryliodonium triflates bearing diverse substituents and mesitylene as the counterligand were successfully synthesized on a gram scale (3 a–l). In particular, the compounds 3 d (80 %) and 3 i (85 %) were obtained in significantly higher yields than previously reported.7, 16 Moreover, the unsymmetrical diaryliodonium triflates bearing electron‐rich substituents (3 c and 3 l) were synthesized for the first time by this one‐step procedure. We believe that this module provides a useful tool to enable the large‐scale preparation of valuable and costly diaryliodonium salts in a safe and time‐efficient fashion.
Table 1.
Scope for the synthesis of (un)symmetrical diaryliodonium salts in flow.[a]
|
[a] Reaction conditions: Syringe 1: 5.0 mmol of aryl iodide (1) and 5.5 mmol of arene (2) in 25 mL DCE at 0.75 mL min−1; syringe 2: 5.5 mmol of m‐CBPA in 25 mL of DCE at 0.75 mL min−1; syringe 3: 10.0 mmol TfOH in 50 mL DCE at 1.5 mL min−1. Added to the reactor by syringe pump. [b] Mesityl iodide was used. [c] 3 mL reactor volume with t r=60 s, 6.5 mmol m‐CBPA, and 15 mmol TfOH. Note: The 3 series refers to unsymmetrical diaryliodonium salts with mesitylene as arene, and the 4 series to symmetrical diaryliodonium salts. m‐CPBA=m‐chloroperbenzoic acid, Tf=trifluoromethanesulfonyl.
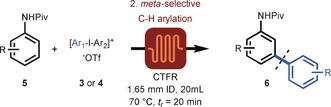
We next addressed the development of module 2, which involves the continuous‐flow meta‐selective C−H arylation of anilines. It is generally accepted that the reported meta‐selective C−H arylation reaction operates by a homogeneous mechanism, thus making the use of heterogeneous catalysts superfluous. Nevertheless, the use of heterogeneous precursor materials, which can serve as cheap and convenient reservoirs for the release of homogeneous catalytically active species, can be of highly added value.17 We hypothesized that the catalytically active species could be readily formed from Cu0 in the presence of highly electrophilic diaryliodonium salts.18 Preliminary batch investigations revealed that inexpensive copper powder enabled the meta‐arylation of N‐(o‐tolyl)pivalamide (5 a) with di‐p‐tolyliodonium triflate (4 a; see Table S2). Moreover, these experiments revealed that copper powder was more active than the benchmark Cu(OTf)2 catalyst source reported by Gaunt, thus reducing the batch reaction time from 24 to 2 hours (see Figure S1). In addition, reactions with surface‐treated copper turnings revealed that both Cu0 and CuI can be used as catalyst source (see Figure S1 b).19 Translating this concept to continuous manufacturing, we speculated that the meta‐selective C−H arylation could highly benefit from the use of copper tube flow reactors (CTFRs),20 which would allow for a significant breakthrough in operational simplicity for C−H activation chemistry.21 To test this hypothesis, a 20 mL CTFR (1.65 mm I.D.) was constructed from cheap and commercially available copper tubing (see Figure S5). After initial optimization of the reaction parameters, full conversion was obtained within only 20 minutes residence time, thus yielding 88 % of 6 a (see Table 2 and Tables S3 and S4). Next, various pivanilides with 4 a as the coupling partner were subjected to our flow protocol (Table 2). Pivanilides bearing either ortho‐alkyl, ortho‐aryl, or ortho‐methoxy substituents were well tolerated and yielded the monoarylated compounds 6 b, 6 c, and 6 d respectively, in excellent yields (86–91 %). In the absence of ortho substituents, both meta positions became highly accessible, thus resulting in a high‐yielding mixture of both mono‐ and diarylated products 6 e/6 e′ (80 % yield, mono/di 1:2.3). Also, the heterocyclic substrate indoline was readily converted into 6 f in our flow reactor, thus yielding the pure compound in 80 % yield upon isolation. Generally, meta‐substituted substrates are perceived as more challenging substrates but could nevertheless be acquired in flow within 20 minutes (6 g,h). More complex ortho,para‐disubstituted pivanilides were also compatible (6 i–l), thus obtaining 6 i and a 6 j/6 j′ mixture in high yields (85–86 %). Modest yields (30–42 %) were obtained for 6 k and 6 l because of incomplete conversion.
