

Abstract
SND1, a subunit of the miRNA regulatory complex RISC, has been implicated as an oncogene in hepatocellular carcinoma (HCC). In this study, we show that hepatocyte-specific SND1 transgenic mice (Alb/SND1 mice) develop spontaneous HCC with partial penetrance and exhibit more highly aggressive HCC induced by chemical carcinogenesis. Livers from Alb/SND1 mice exhibited a relative increase in inflammatory markers and spheroid-generating tumor initiating cells (TIC). Mechanistic investigations defined roles for Akt and NF-κB signaling pathways in promoting TIC formation in Alb/SND1 mice. In human xenograft models of subcutaneous or orthotopic HCC, administration of the selective SND1 inhibitor 3′, 5′-deoxythymidine bisphosphate (pdTp) inhibited tumor formation without effects on body weight or liver function. Our work establishes an oncogenic role for SND1 in promoting TIC formation, and highlights pdTp as a highly selective SND1 inhibitor as a candidate therapeutic lead to treat advanced HCC.
Keywords: Transgenic mouse model, SND1 inhibitor, Inflammation
Introduction
Staphylococcal nuclease and tudor domain containing 1 (SND1) is a multifunctional protein that regulates transcription, mRNA splicing, RNA editing, and miRNA-mediated mRNA degradation as a nuclease in RNA-induced silencing complex (RISC) (1–8). SND1 is overexpressed in multiple cancers, where it functions as an oncogene (9–14). In hepatocellular carcinoma (HCC) SND1 overexpression was identified in ~74% cases (14). Overexpression of SND1 promotes and knockdown of SND1 inhibits proliferation, invasion, angiogenesis and in vivo tumorigenesis by human HCC cells (14–17). Our studies document that SND1 exerts its function in HCC cells by a variety of mechanisms. SND1 overexpression contributed to increased RISC activity in HCC cells resulting in augmented degradation of tumor suppressor mRNAs that are target of oncogenic miRNAs (14). SND1 promotes angiogenesis by activating NF-κB resulting in induction of miR-221 and subsequently angiogenic factors Angiogenin and CXCL16 (16). SND1 binds to 3′-UTR of angiotensin II type 1 receptor (AT1R) mRNA, increases AT1R mRNA stability and increases AT1R protein level (1). We documented that this increase in AT1R by SND1 leads to activation of ERK, Smad2 and subsequently TGFβ signaling pathway promoting epithelial-mesenchymal transition (EMT), migration and invasion by human HCC cells (15). SND1 interacts with monoglyceride lipase (MGLL) resulting in MGLL degradation which leads to activation of Akt and stimulation of cell proliferation and cell cycle progression in HCC cells (17). Overall, SND1 plays a dynamic role in regulating expression of genes crucial to hepatocarcinogenesis by employing diverse transcriptional as well as post-transcriptional molecular mechanisms (15,18).
SND1 is composed of four staphylococcal nuclease (SN) domains and a single fusion domain, consisting of a tudor domain and a nuclease domain (8). The nuclease domains function as RNase while tudor domain is involved in protein-nucleic acid interaction (8). Enzymatic activity of SND1 is required for its function in RISC (7). However, whether the other functions of SND1 require enzymatic activity remains to be determined. Structurally, SND1 is unique in the human proteome with no close homolog as revealed by BLAST search (18). The closest homolog of SND1 is EBNA2, which is not transcribed. SND1 has highly electropositive SN domains to which binds the negatively charged 3′, 5′-deoxythymidine bisphosphate (pdTp) molecule inhibiting SND1 nuclease activity (7,8,19,20). The absence of a close homolog of SND1 and availability of a specific SND1 inhibitor pdTp suggests that pdTp might be developed as a potential HCC therapeutic with little side-effects. Indeed, we showed that pdTp inhibited growth of human HCC cells in vitro (14). However, in vivo anti-tumor efficacy of pdTp remains to be determined. Additionally pdTp might also serve as a valuable tool to distinguish enzymatic and non-enzymatic functions of SND1.
In the present manuscript we employ a hepatocyte-specific transgenic mouse overexpressing SND1 (Alb/SND1) to obtain insights into the oncogenic function of SND1 in an in vivo context. We document that Alb/SND1 mice develop spontaneous HCC with expansion of tumor initiating cells (TICs). We also demonstrate in vivo safety and anti-tumor efficacy of pdTp thereby paving the way for its further characterization as an anti-HCC agent. We thus identify SND1 as a valid molecular target in HCC and present SND1 inhibition as a promising approach to curb hepatocarcinogenesis.
Materials and Methods
Generation of Alb/SND1 mouse and induction of chemical carcinogenesis
Alb/SND1 transgenic mouse in B6CBAF1 background was generated by directing the expression of C-terminal Myc-tagged human SND1 under an upstream enhancer region (−10400 to −8500) fused to the 335-bp core region of mouse albumin promoter (Fig. S1) (21). Microinjection and manipulation procedures were performed according to standard protocols in the VCU Massey Cancer Center Transgenic/Knockout Mouse Core. For induction of chemical carcinogenesis, a single i.p. injection of 10 μg/gm body weight of N-nitrosodiethylamine (DEN) was given at 14 days of age to male WT and Alb/SND1 littermates (22). The animals were sacrificed at 32 weeks of age and liver, internal organs and blood were collected. Serum liver enzymes were analyzed in the Molecular Diagnostic Laboratory, Department of Pathology, VCU using standard procedures. All experiments were performed using sibling littermates, fed regular chow diet during light cycle. All animal studies were approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University, and were conducted in accordance with the Animal Welfare Act, the PHS Policy on Humane Care and Use of Laboratory Animals, and the U.S. Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training.
Cells, culture condition, sphere formation and Matrigel invasion assays and chemicals
Primary mouse hepatocytes were isolated from adult male WT and Alb/SND1 littermates, cultured without passage as described and were mycoplasma-free (22). The human HCC cell line QGY-7703 was developed at Fudan University, Shanghai, obtained from Dr. Zhao-zhong Su in 2008, and cultured as described (23). Generation and characterization of QGY-7703 cells expressing luciferase (QGY-luc) have been described (24,25). Early passage (>5) cultures of QGY-7703 and QGY-luc cells were stored in liquid nitrogen and in vivo studies described in this manuscript were performed with freshly thawed culture of the cells after confirming mycoplasma-free using mycoplasma detection kit (ThermoFisher Scientific). Hepatocytes were cultured in Essential 8 Medium (ThermoFisher Scientific; Catalog # A1517001) for enrichment of tumor initiating cells using ultra-low attachment plates. Sphere formation was monitored and spheres containing more than 50 cells were quantified microscopically. Non proliferative spheres were excluded as abortive spheres. Matrigel invasion assay using primary hepatocytes was performed as described (23,26). pdTp was purchased from Axxora (Catalog # BLG-T012-05). BMS-3445541 (Catalog # B9935) and LY294006 (Catalog # L9908) were purchased from Sigma-Aldrich and U0126 (Catalog # 9903) was purchased from Cell Signaling.
Immunohistochemistry (IHC) and immunofluorescence (IF)
IHC using FFPE sections and IF in primary hepatocytes were performed as described (23). For IHC, the sections were blocked in PBST using 10% normal goat serum for rabbit and mouse polyclonal antibodies. Primary antibodies were diluted in PBST containing 5% blocking serum. The primary antibodies used were: SND1 (rabbit polyclonal; 1:200; Sigma), PCNA (mouse monoclonal; 1:200; Cell Signaling), AFP (rabbit polyclonal; 1:50; Santa Cruz), F4/80 (rat polyclonal; 1:200; Biorad), CD31 (rabbit polyclonal; 1:50; AbCam), cleaved caspase-3 (rabbit polyclonal; 1:300; Cell Signaling), p-p65 (rabbit polyclonal; 1:400; Cell Signaling), EpCAM (rabbit polyclonal; 1:400; Sigma), CD133 (rabbit polyclonal; 1:500; Proteintech) and CD44 (mouse monoclonal; 1:250; AbCam). Biotin conjugated secondary antibodies were diluted in PBST containing corresponding 2.5% blocking serum. Sections were stained using avidin-biotin-peroxidase complexes treated with a DAB substrate solution (Vector laboratories). IHC images were quantified by H-score with WT score normalized to 1. For immunofluorescence the primary antibody was p65 (mouse monoclonal; 1:400; Cell Signaling) and the secondary antibody was Alexa546-conjugated anti-mouse IgG (goat; 1:400; Invitrogen). The slides were mounted in VectaShield fluorescence mounting medium containing 4,6 -diamidino-2- phenylindole (Vector Laboratories). Images were analyzed using a Zeiss confocal laser scanning microscope.
Western Blotting
Lysates were prepared by lysing cells in 1.5% n-dodecyl -D-maltoside (DDM) buffer supplemented with protease and phosphatase inhibitor cocktail (Pierce). Lysates from liver tissue were prepared by mechanical homogenization using the same buffer. Western blot was performed as described (23). Primary antibodies used were SND1 (rabbit polyclonal; 1:1000; Sigma), GAPDH (mouse monoclonal; 1:1000; Santa Cruz), CD133 (rabbit polyclonal; 1:1000; Proteintech), CD44 (mouse monoclonal; 1:1000; Abcam), Myc-Tag, p-Akt, Akt, ERK, p-ERK, p-GSK3β, GSK3β, p-p65, p65 (rabbit polyclonal; 1:1000; Cell Signaling), AT1R (rabbit polyclonal; 1:1000; Abnova), MGLL (rabbit polyclonal; 1:1000; Thermofisher). Densitometric analysis was performed by ImageJ software.
Total RNA extraction, cDNA preparation and quantitative RT-PCR
Total RNA was extracted using the QIAGEN miRNAeasy Mini Kit (QIAGEN, Maryland). cDNA preparation was done using ABI cDNA synthesis kit. Real-time polymerase chain reaction (RT-PCR) was performed using an ABI ViiA7 fast real-time PCR system and Taqman gene expression assays according to the manufacturer’s protocol (Applied Biosystems, Foster City, CA).
Flow cytometry
WT and Alb/SND1 hepatocytes were isolated and washed in PBS. Hepatocytes (1 X 106 per sample) were stained with fluorochrome-conjugated primary antibodies in 2% BSA in PBS at room temperature for 1 hour. CD133 (mouse-APC; 1:50; Miltenyi Biotec), CD44 (rat-FITC; 1:50; Abcam) and EpCAM (rat-PE; 1:20; BD Pharmingen) were used as per manufacturer’s protocol. Cells were washed twice in PBS and resuspended in 2% BSA in PBS for flow cytometric analysis using BD FACSCanto II (San Jose, CA).
Treatment of human HCC xenografts in NSG mice
QGY-7703 cells (6.5 × 105 cells suspended in 50 μl of Matrigel) were injected s.c. in the flanks of adult male NSG mice. Tumor volume was measured twice a week with a caliper and calculated using the formula π/6 × larger diameter × (smaller diameter)2. After the tumors have reached ~100 mm3 (requiring a week) different doses of pdTp (0.16, 0.32 and 0.8 mg/kg) were administered to the mice i.p. twice a week for 4 weeks. Six mice per group were used for each treatment group and eight mice were used for control vehicle-treated group. For orthotopic xenografts, QGY-luc cells (1 × 106) were implanted by intrahepatic injection in adult male NSG mice (24,25). Tumor growth was monitored by bioluminescence imaging (BLI) with a Xenogen IVIS imager once a week. After one week, when well-established tumors were detected by BLI, different doses of pdTp (0.8 and 1.6 mg/kg) were administered to the mice i.v. twice a week for 4 weeks. Six mice per group were used.
Statistical analysis
Data were represented as the mean ± Standard Error of Mean (S.E.M) and analyzed for statistical significance using student’s paired T-test. A p-value of <0.05 was considered as significant.
Results
Alb/SND1 mice manifest aggressive hepatocarcinogenesis
We have created a hepatocyte-specific C-terminal Myc-tagged human SND1-expressing transgenic mouse (Alb/SND1) by using the mouse albumin promoter/enhancer element to drive SND1 expression in a B6CBAF1 background. This particular strain of mouse is very sensitive to N-nitrosodiethylamine (DEN)-induced hepatocarcinogenesis (22). The expression of SND1 in the livers of Alb/SND1 mice was confirmed by Western blot, Taqman Q-RT-PCR and immunohistochemistry (IHC) (Fig. 1A–C, S2A). Alb/SND1 mice develop and reproduce normally and no physiological abnormalities and significant differences in body weight compared to WT littermates were observed. Histopathological analysis at 2 months of age did not reveal any difference in liver architecture between WT and Alb/SND1 mice (Fig. 1C). However, at one year of age, 6 out of 14 (~42%) Alb/SND1 mice developed hepatic nodules, which were confirmed as HCC upon histological examination with loss of hepatic architecture, and AFP expression (Fig. 1D–F, S2A, Table 1). However, no such nodules were observed in WT littermates. Compared to WT, a significant increase in mRNAs for c-Myc, TNFα and IL-6, known drivers of HCC, were detected in Alb/SND1 livers at 2 and 12 months of age, except for IL-6 which showed an increase only at 12 months (Fig. 1G).
Figure 1.
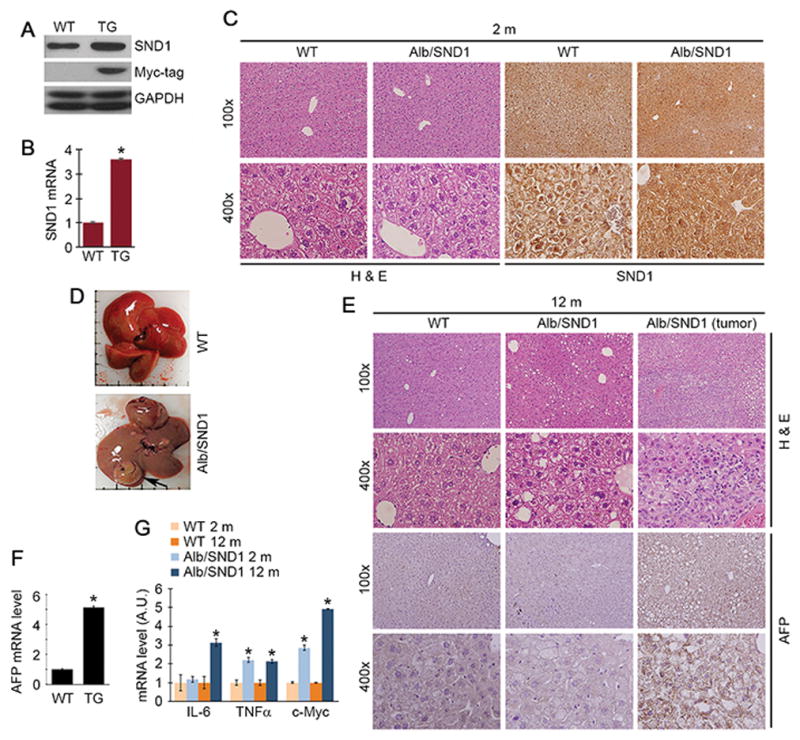
SND1 overexpression in vivo results in neoplastic transformation. SND1 overexpression in Alb/SND1 (TG) liver was confirmed by Western blot (A), Taqman Q-RT-PCR (B) and immunohistochemistry (C). SND1 mRNA levels were normalized by GAPDH mRNA levels. For A–C, liver samples from 2 months old mice were used. D. Photographs of representative livers of one year old mice. Arrow indicates liver nodule. E. Histological analysis and AFP immunostaining of FFPE liver sections from one year old Wildtype (WT) and Alb/SND1 mice with or without tumors. F. Relative expression of AFP mRNA by Taqman Q-RT-PCR in one year old WT and Alb/SND1 mice with tumor (3 mice per group). G. Relative expression of the indicated mRNAs by Taqman Q-RT-PCR in livers of 2 months and one year old mice (3 mice per group). For graphs, data represent mean ± SEM; *: p<0.01 vs WT.
Table 1.
No. of nodules in Wild-type (WT) and Alb/SND1 (TG) mice
| Nodules (mm) | ||||
|---|---|---|---|---|
| ID | 1–2 | >3 | >10 | |
| 1 year old | TG1 | 7 | 0 | 1 |
| TG2 | No nodule | |||
| TG3 | No nodule | |||
| TG4 | No nodule | |||
| TG5 | 3 | 0 | 0 | |
| TG6 | No nodule | |||
| TG7 | No nodule | |||
| TG8 | No nodule | |||
| TG9 | 0 | 0 | 1 | |
| TG10 | 0 | 1 | 0 | |
| TG11 | No nodule | |||
| TG12 | No nodule | |||
| TG13 | 0 | 1 | 0 | |
| TG14 | 0 | 1 | 0 | |
| DEN-treated | WT1 | 0 | 0 | 1 |
| WT2 | 3 | 5 | 0 | |
| WT3 | 0 | 5 | 0 | |
| WT4 | 0 | 0 | 10 | |
| WT5 | No nodule | |||
| WT6 | No nodule | |||
| WT7 | No nodule | |||
| WT8 | No nodule | |||
| TG1 | 4 | 27 | 44 | |
| TG2 | 0 | 0 | 41 | |
| TG3 | Entire liver | |||
| TG4 | Entire liver | |||
| TG5 | Entire liver | |||
| TG6 | Entire liver | |||
| TG7 | Entire liver | |||
| TG8 | Entire liver | |||
| TG9 | Entire liver | |||
| TG10 | Entire liver | |||
We next checked the response of Alb/SND1 mice to DEN-induced HCC. At 32 weeks post DEN-injection, Alb/SND1 mice showed a profound tumorigenic response, with tumorigenesis affecting the entire liver, compared to WT littermates, in which there were either no nodules or nodules that were <5 mm in size (Fig. 2A, Table 1). Liver weight, reflecting higher tumor load, and serum AST, ALT and total protein were significantly elevated in Alb/SND1 mice versus WT (Fig. 2B–C). A marked increase in mRNA of HCC markers AFP and CD36 were detected in DEN-treated Alb/SND1 livers compared to DEN-treated WT (Fig. 2D). Histologically DEN-treated Alb/SND1 livers showed loss of architecture and increased expression of CD31 (angiogenesis marker) and PCNA (proliferation marker) vs DEN-treated WT (Fig. 2E, S2A). Increased activation of ERK, Akt and its downstream GSK3β was observed in DEN-treated and spontaneous Alb/SND1 tumors compared to DEN-treated WT (Fig. 2F, S2B). ERK and Akt are activated by SND1 (15,17) and also play critical role in HCC (27) thus might be crucial in mediating SND1-induced HCC.
Figure 2.
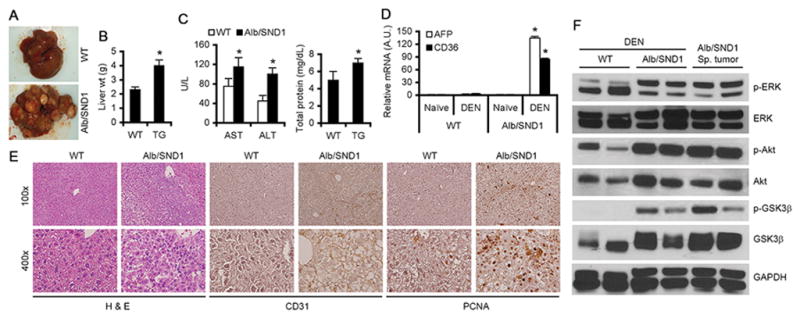
Alb/SND1 mice develop aggressive DEN-induced HCC. WT and Alb/SND1 littermates were injected i.p. with DEN (10μg/gm) at 2 weeks of age and livers were harvested at 32 weeks post-injection when subsequent analyses were performed. A. Photographs of representative livers. Measurement of liver wt (B), and serum AST, ALT and total protein (C) (WT: n = 8; Alb/SND1: n = 10). D. Analysis of AFP and CD36 mRNA levels by Taqman Q-RT-PCR in naïve and DEN-treated livers. Normalized by GAPDH. E. Histological analysis and IHC for CD31 and PCNA in FFPE liver sections. F. Western blot analysis for the indicated proteins in the livers of DEN-treated WT and Alb/SND1 littermates at 32 weeks and spontaneous tumors in Alb/SND1 livers at one year. For graphs, data represent mean ± SEM; *: p<0.01 vs WT.
Alb/SND1 hepatocytes show increased activation of NF-κB
Chronic inflammation is a central event in hepatocarcinogenesis and NF-κB plays a pivotal role in promoting inflammation (28). Increased expression of IL-6 and TNFα (Fig. 1G) indicates that SND1 overexpression results in a chronic inflammatory state leading to HCC. We previously documented activation of NF-κB in SND1-overexpressing human HCC cells (16). NF-κB activation is marked by phosphorylation of serine residue 536 allowing nuclear translocation of p65 subunit, which then functions as a transcription factor to modulate expression of inflammatory genes. We checked nuclear localization of p65 NF-κB in WT and Alb/SND1 hepatocytes, treated or not with lipopolysaccharide (LPS), since serum LPS levels are elevated in HCC patients which promotes inflammation. Alb/SND1 hepatocytes, but not WT, showed nuclear p65 under basal condition indicating constitutive activation of NF-κB (Fig. 3A). LPS treatment resulted in nuclear translocation of p65 in both WT and Alb/SND1 hepatocytes. Inhibition of enzymatic activity of SND1 by pdTp resulted in marked downregulation of p65 levels with simultaneous inhibition of p65 nuclear translocation in both WT and Alb/SND1 hepatocytes suggesting a central role of SND1 in regulating p65 expression (Fig. 3A). Western blot analysis revealed increased basal level of phosphorylated p65 (p-p65) in Alb/SND1 hepatocytes compared to WT (Fig. 3B). In WT hepatocytes, upon LPS treatment, increased p-p65 was observed at 10 minutes which gradually waned down over a period of 2 h (Fig. 3B). However, in Alb/SND1 hepatocytes increased p-p65 level remained sustained during the assay period (Fig. 3B). Increased inflammation was also indicated by increased infiltration of macrophages, detected by staining for F4/80 marker, in DEN-treated and one year old tumor-bearing Alb/SND1 livers versus corresponding WT (Fig. 3C, S2C). Additionally a marked increase in immune checkpoint molecule PD-L1 and a significant increase in its receptor PD-1 were observed in DEN-treated and one year old tumor-bearing Alb/SND1 livers vs corresponding WT indicating a strong carcinogenic response induced by SND1 overexpression (Fig. S2D).
Figure 3.
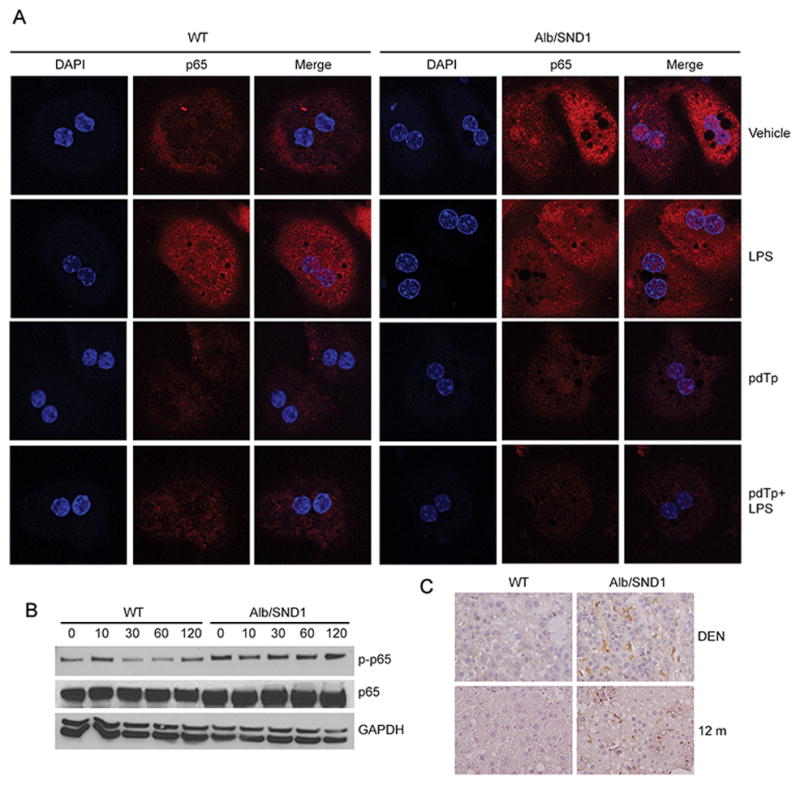
NF-κB activation is exaggerated in Alb/SND1 hepatocytes. A. Hepatocytes were harvested from one year old WT and Alb/SND1 littermates and treated with LPS (200 ng/ml) for 30 minutes. In a second experiment, hepatocytes were treated with pdTp (200 μM) for 18 h before LPS treatment. The hepatocytes were immunostained for p65 NF-κB and immunofluorescence analysis was performed by confocal microscopy. B. Hepatocytes were harvested from 2 months old WT and Alb/SND1 littermates and treated with LPS (200 ng/ml) for the indicated time points. Western blot analysis of the indicated proteins was performed. C. FFPE sections of DEN-treated and one year old WT and Alb/SND1 livers were stained for macrophage marker F4/80.
SND1 overexpression results in expansion of tumor initiating cells (TICs)
Since Alb/SND1 mice develop spontaneous HCC, we checked the effect of SND1 overexpression on TICs. In a sphere formation assay in ultra-low attachment plates, WT hepatocytes formed small abortive spheres while Alb/SND1 hepatocytes formed robust spheres that gradually increased in size and number indicating an expansion of tumor initiating cells (TICs) (Fig. 4A). Indeed, TICs positive for three markers, EpCAM, CD44 and CD133, were significantly more in Alb/SND1 livers versus WT (Fig. 4B). Alb/SND1 hepatocytes isolated from 2 months old mice showed significant increase in mRNA levels of EpCAM (~ 2-fold), CD44 (~4-fold) and CD133 (~4-fold) compared to WT (Fig. S3A). These increases in mRNA levels were further augmented, e.g., EpCAM (~2.5-fold), CD44 (~6-fold) and CD133 (~4-fold), in Alb/SND1 hepatocytes isolated from 12 months old mice versus WT (Fig. S3A). Immunohistochemical analysis showed increased expression of EpCAM, CD44 and CD133 in one year old Alb/SND1 livers, with or without tumor, compared to WT littermates (Fig. S3B–C). Increased CD133 expression was observed in DEN-treated and spontaneous tumors in Alb/SND1 mice when compared to DEN-treated WT livers (Fig. S3D). Treatment with pdTp significantly inhibited sphere formation by both WT and Alb/SND1 hepatocytes indicating that enzymatic function of SND1 is necessary to promote expansion of TICs (Fig. 4C). As expected from studies in human cell lines, naïve Alb/SND1 livers showed increased activation of ERK and Akt (Fig. 3D). To check the effects of the signaling pathways activated by SND1 in regulating TICs we performed sphere formation assay with Alb/SND1 hepatocytes upon treatment with BMS-3445541(IκB kinase inhibitor to block NF-κB activation), LY294006 (PI3K inhibitor to block Akt activation) and U0126 (MEK1/2 inhibitor to block ERK activation) (Fig. 4E–F). Inhibition of NF-κB and Akt activation, but not ERK activation, significantly abrogated sphere formation by Alb/SND1 hepatocytes with corresponding decrease in CD133 expression (Fig. 4E–F, S4A). We interrogated the potential role of ERK activation in SND1-induced phenotypes other than sphere formation. We previously demonstrated a potential role of ERK activation in mediating SND1-induced invasion of human HCC cells (15). While WT hepatocytes did not invade through Matrigel, Alb/SND1 hepatocytes acquired Matrigel invasion property which was significantly abrogated upon treatment with U0126 (Fig. 4G).
Figure 4.
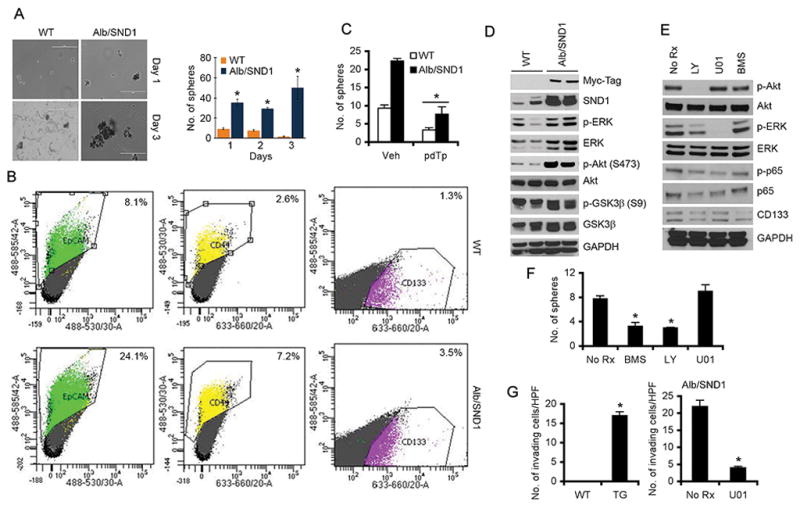
SND1 overexpression results in expansion of tumor initiating cells (TICs). A. Hepatocytes were harvested from one year old WT and Alb/SND1 littermates and cultured under ultra-low attachment conditions to form spheroids and observed over a period of 3 days. Left panel, photomicrograph of representative spheres. Right panel, graphical quantification of the spheres. B. Hepatocytes were harvested from one year old WT and Alb/SND1 littermates, immunostained with fluorochrome-conjugated EpCAM, CD44 and CD133 antibodies and subjected to flow cytometry. C. Hepatocytes were harvested from 2 months old WT and Alb/SND1 littermates, treated with pdTp (200 μM) and sphere formation was measured over a period of 7 days. D. Western blot analysis of the indicated proteins in the livers of one year old WT and Alb/SND1 littermates. Two independent mice per group were used. E–F. Hepatocytes from 2 months old Alb/SND1 mice were harvested and treated with BMS-3445541(5μM), LY294006 (25μM) and U0126 (10μM) in enriching conditions. Western blot analysis using the indicated antibodies was performed (E). Sphere formation was compared between untreated and treated Alb/SND1 hepatocytes over a period of 3 days (F). G. Matrigel invasion using WT and Alb/SND1 (TG) hepatocytes treated or not with U0126 (10 μM) was performed. HPF: high power field. For graphs, data represent mean ± SEM; *: p<0.01 vs WT in panel A and G, Vehicle (Veh) in panel C and No Rx in panel F.
SND1 inhibitor pdTp significantly abrogates human HCC xenografts in vivo
pdTp specifically inhibits the nuclease activity of SND1, but does not affect the oligonucleotide binding function of Tudor domain. We previously documented that pdTp inhibits proliferation of human HCC cells in vitro (14). As yet in vivo efficacy and toxicity has not been tested for pdTp. We injected multiple doses of pdTp (calculated from our in vitro studies) to WT B6CBA mice i.p. twice a week for 4 weeks (a total of 8 doses). At the highest dose of 0.8 mg/kg, no difference in body and liver weights, serum liver enzymes, total protein, albumin and globulin was observed vs vehicle at the end of the treatment cycle (Fig. 5A–B). Bilirubin levels (conjugated and unconjugated) were normal and did not show any increase upon pdTp treatment (data not shown). Histological analysis of internal organs also did not show any abnormality (Fig. 5C). We established s.c. xenografts of QGY-7703 cells in NSG mice and evaluated the effect of i.p. administration of different doses pdTp on tumor development. A significant decrease in tumor volume and tumor weight was observed with 0.32 and 0.8 mg/kg pdTp at the end of the treatment (Fig. 5D). We next established orthotopic xenografts of QGY-luc cells (QGY-7703 cells expressing luciferase) in the livers of NSG mice and evaluated the effect of i.v. administration of pdTp on tumor development by bioluminescence imaging (BLI). A significant inhibitory effect on tumor progression was observed with pdTp treatment compared to vehicle (Fig. 5E–F). Immunohistochemical analysis of s.c. tumor sections revealed that pdTp treatment resulted in a dose-dependent decrease in PCNA, CD133, CD44 and p-p65 staining, and an increase in apoptosis, determined by staining for cleaved caspase 3 (Fig. 6A, S4B). Western blot analysis of tumor samples identified that pdTp treatment resulted in downregulation of CD133 and CD44 levels and decreased phosphorylation of Akt and p65 (Fig. 6B, S4C). Total p65 level was also decreased upon pdTp treatment (Fig. 6B). Interestingly, no change in ERK activation was observed upon treatment with pdTp. We previously documented that increased RISC activity resulting from SND1 overexpression augments oncomiR-mediated degradation of tumor suppressor mRNAs, such as PTEN, target of miR-221 and miR-21, CDKN1C (p57), target of miR-221, CDKN1A (p21), target of miR-106b, SPRY2, target of miR-21, and TGFBR2, target of miR-93 (14). As a corollary, in vivo pdTp treatment resulted in significant increases in PTEN, TGFBR2 and CDKN1C mRNA levels in tumors compared to vehicle (Fig. 6C). Collectively, these findings reveal that pdTp inhibits proliferation and inflammation, induces apoptosis and downregulates tumor initiating cells.
Figure 5.
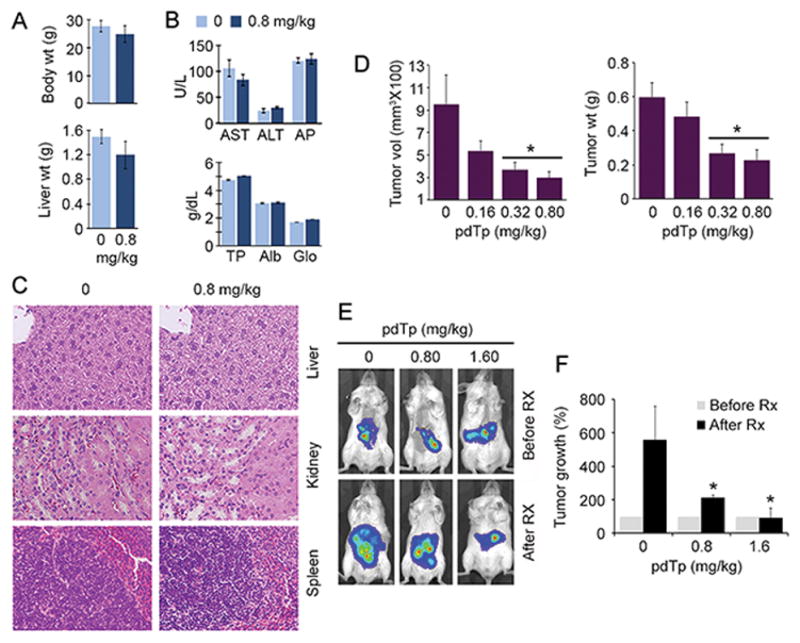
pdTp is non-toxic and inhibits human HCC xenografts. WT B6/CBA mice (2 months old) were injected i.p. with 0.8 mg/kg pdTp twice a week for 4 weeks. Body and liver weights (A), and serum liver enzymes (AST, ALT and AP), total protein (TP), albumin (Alb) and globulin (Glo) (B) were measured at the end of the treatment. C. Histology of internal organs at the end of pdTp treatment. D. QGY-7703 cells were injected s.c. in NSG mice to establish xenografts and treated with the indicated doses of pdTp i.p. twice a week for 4 weeks (n = 6 per dose). Tumor volume and weight were measured at the end of the treatment. E–F. QGY-luc cells were injected orthotopically in the livers of NSG mice to establish xenografts and treated with the indicated doses of pdTp i.v. twice a week for 4 weeks (n = 6 per dose). Bioluminescence imaging (BLI) of the orthotopic tumors before and after pdTp treatment (E). Quantification of photon counts from the mice (F). For graphs, data represent mean ± SEM; A, B and D: *: p<0.01 vs pdTp 0 mg/kg; F: *: p<0.05 vs pdTp 0 mg/kg After Rx.
Figure 6.
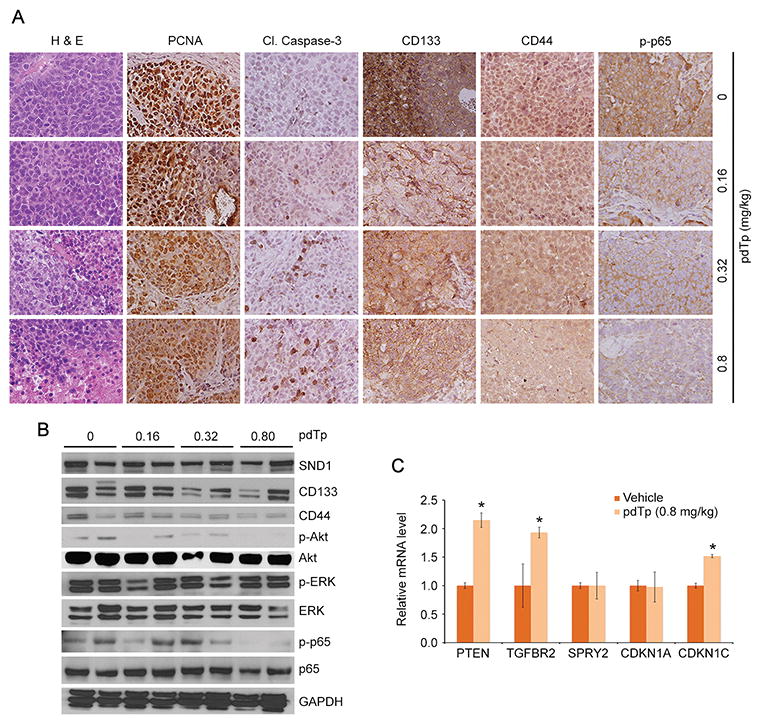
pdTp reduces expression of proliferation, inflammation and TIC markers, and upregulates apoptosis and expression of selective tumor suppressor genes. A. FFPE sections of vehicle- and pdTp-treated s.c. xenograft tumors were analyzed histologically and immunostained for the indicated proteins. B. Western blot analysis of the indicated proteins in vehicle- and pdTp-treated s.c. xenograft tumor samples. Tumor samples from two independent mice per group were used. C. Relative expression of the indicated mRNAs was measured by Taqman Q-RT-PCR in vehicle- and pdTp-treated s.c. xenograft tumors (3 mice per group). Normalized by GAPDH. Data represent mean ± SEM; *: p<0.01.
Discussion
In this research article, we report oncogenic role of SND1 in HCC development and progression by pursuing studies in a novel hepatocyte-specific SND1-overexpressing transgenic mouse model. Our previous in vitro studies established that SND1 overexpression positively regulates multiple hallmarks of cancer, including proliferation, migration, invasion, angiogenesis, epithelial-mesenchymal transition and inhibition of tumor suppressor gene expression (14–17). In the current in vivo studies, we document that SND1 causes spontaneous hepatocarcinogenesis by increasing TICs within the liver and creating a pro-inflammatory microenvironment, and sensitizes hepatocytes towards DEN-induced HCC. As a corollary, chemical inhibition of SND1 enzymatic activity by pdTp reduced tumor-initiating potential of hepatocytes and rescued pro-inflammatory signaling caused by SND1 overexpression resulting in significant abrogation of tumor growth in a xenograft model.
SND1 is a multifunction protein that regulates gene expression at transcriptional as well as post-transcriptional level. Inflammatory cytokine TNFα is constitutively upregulated in Alb/SND1 liver, whereas IL-6 is upregulated in an age-dependent manner. Both these cytokines are known to activate NF-κB signaling and are also induced by NF-κB. As expected, Alb/SND1 hepatocytes manifest an exaggerated NF-κB activation, both constitutive and upon LPS treatment. LPS is detected at notably high levels in HCC patients, thus making SND1-mediated augmentation in NF-κB activation a clinically relevant scenario. Activation of resident Kupffer cells and invasion of liver with macrophages is a crucial event in HCC. Secretion of inflammatory cytokines by SND1-overexpressing hepatocytes might contribute to NF-κB signaling and activation in macrophages. Thus, SND1 overexpression creates an underlying pro-inflammatory condition within liver, which aggravates with age, creating conducive preexisting pathology for tumorigenesis.
We observe that constitutive overexpression of SND1 predisposes Alb/SND1 animals to risk of HCC development in late adulthood, in the absence of any carcinogen exposure, with expansion of CD133+, CD44+ and EpCAM+ TICs. Our inhibitor studies unravel that active Akt and NF-κB signaling is required to maintain tumor initiation potential of SND1 overexpressing hepatocytes. A recent report documented that in a hepatitis B model, AFP-induced upregulation of CD133+, CD44+ and EpCAM+ TICs is dependent on PI3K/Akt signaling (29). Clinical study analyzing protein marker expression in liver from more than 100 HCC patients showed a significant negative correlation with PTEN and positive correlation with Akt levels with TIC marker proteins CD133, EpCAM and CD90 (30). Univariate and multivariate analysis showed significant correlation between loss of PTEN and high AFP, Akt, CD133 and EpCAM levels with overall survival of HCC patients (30). CD133 expression is correlated with Sorafenib resistance in human HCC patients (31,32). CD133 confers chemoresistance and radioresistance via upregulation of PI3K/AKT signaling (32,33). Interaction between hyaluronan and CD44 in extracellular matrix promotes carcinogenic signaling and results in chemoresistance (34). Thus a close interplay between AKT signaling and TIC expansion is predicted in Alb/SND1 livers rendering them therapeutically resistant. It would be interesting to investigate potential of SND1 targeting in overcoming chemoresistance in HCC for future studies.
Studies have shown that IL-8, a NF-κB downstream gene, increases CXCL1 expression, upregulates MAPK signaling and sustains cancer stemness that eventually expands CD133+ population in liver (35). Autocrine IL-6 signaling is also implicated in pro-tumorigenic properties of HCC progenitor cells (36). IL-6 signaling promoted by tumor-associated macrophages is found to promote expansion of CD44+ cells, potentiating growth of xenograft tumors, and inhibition of IL-6/STAT3 signaling in this model, reduced tumorigenic potential of CD44+ cells (37). In SND1 overexpressing HCC, we report an increased invasion of macrophages, which can be predicted to upregulate IL-6 secretion and thus promote expansion of TICs. As yet the molecular mechanism by which SND1 activates NF-κB remains to be determined. pdTp treatment studies clearly demonstrate that SND1 enzymatic activity is required to activate NF-κB (Fig. 3A). However, pdTp treatment also resulted in a decrease in total p65 level indicating that SND1 regulates not only NF-κB activation but also p65 expression. Inflammatory cytokines induce SND1 expression and SND1 promoter contains consensus NF-κB binding sites (38,39). Thus chronic inflammation preceding HCC might result in induction of SND1 which facilitates expansion of TICs and further aggravates inflammation thereby establishing a scenario for the development of HCC.
SND1 overexpression activates Akt, ERK and NF-κB signaling. We document that pdTp treatment inhibited Akt and NF-κB activation but not ERK activation in human HCC cells (Fig. 6B) and inhibited sphere formation by WT and Alb/SND1 hepatocytes (Fig. 4C). Additionally inhibition of Akt and NF-κB, but not that of ERK, abrogated sphere formation by Alb/SND1 hepatocytes (Fig. 4E). These findings suggest that enzymatic activity of SND1 is necessary for expansion of TICs and is not necessary for ERK activation. Binding of SND1 to 3′-UTR of AT1R mRNA increases AT1R mRNA stability and protein translation resulting in increased ERK activation (1,15). This function of SND1 does not require its enzymatic activity and is not inhibited by pdTp treatment. SND1 activates Akt by multiple mechanisms. By augmenting RISC activity it downregulates PTEN, a negative regulator of Akt signaling (14). Protein-protein interaction between SND1 and MGLL results in MGLL degradation (17). MGLL selectively interacts with phosphatidic acid and phosphoinositide derivatives leading to inhibition of PI3K/Akt signaling (40). As such MGLL degradation results in Akt activation. SND1 interacts with MGLL via its SN domains to which pdTp binds (17). pdTp might interfere with SND1/MGLL interaction thereby blocking Akt activation. Thus SND1-induced Akt activation requires enzymatic and non-enzymatic activities of SND1 both of which might be interfered by pdTp. It should be noted that downregulation of MGLL and upregulation of AT1R are also preserved in Alb/SND1 livers compared to WT (Fig. S5).
We present SND1 inhibition as a new avenue for targeted therapeutic research in HCC management. The non-toxicity, specificity for SND1 inhibition and strong therapeutic efficacy make pdTp an attractive reagent for clinical use. More stringent pharmacokinetic and biodistribution studies and medicinal chemistry analysis to generate more potent pdTp analogs should be pursued further. However, pdTp may not block all aspects of SND1 function, especially its non-enzymatic function, thereby requiring an alternative approach to inhibit SND1. We have recently demonstrated therapeutic utility of a hepatocyte-specific nanoparticle delivering siRNA against an oncogene in orthotopic xenograft models of HCC (24). Similar approaches might be employed to knockdown SND1 and in combination with pdTp might bring forth complete and sustained inhibition of SND1 function. These approaches might also be combined with current standard of care chemotherapies for HCC. Our present study opens up a wide avenue of preclinical and clinical research on testing efficacy of a novel targeted treatment approach.
Supplementary Material
Acknowledgments
Financial support
The present study was supported in part by National Cancer Institute Grant R21 CA183954 and The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant 1R01DK107451-01A1 (D. Sarkar). C.L. Robertson is supported by a National Institute of Diabetes And Digestive And Kidney Diseases Grant T32DK007150. Services in support of this project were provided by the VCU Massey Cancer Center Transgenic/Knock-out Mouse Facility and flow cytometry core facility, supported in part with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Abbreviations
- SND1
Staphylococcal nuclease and tudor domain containing 1
- HCC
Hepatocellular carcinoma
- RISC
RNA-induced silencing complex
- pdTp
3′, 5′-deoxythymidine bisphosphate
- ERK
Extracellular Signal-Regulated Kinase
- GSK3β
Glycogen synthase kinase 3β
- PCNA
Proliferating cell nuclear antigen
- AFP
α-fetoprotein
- TIC
Tumor initiating cells
- LPS
Lipopolysaccharide
- DEN
N-nitrosodiethylamine
- FFPE
Formalin-fixed paraffin-embedded
- AST
Aspartate aminotransferase
- ALT
Alanine aminotransferase
- H & E
Hematoxylin and eosin
- PBS
Phosphate buffered saline
- BSA
Bovine serum albumin
- NSG
NOD scid gamma
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed death-ligand 1
Footnotes
Disclosure: All authors have no potential conflicts.
References
- 1.Paukku K, Kalkkinen N, Silvennoinen O, Kontula KK, Lehtonen JY. p100 increases AT1R expression through interaction with AT1R 3′-UTR. Nucleic Acids Res. 2008;36:4474–87. doi: 10.1093/nar/gkn411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paukku K, Yang J, Silvennoinen O. Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Mol Endocrinol. 2003;17:1805–14. doi: 10.1210/me.2002-0256. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Aittomaki S, Pesu M, Carter K, Saarinen J, Kalkkinen N, et al. Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. Embo J. 2002;21:4950–8. doi: 10.1093/emboj/cdf463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Valineva T, Hong J, Bu T, Yao Z, Jensen ON, et al. Transcriptional co-activator protein p100 interacts with snRNP proteins and facilitates the assembly of the spliceosome. Nucleic Acids Res. 2007;35:4485–94. doi: 10.1093/nar/gkm470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Lopez J, de Hourcade JD, Del Mazo J. Reprogramming of microRNAs by adenosine-to-inosine editing and the selective elimination of edited microRNA precursors in mouse oocytes and preimplantation embryos. Nucleic Acids Res. 2013;41:5483–93. doi: 10.1093/nar/gkt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scadden AD. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat Struct Mol Biol. 2005;12:489–96. doi: 10.1038/nsmb936. [DOI] [PubMed] [Google Scholar]
- 7.Caudy AA, Ketting RF, Hammond SM, Denli AM, Bathoorn AM, Tops BB, et al. A micrococcal nuclease homologue in RNAi effector complexes. Nature. 2003;425:411–4. doi: 10.1038/nature01956. [DOI] [PubMed] [Google Scholar]
- 8.Li CL, Yang WZ, Chen YP, Yuan HS. Structural and functional insights into human Tudor-SN, a key component linking RNA interference and editing. Nucleic Acids Res. 2008;36:3579–89. doi: 10.1093/nar/gkn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blanco MA, Aleckovic M, Hua Y, Li T, Wei Y, Xu Z, et al. Identification of staphylococcal nuclease domain-containing 1 (SND1) as a Metadherin-interacting protein with metastasis-promoting functions. J Biol Chem. 2011;286:19982–92. doi: 10.1074/jbc.M111.240077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emdad L, Janjic A, Alzubi MA, Hu B, Santhekadur PK, Menezes ME, et al. Suppression of miR-184 in malignant gliomas upregulates SND1 and promotes tumor aggressiveness. Neuro Oncol. 2015;17:419–29. doi: 10.1093/neuonc/nou220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsuchiya N, Ochiai M, Nakashima K, Ubagai T, Sugimura T, Nakagama H. SND1, a component of RNA-induced silencing complex, is up-regulated in human colon cancers and implicated in early stage colon carcinogenesis. Cancer Res. 2007;67:9568–76. doi: 10.1158/0008-5472.CAN-06-2707. [DOI] [PubMed] [Google Scholar]
- 12.Wan L, Lu X, Yuan S, Wei Y, Guo F, Shen M, et al. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene- and carcinogen-induced mammary tumors. Cancer Cell. 2014;26:92–105. doi: 10.1016/j.ccr.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuruma H, Kamata Y, Takahashi H, Igarashi K, Kimura T, Miki K, et al. Staphylococcal nuclease domain-containing protein 1 as a potential tissue marker for prostate cancer. Am J Pathol. 2009;174:2044–50. doi: 10.2353/ajpath.2009.080776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoo BK, Santhekadur PK, Gredler R, Chen D, Emdad L, Bhutia S, et al. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology. 2011;53:1538–48. doi: 10.1002/hep.24216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santhekadur PK, Akiel M, Emdad L, Gredler R, Srivastava J, Rajasekaran D, et al. Staphylococcal nuclease domain containing-1 (SND1) promotes migration and invasion via angiotensin II type 1 receptor (AT1R) and TGFbeta signaling. FEBS Open Bio. 2014;4:353–61. doi: 10.1016/j.fob.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santhekadur PK, Das SK, Gredler R, Chen D, Srivastava J, Robertson C, et al. Multifunction Protein Staphylococcal Nuclease Domain Containing 1 (SND1) Promotes Tumor Angiogenesis in Human Hepatocellular Carcinoma through Novel Pathway That Involves Nuclear Factor kappaB and miR-221. J Biol Chem. 2012;287:13952–8. doi: 10.1074/jbc.M111.321646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajasekaran D, Jariwala N, Mendoza RG, Robertson CL, Akiel MA, Dozmorov M, et al. Staphylococcal nuclease and tudor domain containing 1 (SND1) promotes hepatocarcinogenesis by inhibiting monoglyceride lipase (MGLL) J Biol Chem. 2016;291:10736–46. doi: 10.1074/jbc.M116.715359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jariwala N, Rajasekaran D, Srivastava J, Gredler R, Akiel MA, Robertson CL, et al. Role of the staphylococcal nuclease and tudor domain containing 1 in oncogenesis (review) Int J Oncol. 2015;46:465–73. doi: 10.3892/ijo.2014.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnone A, Bier CJ, Cotton FA, Hazen EE, Jr, Richardson DC, Richardson JS. The extracellular nuclease of Staphylococcus aureus: structures of the native enzyme and an enzyme-inhibitor complex at 4 A resolution. Proc Natl Acad Sci U S A. 1969;64:420–7. doi: 10.1073/pnas.64.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weber DJ, Mullen GP, Mildvan AS. Conformation of an enzyme-bound substrate of staphylococcal nuclease as determined by NMR. Biochemistry. 1991;30:7425–37. doi: 10.1021/bi00244a009. [DOI] [PubMed] [Google Scholar]
- 21.Ramirez MI, Karaoglu D, Haro D, Barillas C, Bashirzadeh R, Gil G. Cholesterol and bile acids regulate cholesterol 7 alpha-hydroxylase expression at the transcriptional level in culture and in transgenic mice. Mol Cell Biol. 1994;14:2809–21. doi: 10.1128/mcb.14.4.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srivastava J, Siddiq A, Emdad L, Santhekadur PK, Chen D, Gredler R, et al. Astrocyte elevated gene-1 promotes hepatocarcinogenesis: novel insights from a mouse model. Hepatology. 2012;56:1782–91. doi: 10.1002/hep.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoo BK, Emdad L, Su ZZ, Villanueva A, Chiang DY, Mukhopadhyay ND, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest. 2009;119:465–77. doi: 10.1172/JCI36460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rajasekaran D, Srivastava J, Ebeid K, Gredler R, Akiel M, Jariwala N, et al. Combination of Nanoparticle-Delivered siRNA for Astrocyte Elevated Gene-1 (AEG-1) and All-trans Retinoic Acid (ATRA): An Effective Therapeutic Strategy for Hepatocellular Carcinoma (HCC) Bioconjug Chem. 2015;26:1651–61. doi: 10.1021/acs.bioconjchem.5b00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen D, Siddiq A, Emdad L, Rajasekaran D, Gredler R, Shen XN, et al. Insulin-like growth factor-binding protein-7 (IGFBP7): a promising gene therapeutic for hepatocellular carcinoma (HCC) Mol Ther. 2013;21:758–66. doi: 10.1038/mt.2012.282. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Srivastava J, Siddiq A, Gredler R, Shen XN, Rajasekaran D, Robertson CL, et al. Astrocyte elevated gene-1 and c-Myc cooperate to promote hepatocarcinogenesis in mice. Hepatology. 2015;61:915–29. doi: 10.1002/hep.27339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140:1410–26. doi: 10.1053/j.gastro.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 29.Zhu M, Li W, Lu Y, Dong X, Lin B, Chen Y, et al. HBx drives alpha fetoprotein expression to promote initiation of liver cancer stem cells through activating PI3K/AKT signal pathway. Int J Cancer. 2017;140:1346–55. doi: 10.1002/ijc.30553. [DOI] [PubMed] [Google Scholar]
- 30.Su R, Nan H, Guo H, Ruan Z, Jiang L, Song Y, et al. Associations of components of PTEN/AKT/mTOR pathway with cancer stem cell markers and prognostic value of these biomarkers in hepatocellular carcinoma. Hepatol Res. 2016;46:1380–91. doi: 10.1111/hepr.12687. [DOI] [PubMed] [Google Scholar]
- 31.Hagiwara S, Kudo M, Nagai T, Inoue T, Ueshima K, Nishida N, et al. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br J Cancer. 2012;106:1997–2003. doi: 10.1038/bjc.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012;315:129–37. doi: 10.1016/j.canlet.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–58. doi: 10.1038/sj.onc.1210811. [DOI] [PubMed] [Google Scholar]
- 34.Bourguignon LY, Shiina M, Li JJ. Hyaluronan-CD44 interaction promotes oncogenic signaling, microRNA functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv Cancer Res. 2014;123:255–75. doi: 10.1016/B978-0-12-800092-2.00010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, et al. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology. 2012;55:807–20. doi: 10.1002/hep.24739. [DOI] [PubMed] [Google Scholar]
- 36.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–96. doi: 10.1016/j.cell.2013.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393–404. doi: 10.1053/j.gastro.2014.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Armengol S, Arretxe E, Rodriguez L, Ochoa B, Chico Y, Martinez MJ. NF-kappaB, Sp1 and NF-Y as transcriptional regulators of human SND1 gene. Biochimie. 2013;95:735–42. doi: 10.1016/j.biochi.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 39.Arretxe E, Armengol S, Mula S, Chico Y, Ochoa B, Martinez MJ. Profiling of promoter occupancy by the SND1 transcriptional coactivator identifies downstream glycerolipid metabolic genes involved in TNFalpha response in human hepatoma cells. Nucleic Acids Res. 2015;43:10673–88. doi: 10.1093/nar/gkv858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun H, Jiang L, Luo X, Jin W, He Q, An J, et al. Potential tumor-suppressive role of monoglyceride lipase in human colorectal cancer. Oncogene. 2013;32:234–41. doi: 10.1038/onc.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.