

Abstract
Individuals are at risk of exposure to acute ionizing radiation (IR) from a nuclear accident or terrorism, but we lack effective therapies to mitigate the lethal IR effects. In the current study, we exploited an optimized, cell-based, high throughput screening assay to interrogate a small molecule library comprising 3437 known pharmacologically active compounds for mitigation against IR-induced apoptosis. Thirty-three library compounds significantly reduced apoptosis when administered 1 h after 4 Gy IR. Two- or three-dimensional computational structural analyses of the compounds indicated only one or two chemical clusters with most of the compounds being unique structures. The mechanistic target of rapamycin complex 1 (mTORC1) inhibitor, rapamycin, was the most potent compound, and it mitigated apoptosis by 50% at 200 ± 50 pM. Other mTOR inhibitors, namely everolimus, AZD8055, and torin 1, also suppressed apoptosis, providing additional pharmacological evidence for mTOR pathway involvement in regulating cell death after IR. Everolimus and torin 1 treatment after IR decreased the S phase population and enforced both G1 and G2 phase arrest. This prorogation of cell cycle progression was accompanied by decreased IR-induced DNA damage measured by γH2AX phosphorylation at Ser139. RNA interference-mediated knockdown of the respective mTORC1 and mTORC2 subunits, Raptor or Rictor, also mitigated IR-induced apoptosis. Collectively, this study suggests a central role for the mTOR signaling in the cytotoxic response to IR and offers a useful platform to probe for additional agents.
Graphical abstract

Well-known catastrophic events, such as the Three Mile Island, Chernobyl, and Fukushima Daiichi nuclear reactor accidents, dramatically underscore the pervasive possibility for accidental mass ionizing radiation (IR) exposure to populations.1 Additionally, there is the potential that radioactive materials could be exploited as weapons, exposing large numbers of individuals to life-threating levels of IR.2 Whole-body or significant partial-body exposure to 1–8 Gy doses of IR results in acute radiation syndrome, which includes clinical hematopoietic, cutaneous, gastrointestinal, and neurovascular syndromes.3 These clinical manifestations are thought to be due, at least in part, to the death and exhaustion of progenitor cell populations. Serious but delayed loss of cognitive function after IR is believed to be caused by stem cell depletion in neurogenic central nervous system (CNS) cells, leading to impaired neurogenesis.4, 5 Current therapeutic approaches are purely palliative in nature and focus on controlling infection and managing pain or symptoms. Because IR causes prolonged effects on cells, which can mediate the death process, it seems reasonable to hypothesize that there are pharmacological agents that could directly curtail progenitor cell death caused by IR exposure. Indeed, several mitigating agents have been reported to be effective in cell culture models and mice, although in general their mechanism of action has not been firmly established and none have been adopted for clinical use.6–12 Our goal was to expand the number of potential candidate small molecules for further prosecution as radiation mitigators and to exploit the compounds to identify novel targetable pathways.
One powerful approach for identifying lead chemical structures for ultimate therapeutic use has been high throughput screening (HTS).13 It is quite surprising, therefore, that there has been only one reported attempt to exploit HTS and mechanistically unbiased phenotypic assays in the search for mitigators of IR-induced cell death.14 The authors used murine T lymphocytes to examine a library of chemically diverse bioactive small molecules for their ability to reduce IR-induced apoptosis and found the purine nucleosides, adenosine and vidarabine, and the antibiotics, tetracycline and fluoroquinolones, to exhibit efficacy.14 Unfortunately, murine Tlymphocytes are not pluripotent, and reproducibly culturing sufficient numbers of human stem cells represents a formidable obstacle for HTS. Therefore, our group adopted NCCIT cells, which are immortalized and developmentally pluripotent and can differentiate into derivatives of all three embryonic germ layers (i.e., ectoderm, mesoderm, and endoderm) and extraembryonic cell lineages,15 as a screening platform for radiation mitigation, because they have cell cycle characteristics and radiobiological sensitivity relevant to human progenitor cells.16 We initially use NCCIT cells to examine a small interfering RNA (siRNA) library and identified a number of attractive drug targets for IR mitigation, including the phosphoinositide 3-kinase (PI3K) pathway.16 We have now used this optimized cell system to interrogate a chemical library of 3437 pharmacologically active agents for IR mitigation.
RESULTS AND DISCUSSION
Identification of Rapamycin As a Radiomitigator
The human pluripotent NCCIT cell line provides a robust platform to quantify radiosensitivity using an assay measuring caspase 3/7 activation.16 We combined two small molecule libraries comprising pharmaceutically active compounds with only 7% overlap to produce a unified chemical library with 3437 compounds. We focused on chemotypes that are pharmaceutically active, have advanced into clinical testing, or are already approved, hoping to expedite the discovery of additional novel radiation mitigators and attractive therapeutic targets. NCCIT cells were irradiated in suspension with 4 Gy, a radiation dose that we previously found caused a 50% reduction in cell viability,16 were then plated, and 1 h later were treated with vehicle or test compounds (1 or 5 μM) continuously for 47 h until the cells were assayed for apoptosis by measuring caspase 3/7 activation. Compounds with a Z-score of ≤ −1.0 at 1 and 5 μM were rescreened for confirmation. Of the 3437 compounds screened, a total of only 33 (1%) reproducibly decreased caspase 3/7 activation with a Z-score of ≤ −1.0 at either concentration in this primary screen. These compounds were reordered from commercial vendors and tested at 10 different concentrations ranging from 10 nM to 50 μM. We found that 27 of the 33 initial hits generated reproducible concentration-dependent mitigation of IR-induced apoptosis with EC50 values ranging from 0.2 nM to 22 μM (Table 1; Supporting Figure 1). The chemical richness of the identified radiation mitigators was further supported by a two-dimensional chemical analysis of the 27 active compounds, which revealed only one structural cluster of five compounds, containing mycophenolic acid, monobenzone, genistein, emodin, and anisinodione and 22 structurally unique compounds (Figure 1A). When conducting a three-dimensional analysis of similarity, we found four of the 27 compounds, namely cobamamide, hydroxocobalamin, rapamycin, and diadzein, did not have adequate 3D conformers in the PubChem database to permit inclusion in the 3D analyses. With the remaining 23 compounds, we found one small cluster of two pyrimidine nucleosides, 6-azauridine and gemcitabine, and one large cluster of 10 similar structures, namely, chrysoidine, dexindoprofen, genistein, lefunomide, monobenzone, neutral red, emodin, amlexanox, niclosamide, and thiabendazole (Figure 1B). The remaining 11 compounds appear structurally unrelated (Figure 1B).