Table 2.
Scope with respect to anilines[a] and diaryliodonium salts[b] for the meta‐selective C−H arylation in flow.
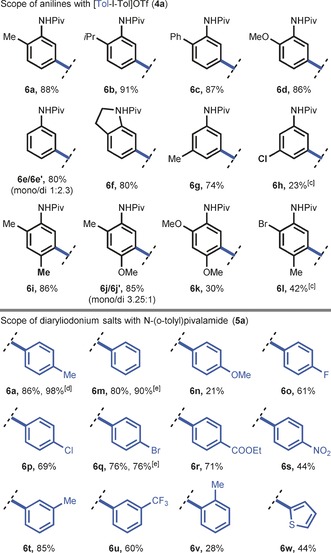
|
[a] Reaction conditions: 0.5 mmol aniline (5) and 2.0 equiv [Tol‐I‐Tol]OTf (4 a) in 5.0 mL DCE. [b] Reaction conditions: 0.5 mmol N‐(o‐tolyl)pivalamide (5 a) and 2.0 equiv [Ar‐I‐Mes]OTf (3) in 5.0 mL DCE. Added to the CTFR by syringe pump. [c] 40 min residence time. [d] 5.03 mmol scale reaction: 1.384 g (98 %) of desired product obtained. [e] Symmetrical [Ar‐I‐Ar]OTf (4) was used. Piv=pivaloyl.
Next, a diverse set of diaryliodonium salts, all prepared on a gram scale in flow by module 1, were evaluated as coupling partners with N‐(o‐tolyl)pivalamide (5 a) as a benchmark substrate (Table 2). The transfer of various aryl groups was effective for a broad range of symmetrical and unsymmetrical diaryliodonium triflates (6 m–w) bearing either electron‐neutral, electron‐donating, or electron‐withdrawing substituents, thus yielding the desired meta‐arylated products in fair to excellent yields (21–90 %). Note that for unsymmetrical diaryliodonium salts, the sterically hindered mesitylene could be successfully used as a “dummy ligand” allowing selective transfer of the functionalized aryl groups. Interestingly, a gram‐scale reaction was readily carried out and resulted in the formation of 6 a in near quantitative yield (1.384 g, 98 %), thus highlighting the excellent scale‐up potential of our continuous‐flow protocol. It should be noted that the complete scope (6 a–w) was performed with only a single copper capillary without any apparent loss of reactivity. Moreover, the accelerated reaction conditions (20 min vs. 24–48 h) and improved yields highlight the potential of these CTFRs, as readily available flow reactors, to enable copper‐catalyzed C−H activation chemistry for gram‐scale drug manufacturing.
Operating the meta‐arylation reaction in a continuous‐flow manner, evidently raised the question of whether significant copper leaching was taking place. Leaching can become apparent at longer operation times, since copper will be progressively chromatographed through the reactor tubing as a result of subsequent leaching/redeposition cycles.17 To investigate the leaching behavior, ICP‐OES analysis was conducted on reaction samples (see Section 2.3 in the Supporting Information). As can be seen from Table 3, the crude reaction sample from module 2 contained about 4720 ppm of Cu, and is in the same range as previously reported transformations in CFTRs.20a To remove the leached copper from the target compound, we considered a continuous‐flow inline extraction module (module 3).22 The extraction module consisted of a 5 mL PFA coil (1.65 mm I.D.) connected to a commercially available Zaiput liquid‐liquid membrane separator (see Figure S6). The aqueous ammonia solution (32 wt %) was merged with the organic stream exiting the CTFR. The combined liquid‐liquid phase provided a rapid extraction of copper through complexation with ammonia, and could be visually confirmed by the deep‐blue colored aqueous phase. The organic stream was subsequently separated from the aqueous stream in the Zaiput device. ICP‐OES analysis revealed that 99.7 % of copper content could be readily extracted with a single pass through the module. This step leads to a residual 14.3 ppm of Cu, and is far below the recommended limit for parenterally administered pharmaceutical substances (<25 ppm according to the European Medicines Agency (EMEA) Guidelines).23
Table 3.
Inline copper extraction and phase separation.[a]
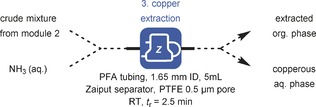
| Entry | Purification | Cu content [ppm] |
|---|---|---|
| Crude mixture from module 2 | n.a. | 4720 |
| Extracted org. phase | 1 extraction | 14.3 |
| Extracted org. phase | 3 extractions | 6.3 |
[a] Crude mixture from module 2 and NH3 (32 wt %) aqueous solution was pumped by syringe pump and mixed together in a T‐mixer. A Zaiput membrane separator was connected at the end of the 5 mL PFA extraction coil. Organic phase was checked for Cu content by ICP‐OES analysis.
The final step in our reaction sequence constitutes the removal of the pivalic protecting group. Notably, the cleavage of pivanilides proved to be extremely challenging. Literature procedures utilize reflux conditions and prolonged reaction times ranging from 1–3 days.7, 24 Such extended reaction times are not suitable for continuous‐flow processing and a thorough screening of potential deprotection strategies was carried out (see Tables S6 and S7). The flow module consisted of a 20 mL PFA capillary (1.65 mm I.D.) equipped with a 140 psi back pressure regulator (BPR) to enable superheated reaction conditions (see Figure S7). Eventually, we found that the deprotection of 6 a could be realized within 40 minutes using an HCl/1,4‐dioxane (1:1) mixture at 130 °C (Table 4), thus yielding the free aniline 7 a in excellent yield (94 %). A final extraction procedure was used to circumvent chromatography (see Section 4.4). This continuous‐flow deprotection protocol appeared to be generally applicable (7 a–h) and constitutes a significant improvement compared to the literature procedures.
Table 4.
Aniline deprotection in flow.[a]
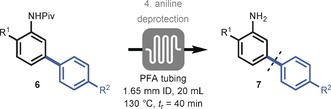
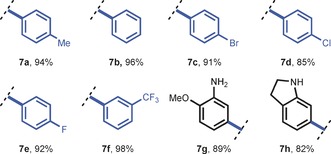
|
[a] Reaction conditions: 0.5 mmol product (6) in 5.0 mL HCl (32 wt %)/1,4‐dioxane (1:1), added to the PFA reactor by syringe pump (for 7 a–f R1=Me, for 7 g,h R2=Me). Aniline obtained after extraction procedure (no chromatography).
Finally, with all the individual modules fully explored and optimized, the different modules were combined to enable direct access to meta‐arylated anilines (Scheme 2). In module 1, 4‐iodotoluene (1 a) and toluene (2 a) where readily converted into the corresponding iodonium salt 4 a within a 2 second residence time. After crystallization, 4 a was combined with N‐(o‐tolyl)pivalamide (5 a) and introduced in module 2. Upon exiting the CTFR, the reaction mixture was merged with the aqueous NH3 solution phase to remove the leached copper in module 3. The organic phase containing 6 a was subsequently evaporated and re‐dissolved in HCl/1,4‐dioxane (1:1). This mixture was fed to the last module to obtain the fully deprotected meta‐arylated aniline 7 a in an overall yield of 80 % and 99 % purity after a final extraction procedure. It should be noted that the overall procedure could be carried out within a 1 hour residence time and required no chromatographic purification.
Scheme 2.
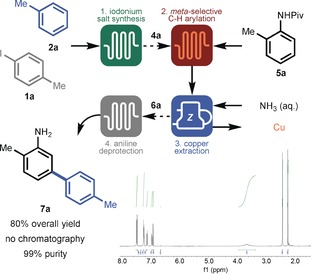
Overview of modular flow experiment for the synthesis of 7 a. Solid arrows indicate direct connections and dashed arrows indicate indirect connections (for example, precipitation or solvent exchange).