Table 1.
Identified Small Molecule Radiation Mitigators Using High-Throughput Screening
| compound | CIDa | EC50 μMb |
|---|---|---|
| rapamycin | 5284616 | 0.0002 ± 0.00005 |
| cycloheximide | 6197 | 0.03 ± 0.01 |
| nitazoxanide | 41684 | 0.11 ± 0.02 |
| leflunomide | 3899 | 0.3 ± 0.2 |
| amlexanox | 2161 | 0.3 ± 0.1 |
| mycophenolic acid | 446541 | 0.4 ± 0.2 |
| hydroxocobalamin | 24892771 | 0.4 ± 0.2 |
| cobamamide | 54740488 | 0.5 ± 0.2 |
| niclosamide | 4477 | 0.5 ± 0.1 |
| gemcitabine | 60750 | 0.6 ± 0.1 |
| AC-93263 | 16078948 | 0.7 ± 0.3 |
| bromindione | 14369 | 0.8 ± 0.3 |
| artesunate | 5464098 | 1.1 ± 0.2 |
| chrysoidine | 10771 | 1.1 ± 0.6 |
| daidzein | 568512 | 1.0 ± 0.4 |
| monobenzone | 7638 | 1.2 ± 0.2 |
| PF-573228 | 11612883 | 1.2 ± 0.1 |
| dexindoprofen | 68700 | 1.4 ± 0.7 |
| quinoline yellow | 24671 | 1.5 ± 0.4 |
| tyrphostin A9 | 5614 | 1.50 ± 0.02 |
| emodin | 3220 | 1.6 ± 0.7 |
| neutral red | 11105 | 1.7 ± 0.2 |
| genistein | 5280961 | 2.1 ± 0.8 |
| sunitinib | 5329102 | 2.9 ± 0.1 |
| thiabendazole | 5430 | 6.8 ± 0.6 |
| 6-azauridine | 5901 | 10.5 ± 1.8 |
| anisindione | 2197 | 22.3 ± 1.1 |
CID is the PubChem chemical identifier.
The mean of four independent determinations ± SEM.
Figure 1.

2D and 3D chemical structural similarity analysis of radiation mitigators. (A) The 2D Tanimoto similarity substructure fingerprint for the 27 identified active radiation mitigators using the PubChem CIDs (listed on the right of the figure) and a single linkage algorithm.40 Compounds with a Tanimoto score of ≥0.68 are highlighted in red. (B) The 23 compounds with ≥10 3D conformers in the PubChem database (listed on the right of the figure) were structurally clustered. Similarity scores of ≥1.1 were considered statistically significantly different,41 and the corresponding compounds are highlighted in red.
The most potent radiation mitigator in our initial screen was the mTORC1 inhibitor rapamycin with an EC50 of 200 ± 50 pM (Table 1; Supporting Figure 1). This immunosuppressant drug was structurally unique among the active small molecules. It is notable that rapamycin was previously found to protect epithelial stem cells from death when given before IR.17 Four other identified compounds, namely genistein,18 emodin,19 cycloheximide,20, 21 and thiabendazole,22 also have been previously reported to protect either cells or mice against IR when given prior to IR exposure. To our knowledge, however, none of these compounds, with the exception of thiabendazole combined with dinitrofluorobenzene,22 have been shown to mitigate toxicity when given after IR. A compound such as the general protein synthesis inhibitor cycloheximide, which has previously been observed to suppress IR-induced apoptosis in human MOLT-4 T-cell leukemia cells20 and skin damage in rats,21 was potent in our assay (EC50 of 30 ± 10 nM) but was of less interest because of its potential systemic toxicity. Thus, we focused on rapamycin.
Initially, mitigation of IR-induced apoptosis with rapamycin was somewhat unexpected as the mTOR pathway is generally viewed as a pro-survival pathway, at least in cancer cells.23 Nonetheless, we probed this chemotype further in part, because we noticed that several of our previously identified radiation mitigators, which were PI3K inhibitors,9 also have some modest capacity to inhibit mTOR. Moreover, three of the compounds identified in our small molecule screen, namely emodin,24 niclosamide,25 and nitazoxanide,26 have been reported to disrupt mTOR signaling. mTOR has recently been implicated in stress-mediated cell death.27 Collectively, these observations lend support for the hypothesis that mTOR signaling could be important in the ultimate lethal effects of irradiation.