In conclusion, we have developed a modular and efficient continuous‐flow approach which allows direct access to valuable meta‐arylated anilines. Module 1 provides a unique, safe, and scalable flow method to prepare highly valuable diaryliodonium salts, within a 2 second residence time (15 examples). In module 2, a copper tube flow reactor was used for the first time to enable copper‐catalyzed meta‐selective C−H arylation of protected anilines within a 20 minute residence time (23 examples). Effective inline copper removal in module 3 led to values suitable for meeting the standards of parenterally administered pharmaceutical substances (i.e., residual Cu content <25 ppm). Finally, deprotection of the pivanilides was realized in module 4, thus delivering the desired meta‐arylated anilines in a straightforward fashion within 40 minutes. Orchestrating all individual modules in an integrated process allowed preparation of meta‐arylated anilines within a total time frame of 1 hour in excellent yield and purity, and without the need of chromatography. We believe that the each of the developed modules are of high value and will find widespread use in both academia and industry.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support is provided by the Dutch Science Foundation (NWO) by an ECHO grant (Grant No. 713.013.001) and a VIDI grant for T.N. (Grant No. 14150). We also acknowledge the support by COST (European Cooperation in Science and Technology) Action (CA15106) CH‐Activation in Organic Synthesis.
H. P. L. Gemoets, G. Laudadio, K. Verstraete, V. Hessel, T. Noël, Angew. Chem. Int. Ed. 2017, 56, 7161.
Contributor Information
Hannes P. L. Gemoets, http://www.noelresearchgroup.com.
Dr. Timothy Noël, Email: t.noel@tue.nl.
References
- 1.
- 1a.Y. Qin, L. Zhu, S. Luo, Chem. Rev 2017, https://doi.org/10.1021/acs.chemrev.6b00657; [DOI] [PubMed]
- 1b. Roudesly F., Oble J., Poli G., J. Mol. Catal. A 2017, 426, 275–296; [Google Scholar]
- 1c.J. He, M. Wasa, K. S. L. Chan, Q. Shao, J.-Q. Yu, Chem. Rev 2016, DOI: httsp://doi.org/10.1021/acs.chemrev.6b00622; [DOI] [PMC free article] [PubMed]
- 1d. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900–2936; [DOI] [PubMed] [Google Scholar]
- 1e. Hartwig J. F., J. Am. Chem. Soc. 2016, 138, 2–24; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Liu C., Yuan J., Gao M., Tang S., Li W., Shi R., Lei A., Chem. Rev. 2015, 115, 12138–12204. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Zhu R.-Y., Farmer M. E., Chen Y.-Q., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 10578–10599; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10734–10756; [Google Scholar]
- 2b. Engle K. M., Mei T.-S., Wasa M., Yu J.-Q., Acc. Chem. Res. 2012, 45, 788–802; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Lyons T. W., Sanford M. S., Chem. Rev. 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Dey A., Agasti S., Maiti D., Org. Biomol. Chem. 2016, 14, 5440–5453; [DOI] [PubMed] [Google Scholar]
- 3b. Yang J., Org. Biomol. Chem. 2015, 13, 1930–1941; [DOI] [PubMed] [Google Scholar]
- 3c. Ackermann L., Li J., Nat. Chem. 2015, 7, 686–687; [DOI] [PubMed] [Google Scholar]
- 3d. Li J., De Sarkar S., Ackermann L., Top. Organomet. Chem. 2015, 55, 217–257; [Google Scholar]
- 3e. Schranck J., Tlili A., Beller M., Angew. Chem. Int. Ed. 2014, 53, 9426–9428; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9580–9582. [Google Scholar]
- 4.
- 4a. Zhang Z., Tanaka K., Yu J. Q., Nature 2017, 543, 538–542; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Yang Y., Gao P., Zhao Y., Shi Z., Angew. Chem. Int. Ed. 2017, 56, 3966–3971; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4024–4029; [Google Scholar]
- 4c. Bag S., Jayarajan R., Mondal R., Maiti D., Angew. Chem. Int. Ed. 2017, 56, 3182–3186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3230–3234; [Google Scholar]
- 4d. Yang Y., Li R., Zhao Y., Zhao D., Shi Z., J. Am. Chem. Soc. 2016, 138, 8734–8737; [DOI] [PubMed] [Google Scholar]
- 4e. Hofmann N., Ackermann L., J. Am. Chem. Soc. 2013, 135, 5877–5884; [DOI] [PubMed] [Google Scholar]
- 4f. Saidi O., Marafie J., Ledger A. E. W., Liu P. M., Mahon M. F., Kociok-Köhn G., Whittlesey M. K., Frost C. G., J. Am. Chem. Soc. 2011, 133, 19298–19301. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Shen P.-X., Wang X.-C., Wang P., Zhu R.-Y., Yu J.-Q., J. Am. Chem. Soc. 2015, 137, 11574–11577; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Wang X. C., Gong W., Fang L. Z., Zhu R. Y., Li S. H., Engle K. M., Yu J. Q., Nature 2015, 519, 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Wang P., Farmer M. E., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 5125–5129; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Wang P., Farmer M. E., Huo X., Jain P., Shen P.-X., Ishoey M., Bradner J. E., Wisniewski S. R., Eastgate M. D., Yu J.-Q., J. Am. Chem. Soc. 2016, 138, 9269–9276; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Dong Z., Wang J., Dong G., J. Am. Chem. Soc. 2015, 137, 5887–5890; [DOI] [PubMed] [Google Scholar]
- 6d. Wan L., Dastbaravardeh N., Li G., Yu J.-Q., J. Am. Chem. Soc. 2013, 135, 18056–18059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6e. Duong H. A., Gilligan R. E., Cooke M. L., Phipps R. J., Gaunt M. J., Angew. Chem. Int. Ed. 2011, 50, 463–466; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 483–486; [Google Scholar]
- 6f. Cornella J., Righi M., Larrosa I., Angew. Chem. Int. Ed. 2011, 50, 9429–9432; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9601–9604; [Google Scholar]
- 6g. Bedford R. B., Mitchell C. J., Webster R. L., Chem. Commun. 2010, 46, 3095–3097. [DOI] [PubMed] [Google Scholar]
- 7. Phipps R. J., Gaunt M. J., Science 2009, 323, 1593–1597. [DOI] [PubMed] [Google Scholar]
- 8. Chen B., Hou X. L., Li Y. X., Wu Y. D., J. Am. Chem. Soc. 2011, 133, 7668–7671. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Fañanás-Mastral M., Synthesis 2017, 49, 1905–1930; [Google Scholar]
- 9b. Yoshimura A., Zhdankin V. V., Chem. Rev. 2016, 116, 3328–3435; [DOI] [PubMed] [Google Scholar]
- 9c. Merritt E. A., Olofsson B., Angew. Chem. Int. Ed. 2009, 48, 9052–9070; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9214–9234. [Google Scholar]
- 10.
- 10a. Movsisyan M., Delbeke E. I. P., Berton J. K. E. T., Battilocchio C., Ley S. V., Stevens C. V., Chem. Soc. Rev. 2016, 45, 4892–4928; [DOI] [PubMed] [Google Scholar]
- 10b. Gemoets H. P. L., Su Y., Shang M., Hessel V., Luque R., Noel T., Chem. Soc. Rev. 2016, 45, 83–117; [DOI] [PubMed] [Google Scholar]
- 10c. Gutmann B., Cantillo D., Kappe C. O., Angew. Chem. Int. Ed. 2015, 54, 6688–6728; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6788–6832. [Google Scholar]
- 11.
- 11a. Su Y., Kuijpers K., Hessel V., Noël T., React. Chem. Eng. 2016, 1, 73–81; [Google Scholar]
- 11b. Teoh S. K., Rathi C., Sharratt P., Org. Process Res. Dev. 2016, 20, 414–431. [Google Scholar]
- 12.
- 12a. Su Y., Straathof N. J., Hessel V., Noel T., Chem. Eur. J. 2014, 20, 10562–10589; [DOI] [PubMed] [Google Scholar]
- 12b. Gemoets H. P. L., Hessel V., Noël T., Org. Lett. 2014, 16, 5800–5803. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Hartman R. L., McMullen J. P., Jensen K. F., Angew. Chem. Int. Ed. 2011, 50, 7502–7519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7642–7661; [Google Scholar]
- 13b. Yoshida J., Nagaki A., Iwasaki T., Suga S., Chem. Eng. Technol. 2005, 28, 259–266. [Google Scholar]
- 14.