mTOR Inhibitors Everolimus, Torin 1, and AZD8055 Inhibit Caspase 3/7 Activation after IR
To confirm the radiation mitigation by the rapamycin structural class, we evaluated an analog, everolimus. At concentrations that inhibited the phosphorylation of the mTORC1 substrate p70S6 kinase (Supporting Figure 2), everolimus also profoundly suppressed IR-induced caspase 3/7 activation at drug concentrations comparable to rapamycin (Figure 2A, B). Although rapamycin and everolimus target the mTOR signaling pathway, these first generation allosteric inhibitors only block mTORC1 and not mTORC2.28 Thus, we next evaluated torin 1 and AZD8055, second-generation, structurally distinct, ATP competitive mTOR inhibitors that target both mTORC1 and mTORC2 for IR mitigative effects.28 Torin 1 and AZD8055 treatment essentially restored caspase 3/7 levels almost to baseline nonirradiated levels when given 1 h after IR (Figure 2C, D). The mitigation with torin 1 was independent of the IR dose (Figure 2E). None of the compounds significantly activated NCCIT caspase 3/7 under non-IR conditions. Treatment of cells 3 or 6 h after IR with rapamycin or AZD8055 also reduced apoptosis as measured by caspase 3/7 activation but not after 24 h (Figure 3A, C). In contrast, torin 1 mitigated caspase 3/7 activation at all three time points, albeit rather modestly when given 24 h after IR (Figure 3B). Based on the observed reduction in apoptosis, we next evaluated the ability of everolimus to enhance cell replicative capacity as measured by in vitro colony formation. When NCCIT cells were treated with only 10 nM everolimus 1 h after IR (0−8 Gy), we observed a modest but reproducible increase in NCCIT survival, as indicated by the increased shoulder on the radiation survival curves versus the vehicle control irradiated cells (vehicle ñ = 3.3 ± 0.4 vs everolimus ñ = 9.4 ± 1.6, p = 0.018, N = 3; Figure 2F).
Figure 2.

Radiation mitigation with mTOR inhibitors. NCCIT cells were exposed to 0 (○) or 4 Gy (■) IR. One hour later, cells were treated with 0.1% DMSO vehicle control or various concentrations of rapamycin (A), everolimus (B), torin 1 (C), or AZD8055 (D). After 48 h, caspase 3/7 activity was quantified (N = 3 or 4, SEM indicated by bars unless smaller than the symbol). Data analyzed using ANOVA. *p < 0.05 between cells exposed to 0 or 4 Gy IR. (E) NCCIT cells were exposed to various IR doses and 1 h later treated with DMSO control (○) or 200 nM torin 1 (■) and incubated for 48 h, at which time caspase 3/7 activity was determined (N = 3, SEM indicated by bars unless smaller than the symbol). (F) NCCIT cells were exposed to 0−8 Gy and 1 h later treated with DMSO (○) or 10 nM everolimus (Δ). Cells were incubated at 37 °C for 7 days with everolimus or DMSO, at which time surviving colonies were counted. The data were fitted using a single-hit, multitarget model. N = 3, SEM indicated by bars unless smaller than the symbol.
Figure 3.

Kinetics of radiation mitigation by mTOR inhibitors and mitigation with genetic knockdown of mTOR subunits Rictor and Raptor. Cells were exposed to 0 (open symbols) or 4 (closed symbols) Gy IR, and 3, 6, or 24 h later, cells were treated with 0.1% DMSO vehicle control or various concentrations of rapamycin (A), torin 1 (B), or AZD8055 (C). Forty-eight hours after IR exposure, cellular caspase 3/7 activity was quantified (N = 9−14 samples, SEM indicated by bars unless smaller than the symbol). Data analyzed using ANOVA. *p < 0.05 between irradiated cells exposed to vehicle or compound. (D and E) NCCIT cells were transfected with Raptor, Rictor, and/or scrambled siRNA then exposed to 4 Gy IR with a nonirradiated sample set run in parallel. Total siRNA added was held at a constant 600 ng with 300 ng of Raptor, Rictor, or scrambled siRNA. Forty-eight hours later, caspase 3/7 activity was quantified. Shown is a representative experiment with three samples. The experiment has been repeated three times with similar results. *Statistical significance p < 0.05 (ANOVA).
Genetic Knockdown of Rictor and Raptor with siRNA Inhibits IR-Induced Caspase 3/7 Activation
To further document the radiation mitigation effects of mTOR inhibition, we performed genetic knockdown studies targeting the respective mTORC1 and mTORC2 subunits, Raptor and Rictor. NCCIT cells were transfected with various combinations of scrambled, Raptor, and Rictor siRNA and then were exposed to IR. Following a 47 h incubation, siRNA knockdown of Raptor or Rictor modestly but reproducibly inhibited caspase 3/7 activation in irradiated cells (p < 0.05, ANOVA; Figure 3D, E). Similarly, a combination of Raptor and Rictor siRNA also significantly inhibited IR-induced caspase 3/7 activation (p < 0.05 ANOVA). RNA knockdown was confirmed by quantitative-PCR and Western blot, respectively (Supporting Figure 3).
Everolimus and Torin 1 Suppresses IR-Induced Annexin V Expression
Inhibition of caspase 3/7 activity suggested that everolimus and torin 1 suppress IR-induced apoptosis. To confirm this potential radiation mitigation response, we next examined the effects of everolimus and torin 1 treatment on phosphatidylserine cell surface expression, which reflects later stages of apoptosis. NCCIT cells were exposed to 0 or 4 Gy IR, then 1 h later, they were treated with 0.1% DMSO, 12.5 nM everolimus, or 200 nM torin 1 for 48 h. In the DMSO treated cells, as anticipated, IR exposure significantly increased phosphatidylserine cell surface expression as quantified by annexin V staining and flow cytometry (Figure 4). After exposure to 4 Gy IR, treatment with either everolimus or torin 1 significantly, albeit incompletely, suppressed phosphatidylserine cell surface expression (Figure 4; *p < 0.05 ANOVA).
Figure 4.
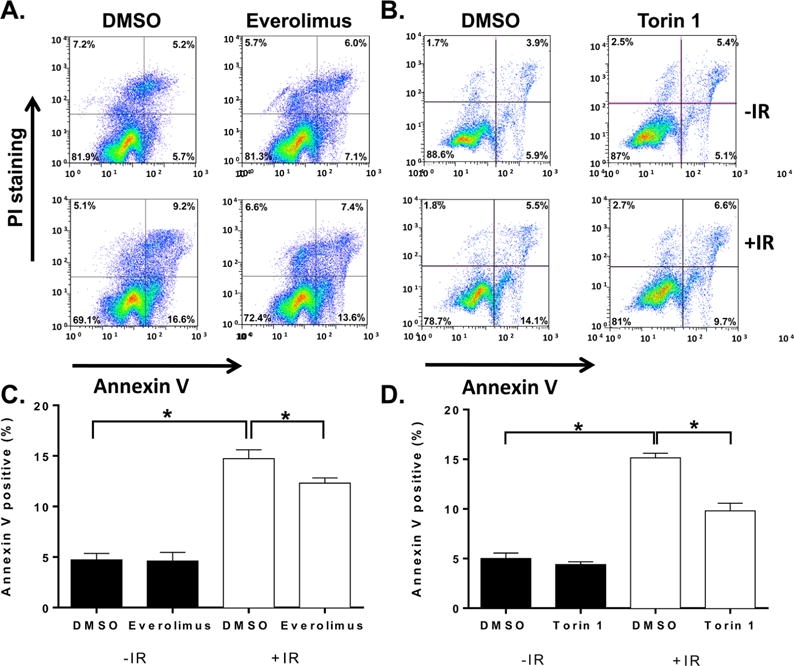
Reduction of phosphatidylserine expression on IR exposed NCCIT cells as a result of everolimus and torin 1 treatment. NCCIT cells were exposed to 0 (black bars) or 4 (white bars) Gy IR, then treated one h later with either 12.5 nM everolimus or 200 nM torin 1 for 47 h. Effects on phosphatidylserine cell surface expression were quantified by annexin V staining via flow cytometry. (A and B) Representative flow cytometry profiles. (C and D) Data are presented as average ± SEM (N = 3 independent experiments) and were analyzed using ANOVA. *Statistically significant (p < 0.05).
Inhibition of the mTOR Pathway Enforces a G1 and G2 Phase Arrest after IR
Nonirradiated NCCIT cells, similar to embryonic stem cells,29 maintained a high percentage (73.5 ± 2.6%) of the total population in S phase with 19.6 ± 2.2% and 6.9 ± 3.9% in G1 and G2/M phases, respectively (Figure 5 and Supporting Figure 4). These data also confirmed our previously reported results using NCCIT cells.9 After exposure to 4 Gy IR, NCCIT cells displayed a prominent but transient G2 phase arrest (Figure 5 and Supporting Figure 4). Twenty-eight hours after IR exposure, 62.0 ± 3.9% of the total cell population was in the G2/M phase. By 48 h, the G2/M phase population had declined to 17.2 ± 1.9% and was comparable to the nonirradiated levels (Figure 5). The G1 phase cells comprised 11.5% ± 3.7% and 18.5% ± 4.2% of the total population 28 and 48 h after IR, respectively. Treatment of nonirradiated cells with a radiation mitigation concentration of torin 1 (200 nM) increased the percentage of nonirradiated cells in the G1 phase at 24 and 48 h and significantly (p < 0.05) decreased the percentage of S phase cells to 44.2 ± 3.5% and 42.9 ± 0.7%, respectively, compared to the vehicle treated control cell population (Figure 5). After IR, torin 1 maintained a relatively stable S phase population at 46.6% ± 08%, while producing an increase in the fraction of cells in G2/M phase to 25.1% ± 2.9% at 24 h (Figure 5C–D). At 48 h, torin 1 decreased S phase cells (27.7 ± 1.8%) and increased the G2/M population (Figure 5C–D). Similar results were observed with radiation mitigation concentration of everolimus (12.5 nM) (Supporting Figure 4). Thus, both compounds markedly reduced the S phase population in irradiated cells. Mechanistically this may be important as a delay in entry into S phase or mitosis would provide additional time to repair any DNA damage that could stimulate cell death. We therefore examined the DNA integrity of the cells.
Figure 5.

Cell cycle analyses after torin 1 treatment. NCCIT cells were exposed to 0 or 4 Gy IR, plated, and incubated at 37 °C for 1 h before treatment with DMSO vehicle or torin 1 (200 nM). Cells were incubated for 28 or 48 h and treated with 10 μM 5-ethynyl-2′-deoxyuridine (EdU) and, after an additional 6 h incubation, were harvested and analyzed with FACSCalibur. (A,B). Representative flow cytometry profiles. Quantitative cell cycle distribution 28 h (C) or 48 h (D) after γIR exposure. Sub-G1 DNA content was gated out. Black columns indicate nonirradiated cells, and white columns indicate irradiated cells. N = 3 independent experiments, bars = mean ± SEM; * p < 0.05 Student t test.
DNA Integrity Improves with mTOR Inhibitors and Radiation Mitigation Occurs with Neural Progenitor Cells (NPCs)
Exposure of cells to IR produces double-strand DNA breaks that rapidly resulted in the phosphorylation of histone γH2AX on Ser39, which is a reliable marker for DNA damage.30 Thus, as anticipated, treatment of NCCIT cells with 4 Gy IR produced >3-fold increase in histone γH2AX phosphorylation at 3 h (Figure 6A, B). Neither torin 1 nor everolimus altered the basal γH2AX phosphorylation but decreased DNA damage as measured by histone γH2AX phosphorylation (Figure 6A, B). Control of DNA integrity is a complex process, and the decreased histone γH2AX phosphorylation could reflect increased DNA repair or less secondary DNA damage that occurs subsequent to IR. Because IR has been reported to accelerate cellular senescence and mTOR inhibition protects against stem cell depletion,17 we examined NCCIT cell senescence after IR and everolimus treatment. Within 24 h after 4 Gy IR, approximately 20% of the cell population expressed senescence-associated β-galactosidase (Figure 6D). Treatment with everolimus 1 h after IR did not alter the β-galactosidase activity excluding altered cellular senescence as a factor in the mitigation. Finally, we examine another cell population, NPCs, for mitigation of IR induced apoptosis by mTOR pathway inhibition. Because torin 1 was the most potent and effective IR mitigator with NCCIT cells, we treated NPCs 1 h after 1 Gy IR with 1−10 nM torin 1 and, similar to our results with NCCIT cells, torin 1 treatment of NPCs decreased IR induced caspase 3/7 activation in a concentration-dependent manner with complete suppression observed with 10 nM torin 1 (Figure 6E). We examined mTOR signaling in NPCs after IR treatment using immunofluorescence (Supporting Figure 5). An antibody to Rac1, which is a small plasma membrane associated GTPase involved in the PI3K/mTOR signaling pathway, was used to define the plasma membrane. We found evidence for transient phosphorylation and activation of the mTORC1 substrate pP70S6K within 30 min after NPCs were exposed to 1 Gy IR (Supporting Figure 5A and B).
Figure 6.

Reduction of DNA damage with everolimus and torin 1. NCCIT cells were treated with 0 or 4 Gy IR, plated, and incubated at 37 °C for 1 h before treatment with DMSO vehicle, everolimus (EV) (12.5 nM), or torin 1 (200 nM) for 3 h. (A,C) Representative Western blots of γH2AX and GAPDH are shown. Western blots were quantified using the LI-COR Odyssey, and the data were normalized to the non-IR control and indicated above the blot. (B) Quantification of the GAPDH normalized γH2AX blot intensity in cells exposed to 0 (black bars) or 4 (white bars) 4 Gy IR with 200 nM torin 1 or vehicle control. N = 3, bars = mean ± SEM. *p < 0.05 Student t test. (D) Quantification of senescence-associated β-galactosidase (β-gal) 24 h after cells were exposed to 0 (black bars) or 4 (white bars) 4 Gy IR and treated with 12.5 nM everolimus or vehicle control. N = 3, bars = mean ± SEM. *p < 0.05 Student t test. (E) Human NPCs were exposed to 1 Gy IR and 1 h later treated with 0−10 nM torin 1. Forty-eight hours later, caspase 3/7 activity was measured. Bars = mean of three independent studies ± SEM. *p < 0.05 Student t test.
The HTS Platform Provides New Lead Compounds for Radiation Mitigation
The compounds identified in our initial small molecule screen represent a rich, chemically diverse, and previously unrecognized potential set of compounds for further investigation. The only other previously described radiation migitators from HTS, namely adenosine, vidarabine, tetracycline, and fluoroquinolones, were absent from our library and, thus, not detected. These compounds, however, were not very potent, and the mechanisms responsible for their radiation mitigation effects are unclear. In contrast, several of the compounds we identified, such as the mTOR inhibitors, were substantially more potent and have a well-defined mechanism of action in eukaryotic cells, which should enhance their therapeutic potential.
During a previous siRNA screening study, we identified PI3K as a molecular target with a role in mitigating the in vitro and in vivo lethal effects after exposure to IR.9, 16 Interestingly, most of the PI3K inhibitors with radiation mitigative effects also inhibit mTOR, which is a master regulator of cell growth and metabolism and points to a potential role for mTOR in mitigating the effects of IR in NCCIT cells. This hypothesis was supported with the identification of rapamycin in the current screen. Rapamycin is FDA-approved for prevention of organ rejection after transplantation.31, 32 It is a potent allosteric inhibitor of mTORC1, which acts as an energy sensor and regulator of protein synthesis.33 Under normal cellular growth conditions, mTORC1 activation would signify that the cells have the appropriate resources (i.e., energy, nutrients, growth factors) available to initiate protein translation leading to cell growth and/or sustained viability.33 Under potential catastrophic cellular stress, however, such as radiation exposure, activation of mTOR leads to accelerated cell death as the necessary cellular nutrients for productive protein translation are not available or have been diverted to other cell survival or maintenance mechanisms. Thus, inhibiting the mTOR pathway may prevent the damaged cell from progressing through a signaling program that would ultimately lead to cell death. Significantly, rapamycin protects epithelial stem cells against IR17 and prevents stem cell exhaustion.23 Rapamycin was recently found to block Fyn- and ER-stress induced cell death, which is mediated at least in part by mTOR.27
The rapamycin analog everolimus, which also allosterically targets mTORC1, and the dual mTORC1/mTORC2 ATP competitive inhibitors, torin 1 and AZ8055, inhibited IR induced cell apoptosis after IR exposure. Despite their differential targeting of mTORC subcomplexes and mechanism of action (i.e., allosteric versus ATP competitive inhibitor), both everolimus and torin 1 arrested the IR damaged NCCIT cell in G1/G2, presumably enabling the cells to recover from DNA damage (i.e., γH2AX phosphorylation) sustained after IR. Both pharmacological and genetic inhibition of mTOR support mTOR as a potential radiation mitigation “node.”
Our results suggest HTS with model systems can provide valuable new potential radiation mitigation agents. It is worth noting, however, that the NCCIT cells originated from an extragonadal mediastinal human germ cell cancer and, thus, may not faithfully replicate all of the responses of the human stem cell populations most seriously damaged by IR. Nonetheless, it is well established that NCCIT cells are developmentally pluripotent and retain the ability to differentiate into all three embryonic germ layers.15 Our observation that caspase 3/7 activation was reduced in human NPCs treated with torin 1 after IR suggests mitigation is not unique to NCCIT cells. Clearly, other stem cell populations should be investigated to determine the generality of the phenomenon. The regulation of cellular responses by the mTOR pathway is quite complex, and additional studies are warranted to determine the precise mechanisms responsible for the observed mitigation. Our observation that mTOR inhibitors have radiomitigative effects complements our previous work describing the pharmacological inhibition of the PI3K signaling pathway. Rapamycin and its analogs are reasonably well-tolerated in animals and humans,34 making them attractive candidates for further studies to determine if they can reduce the toxic effects of IR in whole animals. In addition, our results suggest that we can target effectors downstream of PI3K, thereby minimizing possible adverse effects that may be associated with inhibiting a kinase that is involved with a multitude of vital cellular functions.
METHODS
Reagents
CaspaseGlo 3/7 caspase activity assays were purchased from Promega. White and black 384-well microtiter plates were from Greiner Bio-One and six-well plates from Corning. RPMI-1640 medium and fetal bovine sera (FBS) were from MediaTech, Inc. Rapamycin and everolimus were from Selleck Chemicals, and torin 1 was from Tocris Bioscience. The Library of Pharmacologically Active Compounds (LOPAC) set comprising 1280 compounds was purchased from Sigma-Aldrich. The PharmSub library was assembled at the University of Virginia and consisted of 2157 compounds with 7% overlapping compounds with LOPAC. Rictor and Raptor siRNA expression vectors were obtained from Addgene.
Cell Culture and HTS
Human pluripotent embryonal carcinoma NCCIT cells (American Type Culture Collection) were cultured as previously described.9, 16 Our previously described caspase 3/7 assay9, 16 was adapted to an automated screening format. Briefly, we seeded 3500 NCCIT cells/well in 384 well microtiter plates after 0 or 4 Gy IR using a JL Shepard Mark I Model 68 cesium source (75 cGy/min). After a 1 h incubation at 37 °C, DMSO vehicle control or one of the 3437 library compounds (1 or 5 μM) were added to the cells. After a 47 h incubation, CaspaseGlo 3/7 was added, and after 1 h at RT, luminescence was measured on a Molecular Devices SpectraMax M5 plate reader. Primary actives were compounds that displayed a Z-score ≤ −1.0 at both concentrations for decreased caspase 3/7 activity, were resupplied from commercial vendors (primarily Sigma-Aldrich and Tocris), and were confirmed using concentration response assays in the same caspase 3/7 screening assay format (0−50 μM).
Human NPC Culture and IR
NPCs (BJ2B) were maintained in Dulbecco’s Modified Medium/Ham’s F12 (1:1) with Glutamax supplement, 1X N2 (Invitrogen), 1X B27 without vitamin A (Invitrogen), murine laminin (1 μg/mL; Invitrogen), and FGF2 (1:1000; PeproTech). NPCs were cultured on Matrigel coated (1.7% in growth medium) tissue cultureware, harvested using Accutase (Innovative Cell Technologies), and maintained at 37 °C with 5% CO2. NPCs were carried for ≤8 passages. All IR exposures were performed with suspended NPCs. NPCs were plated and incubated for 1 h prior to compound addition. Caspase 3/7 levels were quantified 48 h post-IR and as described above. Immunohistochemistry of NPCs was as previously described35 on a Zeiss Axiovert 100 microscope.
Cell Cycle, Annexin V, and Colony Formation Assays
NCCIT cells were exposed to 0 or 4 Gy IR as a single cell suspension in a 50 mL conical tube and plated in six-well plates at a density of 2 × 105 cells/well. After 1 h, cells were treated with DMSO vehicle, everolimus (12.5 nM), or torin 1 (200 nM) and incubated for the indicated time. We used our previously described flow cytometry assays9, 16 to determine cell cycle distribution, apoptosis with annexin V, and live or dead cells with propidium iodide as described by Firat et al.36 Colony formation was determined after 7 days with continuous compound treatment and data analyzed as previously described.37, 38
Western Blotting
Protein lysates from NCCIT cells were isolated and examined by Western blotting as previously described.39 Membranes were blotted in PBS containing 0.1% Tween 20 with Odyssey Blocking Buffer (LI-COR) diluted 1:2 (vol:vol) at 4 °C overnight with the following antibodies: anti-P70S6K, anti-pP70S6K, anti-p4EBP1, anti-4EBP1, anti-γH2AX, or anti-GAPDH (Cell Signaling Technology). Positive antibody reactions were visualized using goat antirabbit DyLight 680 or 800 (Thermo Fisher Scientific). Membranes were imaged using a LI-COR Odyssey scanner, and bands were analyzed using Odyssey 3.0 analytical software (LI-COR).
siRNA Knockdown of Rictor and Raptor
We seeded 3500 NCCIT cells/well in 384-well microtiter plates. siRNA (total 600 ng) transfection reactions were constructed using Lipofectamine LTX reagent (Life Technologies) following the manufacturer’s instructions. siRNA transfection reactions were added to cells and allowed to incubate for 24 h at 37 °C in 5% CO2. Transfected cell cultures were then exposed to 0 or 4 Gy IR. Cells were incubated for 47 h at 37 °C in 5% CO2, at which time the CaspaseGlo 3/7 reagent (Promega) was added into the wells containing phenol red-free RPMI-1640 medium with 10% FBS (1:2 dilution). After a 1 h incubation at RT, the luminescence was measured on a SpectraMax M5 plate reader.
Chemical Structural Analysis
We performed 2D and 3D similarity analyses of the identified high quality radiation mitigators using public domain software (http://pubchem.ncbi.nim.nih.gov/). Briefly, we generated a 2D Tanimoto similarity substructure fingerprint for the 27 identified active radiation mitigators using the PubChem CIDs and the single linkage algorithm.40 A Tanimoto score of ≥0.68 was considered statistically significant at the 95% confidence interval. We found 3D conformers for 23 of the 27 compounds from the PubChem database (i.e., PubChem CIDs). For each parental chemotype, up to 10 conformers were calculated. Structural clustering was based on the parental chemotypes, with the most similar conformer pairs used to represent a pair of compounds when conformer sets were compared. For calculations of 3D similarity, 3D superposition was optimized by shape with 3D similarity based on the sum of the shape and feature similarity scores. Similarity scores of ≥1.1 using up to 10 calculated conformers were considered statistically significantly different.41
Data Analysis and Statistics
Data were analyzed using GraphPad Prism 6.0. Data were expressed as means ± SEM of at least three determinations. Statistical comparisons between different groups were performed with Student’s t test or ANOVA unless otherwise noted. p ≤ 0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health National Institute of Allergy and Infectious Diseases [1U19AI68021], the Fiske Drug Discovery Fund, the Owens Family Foundation, and the Cure Alzheimer’s Fund. M.J.M. receives support from the University of Virginia School of Medicine and the Department of Biochemistry and Molecular Genetics for establishing a hIPS cell core. We are very grateful for the technical assistance of J. Bryant, H. Wallrabe, and the UVA Flow Cytometry Core Facility.
Footnotes
Supporting Information
This material is available free of charge via the Internet. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.5b00909.
Supporting Figures 1–5 (PDF)
Supporting figure cations (PDF)
Author Contributions
E.R.S., G.S.B., J.S.G., and J.S.L. conceived the experimental question and design and assisted in data analyses. E.R.S. and J.S.L. conducted the chemical structural analyses. S.L., A.L., and M.W.E. conducted the IR mitigation assays with NCCIT cells and caspase, flow cytometry, colony formation, γH2AX, and β-galactosidase. M.J.M., E.R.S., and A.N. performed the experiments with NPCs.
Notes
The authors declare no competing financial interest.
References
- 1.Fushiki S. Radiation hazards in children – lessons from Chernobyl, Three Mile Island and Fukushima. Brain Dev. 2013;35:220–227. doi: 10.1016/j.braindev.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Rios P, Li X, Kohn M. Molecular mechanisms of the PRL phosphatases. FEBS J. 2013;280:505–524. doi: 10.1111/j.1742-4658.2012.08565.x. [DOI] [PubMed] [Google Scholar]
- 3.Christensen DM, Iddins CJ, Sugarman SL. Ionizing radiation injuries and illnesses. Emerg Med Clin North Am. 2014;32:245–265. doi: 10.1016/j.emc.2013.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Tseng BP, Giedzinski E, Izadi A, Suarez T, Lan ML, Tran KK, Acharya MM, Nelson GA, Raber J, Parihar VK, Limoli CL. Functional consequences of radiation-induced oxidative stress in cultured neural stem cells and the brain exposed to charged particle irradiation. Antioxid Redox Signaling. 2014;20:1410–1422. doi: 10.1089/ars.2012.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lan ML, Acharya MM, Tran KK, Bahari-Kashani J, Patel NH, Strnadel J, Giedzinski E, Limoli CL. Characterizing the radioresponse of pluripotent and multipotent human stem cells. PLoS One. 2012;7:e50048. doi: 10.1371/journal.pone.0050048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greenberger JS, Clump D, Kagan V, Bayir H, Lazo JS, Wipf P, Li S, Gao X, Epperly MW. Strategies for discovery of small molecule radiation protectors and radiation mitigators. Front Oncol. 2011;1:1–12. doi: 10.3389/fonc.2011.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moulder JE, Cohen EP, Fish BL. Captopril and losartan for mitigation of renal injury caused by single-dose total-body irradiation. Radiat Res. 2011;175:29–36. doi: 10.1667/RR2400.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee CL, Lento WE, Castle KD, Chao NJ, Kirsch DG. Inhibiting glycogen synthase kinase-3 mitigates the hematopoietic acute radiation syndrome in mice. Radiat Res. 2014;181:445–451. doi: 10.1667/RR13692.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazo JS, Sharlow ER, Epperly MW, Lira A, Leimgruber S, Skoda EM, Wipf P, Greenberger JS. Pharmacologic profiling of phosphoinositide 3-kinase inhibitors as mitigators of ionizing radiation-induced cell death. J Pharmacol Exp Ther. 2013;347:669–680. doi: 10.1124/jpet.113.208421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh VK, Romaine PL, Seed TM. Medical Countermeasures for Radiation Exposure and Related Injuries: Characterization of medicines, FDA-approval status and inclusion into the strategic national stockpile. Health Phys. 2015;108:607–630. doi: 10.1097/HP.0000000000000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taniguchi CM, Miao YR, Diep AN, Wu C, Rankin EB, Atwood TF, Xing L, Giaccia AJ. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med. 2014;6:236r–a264. doi: 10.1126/scitranslmed.3008523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patil R, Szabó E, Fells JI, Balogh A, Lim KG, Fujiwara Y, Norman DD, Lee SC, Balazs L, Thomas F, Patil S, Emmons-Thompson K, Boler A, Strobos J, McCool SW, Yates CR, Stabenow J, Byrne GI, Miller DD, Tigyi GJ. Combined mitigation of the gastrointestinal and hematopoietic acute radiation syndromes by an LPA2 receptor-specific nonlipid agonist. Chem Biol. 2015;22:206–216. doi: 10.1016/j.chembiol.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discovery. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 14.Kim K, Damoiseaux R, Norris AJ, Rivina L, Bradley K, Jung ME, Gatti RA, Schiestl RH, McBride WH. High throughput screening of small molecule libraries for modifiers of radiation responses. Int J Radiat Biol. 2011;87:839–845. doi: 10.3109/09553002.2011.560994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damjanov I, Horvat B, Gibas Z. Retinoic acid-induced differentiation of the developmentally pluripotent human germ cell tumor-derived cell line, NCCIT. Lab Invest. 1993;68:220–232. [PubMed] [Google Scholar]
- 16.Zellefrow CD, Sharlow ER, Epperly MW, Reese CE, Shun T, Lira A, Greenberger JS, Lazo JS. Identification of druggable targets for radiation mitigation using a small interfering RNA screening assay. Radiat Res. 2012;178:150–159. doi: 10.1667/rr2810.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iglesias-Bartolome R, Patel V, Cotrim AP, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012;11:401–414. doi: 10.1016/j.stem.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Landauer MR, Srinivasan V, Seed TM. Genistein treatment protects mice from ionizing radiation injury. J Appl Toxicol. 2003;23:379–385. doi: 10.1002/jat.904. [DOI] [PubMed] [Google Scholar]
- 19.Sharma R, Tiku AB. Emodin, an anthraquinone derivative, protects against gamma radiation-induced toxicity by inhibiting DNA damage and oxidative stress. Int J Radiat Biol. 2014;90:275–283. doi: 10.3109/09553002.2014.884292. [DOI] [PubMed] [Google Scholar]
- 20.Ito A, Morita A, Ohya S, Yamamoto S, Enomoto A, Ikekita M. Cycloheximide suppresses radiation-induced apoptois in MOLT-4 cells with Arg72 variant of p53 through translational inhibition of p53 accumulation. J Radiat Res. 2011;52:342. doi: 10.1269/jrr.10151. [DOI] [PubMed] [Google Scholar]
- 21.Weissberg JB, Moulder JE, Fischer JJ, Parcells V. Effect of cycloheximide on skin and renal radiation tolerance in the rat. Radiat Res. 1983;96:173–179. [PubMed] [Google Scholar]
- 22.Elgebaly SA, Barton R, Forouhar F. Reversal of gamma-radiation-induced leukemogenesis in mice by immunomodulation with thiabendazole and dinitrofluorobenzene. J Natl Cancer Inst. 1985;74:811–815. [PubMed] [Google Scholar]
- 23.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu K, Park C, Li S, Lee KW, Liu H, He L, Soung NK, Ahn JS, Bode AM, Dong Z, Kim BY, Dong Z. Aloe-emodin suppresses prostate cancer by targeting the mTOR complex 2. Carcinogenesis. 2012;33:1406–1411. doi: 10.1093/carcin/bgs156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fonseca BD, Diering GH, Bidinosti MA, Dalal K, Alain T, Balgi AD, Forestieri R, Nodwell M, Rajadurai CV, Gunaratnam C, Tee AR, Duong F, Andersen RJ, Orlowski J, Numata M, Sonenberg N, Roberge M. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2012;287:17530–17545. doi: 10.1074/jbc.M112.359638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lam KKY, Zheng X, Forestieri R, Balgi AD, Nodwell M, Vollett S, Anderson HJ, Raymond J, Andersen RJ, Av-Gay Y, Roberge M. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8:e1002691. doi: 10.1371/journal.ppat.1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Yamada E, Zong H, Pessin JE. Fyn activation of mTORC1 stimulates the IRE1α-JNK pathway, leading to cell death. J Biol Chem. 2015;290:24772–24783. doi: 10.1074/jbc.M115.687020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang XQ, Liu VCH, Poon RTP, Lu P, Poon RYC. DNA damage mediated S and G2 checkpoints in human embryonal carcinoma cells. Stem Cells. 2009;27:568–576. doi: 10.1634/stemcells.2008-0690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol. 2012;920:613–626. doi: 10.1007/978-1-61779-998-3_40. [DOI] [PubMed] [Google Scholar]
- 31.Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1:27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders RN, Metcalfe MS, Nicholson ML. Rapamycin in transplantation: a review of the evidence. Kidney Int. 2001;59:3–16. doi: 10.1046/j.1523-1755.2001.00460.x. [DOI] [PubMed] [Google Scholar]
- 33.Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203:563–574. doi: 10.1083/jcb.201306041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS. Amyloid-β signals through tau to drive ectopic neuronal cell cycle reentry in Alzheimer’s disease. J Cell Sci. 2013;126:1278–1286. doi: 10.1242/jcs.1125880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS One. 2012;7:e47357. doi: 10.1371/journal.pone.0047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang J, McDonald PR, Dixon TM, Franicola D, Zhang X, Nie S, Epperly LD, Huang Z, Kagan VE, Lazo JS, Epperly MW, Greenberger JS. Synthetic protection short interfering RNA screen reveals glyburide as a novel radioprotector. Radiat Res. 2009;172:414–422. doi: 10.1667/RR1674.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rwigema JCM, Beck B, Wang W, Doemling A, Epperly MW, Shields D, Goff JP, Franicola D, Dixon T, Frantz M-CÈ, Wipf P, Tyurina Y, Kagan VE, Wang H, Greenberger JS. Two strategies for the development of mitochondrion-targeted small molecule radiation damage mitigators. Int J Radiat Oncol, Biol, Phys. 2011;80:860–868. doi: 10.1016/j.ijrobp.2011.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thaker NG, Zhang F, McDonald PR, Shun TY, Lewen MD, Pollack IF, Lazo JS. Identification of survival genes in human glioblastoma cells by small interfering RNA screening. Mol Pharmacol. 2009;76:1246–1255. doi: 10.1124/mol.109.058024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson C. Parallel algorithms for hierarchical clustering. Parallel Computing. 1995;21:1313–1325. [Google Scholar]
- 41.Kim SO, Bolton EE, Bryant SH. Effects of multiple conformers per compound upon 3-D similarity search and bioassay data analysis. J Cheminf. 2012;4:28–58. doi: 10.1186/1758-2946-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.