- 14a. Watts K., Gattrell W., Wirth T., Beilstein J. Org. Chem. 2011, 7, 1108–1114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Bielawski M., Zhu M., Olofsson B., Adv. Synth. Catal. 2007, 349, 2610–2618. [Google Scholar]
- 15. Tolnai G. L., Nilsson U. J., Olofsson B., Angew. Chem. Int. Ed. 2016, 55, 11226–11230; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 11392–11396. [Google Scholar]
- 16.
- 16a. Gonda Z., Novak Z., Chem. Eur. J. 2015, 21, 16801–16806; [DOI] [PubMed] [Google Scholar]
- 16b. Aradi K., Novák Z., Adv. Synth. Catal. 2015, 357, 371–376; [Google Scholar]
- 16c. Sinai Á., Mészáros Á., Gáti T., Kudar V., Palló A., Novák Z., Org. Lett. 2013, 15, 5654–5657; [DOI] [PubMed] [Google Scholar]
- 16d. Bigot A. L., Williamson A. E., Gaunt M. J., J. Am. Chem. Soc. 2011, 133, 13778–13781. [DOI] [PubMed] [Google Scholar]
- 17. Cantillo D., Kappe C. O., ChemCatChem 2014, 6, 3286–3305. [Google Scholar]
- 18.
- 18a. Novák Z., Székely A., Sinai Á., Tóth E., Synthesis 2014, 46, 1871–1880; [Google Scholar]
- 18b. Lee E. Y., Park J., ChemCatChem 2011, 3, 1127–1129. [Google Scholar]
- 19. Fuchs M., Goessler W., Pilger C., Kappe C. O., Adv. Synth. Catal. 2010, 352, 323–328. [Google Scholar]
- 20.
- 20a. Zhang Y., Jamison T. F., Patel S., Mainolfi N., Org. Lett. 2011, 13, 280–283; [DOI] [PubMed] [Google Scholar]
- 20b. Bao J., Tranmer G. K., Chem. Commun. 2015, 51, 3037–3044; [DOI] [PubMed] [Google Scholar]
- 20c. Bao J., Tranmer G. K., Tetrahedron Lett. 2016, 57, 654–657; [Google Scholar]
- 20d. Zhang P., Russell M. G., Jamison T. F., Org. Process Res. Dev. 2014, 18, 1567–1570; [Google Scholar]
- 20e. Bogdan A. R., James K., Chem. Eur. J. 2010, 16, 14506–14512; [DOI] [PubMed] [Google Scholar]
- 20f. Ceylan S., Klande T., Vogt C., Friese C., Kirschning A., Synlett 2010, 2009–2013; [Google Scholar]
- 20g. Bogdan A. R., Sach N. W., Adv. Synth. Catal. 2009, 351, 849–854. [Google Scholar]
- 21.
- 21a. Zakrzewski J., Smalley A. P., Kabeshov M. A., Gaunt M. J., Lapkin A. A., Angew. Chem. Int. Ed. 2016, 55, 8878–8883; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9024–9029; [Google Scholar]
- 21b. Moschetta E. G., Negretti S., Chepiga K. M., Brunelli N. A., Labreche Y., Feng Y., Rezaei F., Lively R. P., Koros W. J., Davies H. M. L., Jones C. W., Angew. Chem. Int. Ed. 2015, 54, 6470–6474; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6570–6574. [Google Scholar]
- 22. Varas A. C., Noël T., Wang Q., Hessel V., ChemSusChem 2012, 5, 1703–1707. [DOI] [PubMed] [Google Scholar]
- 23.European Medicines Agency (EMEA), “Guidelines on the specification limits for residues of metal catalysts or metal reagents”, can be found under http://www.ema.europa.eu/ema/, 2008.
- 24.
- 24a. Tian T., Zhong W.-H., Meng S., Meng X.-B., Li Z.-J., J. Org. Chem. 2013, 78, 728–732; [DOI] [PubMed] [Google Scholar]
- 24b. Clayden J., Lemiègre L., Pickworth M., Tetrahedron: Asymmetry 2008, 19, 2218–2221. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary