

Abstract
Aims/hypothesis
Although it is known that lipid metabolism plays a role in insulin resistance in type 2 diabetes and in obesity, the mechanism is still largely unknown. Apolipoprotein E (ApoE) regulates plasma lipid levels and also plays a role in the uptake of lipids into various tissues. To investigate whether the suppression of whole-particle lipoprotein uptake into tissues affects insulin responsiveness and the diabetic condition, we examined the effect of an ApoE (also known as Apoe) gene deletion in MKR mice, a mouse model of type 2 diabetes.
Methods
ApoE−/−, MKR, ApoE−/−/MKR and control mice were placed on a high-fat, high-cholesterol diet for 16 weeks. Glucose tolerance, serum insulin, blood glucose, insulin tolerance, tissue triacylglycerol content and atherosclerotic lesions were assessed.
Results
ApoE−/−/MKR and ApoE−/− mice showed significantly improved blood glucose, glucose tolerance and insulin sensitivity. Reduced triacylglycerol content in liver and reduced fat accumulation in liver and adipose tissue were found in ApoE−/−/MKR and ApoE−/− mice compared with control and MKR mice. ApoE−/− and ApoE−/−/MKR mice demonstrated similarly large atherosclerotic lesions, whereas MKR and control mice had small atherosclerotic lesions.
Conclusions/interpretation
We demonstrated that ApoE deficiency abrogates insulin resistance in a mouse model of type 2 diabetes, suggesting that lipid accumulation in tissue is a major cause of insulin resistance in this mouse model.
Keywords: ApoE gene deletion, Atherosclerosis, MKR mouse model of type 2 diabetes
Introduction
The incidence of type 2 diabetes mellitus is dramatically increasing and represents one of the world’s chief economic and healthcare challenges [1]. Associated with obesity, type 2 diabetes mellitus results from impaired insulin secretion and peripheral insulin responsiveness. Adipose tissue stores and mobilises triacylglycerol, and plays a central role in regulating whole-body metabolism and glucose homeostasis [2]. High circulating NEFA and triacylglycerol, present in obesity and the metabolic syndrome, are associated with impaired insulin sensitivity in skeletal muscle and decreased glucose tolerance [3, 4]. Studies showing an increased triacylglycerol content in liver and muscle have revealed a strong correlation with whole-body insulin resistance [5, 6].
Apolipoprotein E (ApoE) is a 34 kDa glycoprotein that is synthesised by liver, adipocyte, brain and other tissues, and is a ligand for receptors that clear remnants of chylomicrons and VLDL. Lack of ApoE causes accumulation of cholesterol-rich remnants in plasma, leading eventually to atherosclerosis [7]. ApoE-deficient mice (ApoE [also known as Apoe]−/−) fed a high-cholesterol diet have been used as a mouse model to study atherosclerosis. These mice exhibit reduced lipid accumulation in adipose tissue, improved insulin sensitivity and resistance to the development of obesity [8–10].
We have previously developed a mouse model of type 2 diabetes by overproducing a dominant negative IGF-1 receptor specifically in skeletal muscle (MKR mice) [11]. Hybrid formation of the mutated IGF-1 receptor with the endogenous IGF-1 and insulin receptors caused impaired insulin and IGF-1 receptor signalling pathways specifically in skeletal muscle. This primary defect in skeletal muscle resulted in hyperinsulinaemia, dyslipidaemia and beta cell dysfunction, leading eventually to hyperglycaemia. Whole-body insulin resistance was associated with impaired insulin sensitivity in skeletal muscle as well as in adipose tissue and liver. Following treatment with WY-14643, a peroxisome proliferator-activated receptor (PPAR)α agonist, or with leptin, MKR mice showed a marked reduction in serum lipids (NEFA and triacylglycerol) and in the lipid content of liver. This was also associated with improved insulin sensitivity and diabetes of the MKR mice [12, 13], suggesting that lipotoxicity plays a major role in the insulin resistance seen in this model.
To investigate whether the suppression of whole-particle lipoprotein uptake affects insulin responsiveness and the diabetic condition, we examined the effect of ApoE gene deletion in MKR mice.
Methods
Mouse models and diet
The generation of the MKR mice (FVB/N background) has been described previously [11]. ApoE−/− (FVB/N background) mice were obtained from J. L. Breslow of the Rockefeller Institute (New York, USA) [14] and were crossed with MKR mice to generate the double gene-disrupted mice (ApoE−/−/MKR). Mice that were heterozygous for any of the alleles ApoE or MKR were not included in the study.
Atherosclerosis was induced by feeding a high-cholesterol diet containing 1.25% cholesterol (wt/wt) and 20% fat (wt/wt) for 16 weeks (D12108; Research Diets, New Brunswick, NJ, USA) starting from 4 weeks of age. Food intake was measured weekly, from 1 to 7 weeks after starting the diet. Food intake was normalised to body weight and expressed as: g food (g body weight)−0.75 day−1. Both male and female mice were followed.
The animal experiments were conducted in accordance with the principles of laboratory care, as designated by the Mount Sinai School of Medicine, Animal Care and Use Committee.
Body composition
Whole-body fat and lean mass were measured in conscious mice using a quantitative magnetic resonance method (Echo MRI 3-in-1; Echo Medical Systems, Houston, TX, USA).
Serum analysis
Serum was obtained from the vein between 10:00 and 12:00 hours in the non-fasting state. Serum levels of NEFA were measured using a fatty acid assay kit (Roche, Indianapolis, IN, USA). Serum insulin levels were determined using a radioimmunoassay kit (Linco Research, St Charles, MO, USA). Serum IL-6 and TNF-α levels were measured by ELISA (R & D Systems, Minneapolis, MN, USA). Blood glucose was measured using a glucose monitoring system (FreeStyle Lite; Abbott, Abbott Park, IL, USA). Serum total cholesterol and triacylglycerol levels were measured by ELISA (BioVision, Mountain View, CA, USA).
Glucose tolerance and insulin tolerance tests
For glucose tolerance tests, mice were fasted for 8 h and blood glucose levels were measured at the indicated time points following an intraperitoneal injection of glucose (2 mg/g body weight). For insulin tolerance tests, mice were injected intraperitoneally with insulin (0.75 U/kg body weight) and blood glucose levels were measured from the tail vein at the indicated time points.
Tissue triacylglycerol assay
Tissue triacylglycerol was extracted with chloroform/methanol as previously described [15]. After hydrolysis with KOH base, triacylglycerol was measured radiometrically using a glycerol kinase assay [15].
Characterisation of atherosclerosis
Following 16 weeks of a high-cholesterol diet, mice were killed and the aorta removed and perfusion-fixed in phosphate-buffered paraformaldehyde (4% [wt/vol.], pH 7.4). Fat from the isolated aorta was removed under the microscope and the aortas were fixed overnight in 4% paraformaldehyde (pH 7.4). The fixed aortas were cut into four regions: (1) at the region of valve attachment; (2) at the level of the branching of the innominate artery; (3) at the most proximal part of the descending aorta at the level of the subcutaneous artery; and (4) at the descending aorta. Segments of the aorta were paraffin-embedded and stained with haematoxylin and eosin. Four sections per region were used for histomorphometric analysis. The neointimal area, total vessel wall area and medial area in each section were measured using MicroSuite Five (Olympus America, Center valley, PA) and the ratios of neointimal area:medial area and neointimal area:total vessel-wall area were calculated for evaluation of neointimal formation (Fig. 5f). The average of neointimal area:medial area and neointimal area:total vessel-wall area in the above four sections was used to represent the lesion size of each animal.
Fig. 5.
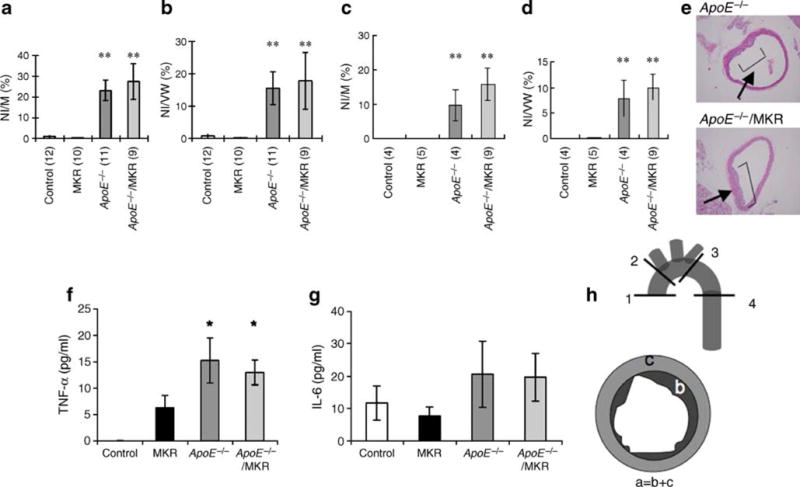
ApoE−/− and ApoE−/−/MKR mice develop atherosclerotic lesions when fed a high-cholesterol diet. The aorta was removed at 22–24 weeks of age. The areas of neointima (NI), of total vessel wall (VW) and of media (M) in each section of (a, b) male and (c, d) female mice were measured using MicroSuite and the NI:M and NI:VW ratios (%) calculated for evaluation of neointimal formation. Values in parentheses are n. Data are expressed as mean±SE. **p<0.01 compared with control mice. e Histology of the aorta for ApoE−/− and ApoE−/−/MKR mice with haematoxylin–eosin. f Serum levels of TNF-α and IL-6 (g) were determined at 22–24 weeks. Data are expressed as mean±SE; *p<0.05 compared with control mice. h The schema for analysis of atherosclerotic lesion. Fixed aorta were cut into four regions as indicated: 1, at the region of valve attachment; 2, at the level of the branching of the innominate artery; 3, at the most proximal part of the descending aorta at the level of the subcutaneous artery; 4, at the descending aorta. a, total vessel wall; b, neointimal area; c, medial area
Histology
The left epididymal fat pad and the right lobe of the liver were isolated and fixed overnight in 4% paraformaldehyde in PBS. The tissues were then embedded in paraffin and 5 μm sections were stained with haematoxylin and eosin.
Statistics
All values are given as mean±SEM. Statistical differences were determined using one-factor ANOVA followed by a t test. Significance was accepted at p< 0.05.
Results
Cholesterol diet increases body weight of ApoE−/− and MKR mice with no increase in body adiposity
Over the 16 weeks on a high-cholesterol diet, all groups of mice increased their body weights (Fig. 1a). We found no differences in food intake between the four groups (Fig. 1b). MKR mice are generally smaller than control mice [16], a difference that was still apparent on the high-cholesterol diet. Interestingly, in the current study, ApoE−/− and ApoE−/−/MKR mice on the diet had significantly higher body weights than control mice, despite previous reports that the former are resistant to obesity [9, 10, 17]. All four groups had similar body adiposity, measured by MRI (Fig. 1c). Gonadal fat pads, obtained at the end of the study, were also similar between all groups (Fig. 2a). However, histological analysis showed smaller adipocytes in ApoE−/−/MKR and ApoE−/− mice than in MKR or control mice (Fig. 2b). While obese mice usually show increased fat mass measured as per cent of body weight, ApoE−/− mice and ApoE−/−/MKR mice on the high-cholesterol diet did not have increased relative fat mass, despite increased body weight. ApoE−/− mice and ApoE−/−/MKR mice exhibited marked skin xanthomata; however, its contribution to the increased body weight requires more in-depth studies.
Fig. 1.
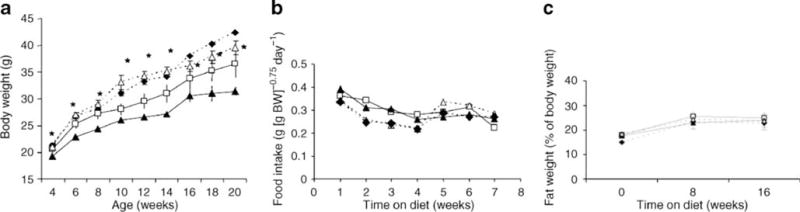
Body weight, food intake and body composition on a high-cholesterol diet. a Body weight was measured every 2 weeks from 4 to 20 weeks of age (n = 10–15 mice per group). Data are expressed as mean±SE; *p<0.05 for ApoE−/−/MKR mice compared with MKR mice from 12 weeks of age. b Food intake was measured weekly in ApoE−/−, MKR, ApoE−/−/MKR and control mice from 5 to 12 weeks following the high-cholesterol diet. c Whole-body fat and lean mass was measured in conscious mice using the quantitative magnetic resonance imaging method at the onset, and at 8 and 16 weeks on the diet (n=10–15 mice per group). Squares, control; black triangles, MKR; diamonds, ApoE−/−; white triangles, ApoE−/−/MKR. Data are expressed as mean±SE. BW, body weight
Fig. 2.
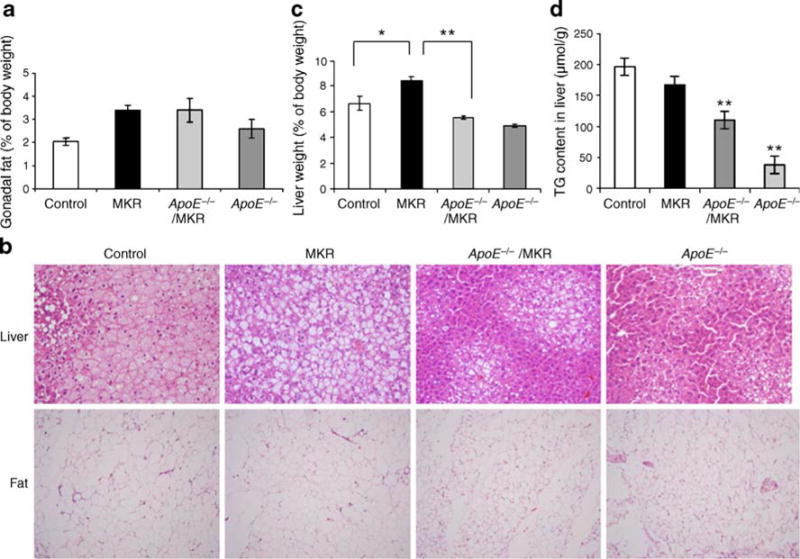
ApoE deficiency reversed fat accumulation in MKR mice. a Relative weight of gonadal fat to body weight at 22–24 weeks of age. b Histology of the liver and fat stained with haematoxylin–eosin. c Liver weight was measured at 22–24 weeks of age and is expressed as per cent of body weight. *p < 0.05; **p < 0.01. d Liver triacylglycerol (TG) content. At the end of 16 weeks on the high-cholesterol diets, livers were removed and triacylglycerol content measured as described. **p < 0.01 compared with MKR mice. Data (a, c, d) are expressed as mean±SE
ApoE gene ablation in MKR mice led to reduced liver triacylglycerol content
Consistent with our findings from previous studies [12], liver weights of MKR mice were increased compared with control mice (Fig. 2c). However, when the ApoE gene was deleted in MKR mice (i.e. ApoE−/−/MKR), liver weights were reduced compared with MKR mice alone (Fig. 2c). Histological analyses of the liver showed reduced lipid accumulation in ApoE−/−/MKR and ApoE−/− mice, while there were abundant lipid droplets in MKR mice on the high-cholesterol diet (Fig. 2b). Liver triacylglycerol content was lower in ApoE−/− and ApoE−/−/MKR mice than in MKR and control mice (Fig. 2d), despite markedly high levels of triacylglycerol in serum (Fig. 3a).
Fig. 3.
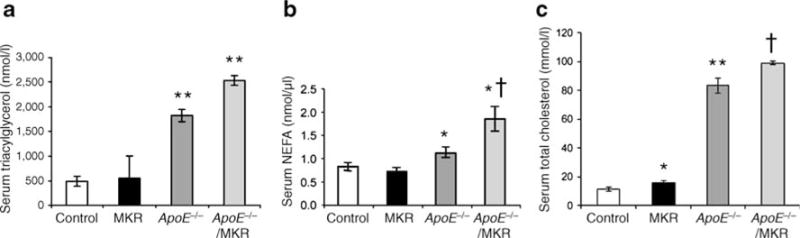
Serum levels of triacylglycerol, NEFA and cholesterol in ApoE deficient mice. a Serum triacylglycerol levels were determined at 22–24 weeks. **p< 0.01 compared with control and MKR mice. b Serum NEFA levels were determined at 22–24 weeks. Data are expressed as mean±SE. *p < 0.05 for control and MKR mice vs ApoE−/− and ApoE−/−/MKR mice; †p< 0.05 for ApoE−/− mice vs ApoE−/−/MKR mice. c Serum cholesterol levels were determined at 22–24 weeks. *p<0.05 compared with control; **p<0.01 compared with control and MKR mice; †p<0.05 compared with ApoE−/− mice
Effects of ApoE deficiency on serum cholesterol, triacylglycerol, insulin, glucose and insulin sensitivity in MKR mice
ApoE−/− and ApoE−/−/MKR mice on the diet showed striking increases in serum levels of cholesterol, triacylglycerol and NEFA compared with MKR and control mice (Fig. 3), which was consistent with previous reports [9]. On a regular chow diet, MKR mice have higher serum triacylglycerol and NEFA levels than control mice [11, 13]. On the high-cholesterol diet, however, MKR mice exhibited similar NEFA and triacylglycerol levels to controls, although the former had slightly, but significantly increased serum cholesterol levels. Interestingly, ApoE−/−/MKR mice had significant increases in serum levels of cholesterol, triacylglycerol and NEFA compared with ApoE−/− mice (p= 0.03, p=0.048, p=0.02 respectively) (Fig. 3a,b,c). Deletion of the ApoE gene in the MKR mice (ApoE−/−/MKR) resulted in lower fasting glucose and insulin levels than in MKR mice at all time points tested (Fig. 4a,b,c). MKR mice on the high-cholesterol diet had severe glucose and insulin intolerance (Fig. 4g,h). Following 12 weeks on the high-cholesterol diet, ApoE−/−/MKR mice showed a significant improvement in glucose and insulin tolerance compared with MKR mice (Fig. 4g,h). Together, our data show that ApoE−/−/MKR have improved insulin sensitivity and glucose tolerance, despite markedly high levels of cholesterol, triacylglycerol and NEFA in serum.
Fig. 4.
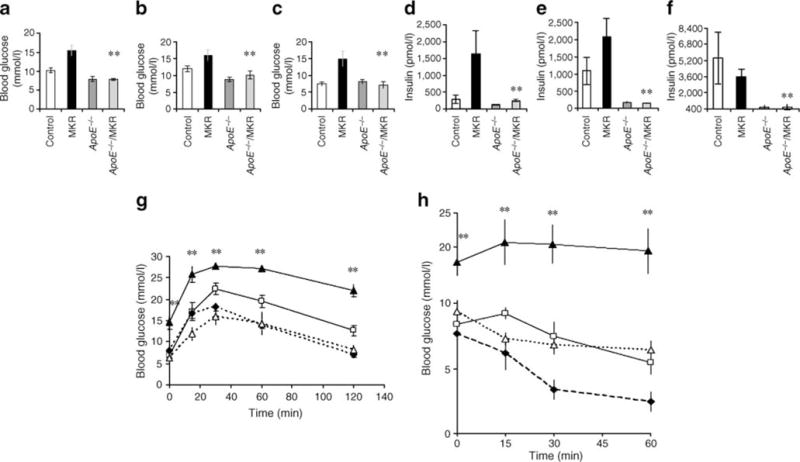
ApoE deficiency improved hyperglycaemia, hyperinsulinaemia and insulin tolerance of MKR mice. a–f Mice were fed a high-cholesterol diet starting from 4 weeks of age for a total of 16–20 weeks. Blood glucose and serum insulin levels were measured at 4 (a, d), 12 (b, e) and 22–24 (c, f) weeks of age. Data are expressed as mean±SE. **p<0.01 compared with MKR. Glucose tolerance test (g) of mice fed high-cholesterol diets was performed i.p. after 8 h of fasting on 3–5 mice per group at 12 weeks on the diet. h Insulin tolerance tests of mice fed high-cholesterol diets were performed on 3–5 mice per group after 8 h of fasting at 12 weeks on the diet. Data (g, h) are expressed as mean±SE; **p<0.01 compared with ApoE−/−/MKR mice. The percentage fall in blood glucose following insulin injection was 0% in MKR, ~25% in control mice, ~60% in ApoE−/− mice and >25% in ApoE−/−/MKR mice. g, h Squares, control; black triangles, MKR; diamonds, ApoE−/−; white triangles, ApoE−/−/MKR
Atherosclerotic changes after exposure to high-cholesterol diet
To evaluate the severity of atherosclerotic lesions in the different mouse groups, we used an average of the neointimal area:medial area and neointimal area:total vessel-wall area in four regions. ApoE−/− and ApoE−/−/MKR male and female mice had markedly increased atherosclerotic lesions compared with MKR and control mice (Fig. 5a–d), but compared with each other, showed similar degrees of atherosclerotic lesions (Fig. 5e). Taken together, despite improvement in insulin sensitivity, blood glucose and serum insulin levels, ApoE−/−/MKR mice fed high-cholesterol diet did not exhibit any reductions in atherosclerotic lesions. Serum levels of the inflammatory cytokine IL-6 and TNF-α, which are associated with the development of atherosclerotic lesions [18], were increased in ApoE−/− and ApoE−/−/MKR to a similar degree compared with control or MKR mice (Fig. 5f,g).
Discussion
Our study demonstrates that ApoE gene deletion in the MKR mouse model of type 2 diabetes improved glycaemic control, as evidenced by reduced hyperinsulinaemia and hyperglycaemia. ApoE−/− and ApoE−/−/MKR mice showed reduced body adiposity and smaller adipocytes, probably due to reduced triacylglycerol and NEFA content in fat tissue [8]. These reductions were attributed to impaired adipocyte differentiation in ApoE−/− mice, based on in vitro findings showing that levels of adipocyte fatty acid binding protein (AFABP/aP2) and PPAR-γ were markedly reduced in co-cultures of bone marrow stromal cells and 3T3-L1 cells [19].
Liver triacylglycerol content, which was increased in MKR and control mice fed a high-cholesterol diet, decreased in ApoE−/− and ApoE−/−/MKR mice, a finding also evident in histological examination. Previous studies with MKR mice have shown that leptin [13], PPARα agonist [12] and the β3 adrenergic agonists [20] treatment reduced triacylglycerol content in liver and muscle, and improved insulin sensitivity of MKR mice. However, treatment with thiazolidinediones [21], which improved insulin sensitivity solely in adipose tissue of MKR mice, was insufficient to improve whole-body insulin sensitivity. In these previous studies, we used the euglycaemic–hyperinsulinaemic clamp technique and demonstrated that when insulin sensitivity improved, the liver was the major organ to show improved insulin responsiveness. Skeletal muscle is very unlikely to play a major role, since the function of the insulin and IGF-1 receptors was interfered with (genetically affected). In the current study, we show that ApoE gene deletion in MKR mice led to reduced lipid accumulation not only in fat but also in liver, resulting in improved insulin sensitivity. Together, these studies suggest that the liver plays a major role in improvement of whole-body insulin sensitivity in MKR mice.
Our studies are in line with studies published by Hofmann et al., who found that ApoE−/− mice have impaired lipid uptake in adipose tissue and improved glucose tolerance [22]. Gao et al. found that ApoE−/− mice prevented the development of genetically induced obesity [9]. These ApoE−/−/KKAy mice had decreased fat accumulation in liver and adipose tissue, and improved insulin sensitivity despite increased plasma NEFA levels [9]. Adipocytes of ApoE−/−/KKAy mice were smaller and the serum cytokine profile (leptin, TNF-α, adiponectin) was improved.
Several molecular mechanisms have been presented to explain the relationship between insulin resistance and increased cellular accumulation of triacylglycerol. In muscle, increased triacylglycerol content leads to increases in diacylglycerol and ceramide that activate a serine kinase cascade, decreasing insulin-stimulated activity of IRS-1-associated phosphatidylinositol-3-kinase [23, 24], leading eventually to reduced glucose transport [25] and glycogen synthesis [26]. Moreover, increased intramyocellar triacylglycerol, thought to be more prone to oxidative stress, generates lipid peroxidation products that potentially inhibit signalling [27]. Studies have shown that cellular triacylglycerol accumulation in liver as such is not initially toxic [28, 29]; however, intermediate metabolites such as acyl-CoAs and diacylglycerol are determinants of the development of insulin resistance [30, 31]. Studies in mouse models deficient in the enzymes acetyl coenzyme A carboxylase, stearoyl-CoA desaturate, or the elongation of long-chain fatty acids family member 6, which is related to fatty acid and triacylglycerol synthesis in liver, showed decreased lipogenesis in liver and improved insulin resistance and resistance to diet-induced obesity [31–34].
Several studies have shown that increased secretion of proinflammatory cytokines, decreased secretion of adiponectin, increased circulating levels of NEFA and postprandial hyperglycaemia can all affect endothelial function [35, 36]. ApoE−/− mice with deletion of the gene encoding endothelial nitric oxide synthase showed accelerated atherosclerosis [37] and ApoE−/− mice with deletion of the receptor for advanced glycation end-products or with the deletion of TNF-α showed reduced atherosclerotic lesions [38]. In our study, ApoE−/− and ApoE−/−/MKR male and female mice had markedly increased atherosclerotic lesions compared with MKR and control mice, despite improvement in insulin sensitivity. These findings strongly suggest that lipid metabolism and increase in inflammatory cytokines in serum play a major role in causing atherosclerosis and that improvement of hyperglycaemia or insulin resistance in liver is not necessarily a prerequisite for the treatment of atherosclerosis. Interestingly, recent outcomes in studies of type 2 diabetes have found that improved blood glucose control was associated with reduced microvascular complications, but was not associated with clear-cut improvement in macrovascular complications [39–41]. Our study is in line with these findings, showing that improved insulin sensitivity and blood glucose, in the face of an atherogenic phenotype, are unable to reverse macrovascular disease.
Acknowledgments
We thank N. Mall (Mt Sinai School of Medicine, New York) for help with histological experiments, and W. Jou and O. Gavrilova (NIDDK, Bethesda, MD, USA) for assistance with liver triacylglycerol assays. This study was supported by an American Diabetes Association Mentorship Grant, awarded to Y. Kawashima and D. LeRoith.
Abbreviations
- ApoE
Apolipoprotein E
- PPAR
Peroxisome proliferator-activated receptor
Footnotes
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contributor Information
Y. Kawashima, Division of Endocrinology, Diabetes, and Bone Diseases, Mount Sinai School of Medicine, 1 Gustave Levy Place (1055), New York, NY 10029, USA
J. Chen, Cardiovascular Research Center, Mount Sinai School of Medicine, New York, NY, USA
H. Sun, Division of Endocrinology, Diabetes, and Bone Diseases, Mount Sinai School of Medicine, 1 Gustave Levy Place (1055), New York, NY 10029, USA
D. Lann, Division of Endocrinology, Diabetes, and Bone Diseases, Mount Sinai School of Medicine, 1 Gustave Levy Place (1055), New York, NY 10029, USA
R. J. Hajjar, Cardiovascular Research Center, Mount Sinai School of Medicine, New York, NY, USA
S. Yakar, Division of Endocrinology, Diabetes, and Bone Diseases, Mount Sinai School of Medicine, 1 Gustave Levy Place (1055), New York, NY 10029, USA
D. LeRoith, Division of Endocrinology, Diabetes, and Bone Diseases, Mount Sinai School of Medicine, 1 Gustave Levy Place (1055), New York, NY 10029, USA
References
- 1.Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12:75–80. doi: 10.1038/nm0106-75. [DOI] [PubMed] [Google Scholar]
- 2.Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- 3.Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest. 1983;72:1737–1747. doi: 10.1172/JCI111133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93:2438–2446. doi: 10.1172/JCI117252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim JK, Fillmore JJ, Chen Y, et al. Tissue-specific overexpression of lipoprotein lipase causes tissue-specific insulin resistance. Proc Natl Acad Sci U S A. 2001;98:7522–7527. doi: 10.1073/pnas.121164498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryysy L, Hakkinen AM, Goto T, et al. Hepatic fat content and insulin action on non-esterified fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes. 2000;49:749–758. doi: 10.2337/diabetes.49.5.749. [DOI] [PubMed] [Google Scholar]
- 7.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 8.Huang ZH, Reardon CA, Mazzone T. Endogenous ApoE expression modulates adipocyte triglyceride content and turnover. Diabetes. 2006;55:3394–3402. doi: 10.2337/db06-0354. [DOI] [PubMed] [Google Scholar]
- 9.Gao J, Katagiri H, Ishigaki Y, et al. Involvement of apolipoprotein E in excess fat accumulation and insulin resistance. Diabetes. 2007;56:24–33. doi: 10.2337/db06-0144. [DOI] [PubMed] [Google Scholar]
- 10.Schreyer SA, Vick C, Lystig TC, Mystkowski P, LeBoeuf RC. LDL receptor but not apolipoprotein E deficiency increases diet-induced obesity and diabetes in mice. Am J Physiol Endocrinol Metab. 2002;282:E207–E214. doi: 10.1152/ajpendo.2002.282.1.E207. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez AM, Kim JK, Yakar S, et al. Functional inactivation of the IGF-I and insulin receptors in skeletal muscle causes type 2 diabetes. Genes Dev. 2001;15:1926–1934. doi: 10.1101/gad.908001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H, Haluzik M, Asghar Z, et al. Peroxisome proliferator-activated receptor-alpha agonist treatment in a transgenic model of type 2 diabetes reverses the lipotoxic state and improves glucose homeostasis. Diabetes. 2003;52:1770–1778. doi: 10.2337/diabetes.52.7.1770. [DOI] [PubMed] [Google Scholar]
- 13.Toyoshima Y, Gavrilova O, Yakar S, et al. Leptin improves insulin resistance and hyperglycaemia in a mouse model of type 2 diabetes. Endocrinology. 2005;146:4024–4035. doi: 10.1210/en.2005-0087. [DOI] [PubMed] [Google Scholar]
- 14.Dansky HM, Charlton SA, Sikes JL, et al. Genetic background determines the extent of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 1999;19:1960–1968. doi: 10.1161/01.atv.19.8.1960. [DOI] [PubMed] [Google Scholar]
- 15.Gavrilova O, Leon LR, Marcus-Samuels B, et al. Torpor in mice is induced by both leptin-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 1999;96:14623–14628. doi: 10.1073/pnas.96.25.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H, Barton E, Muja N, Yakar S, Pennisi P, Leroith D. Intact insulin and insulin-like growth factor-I receptor signaling is required for growth hormone effects on skeletal muscle growth and function in vivo. Endocrinology. 2005;146:1772–1779. doi: 10.1210/en.2004-0906. [DOI] [PubMed] [Google Scholar]
- 17.Chiba T, Shinozaki S, Nakazawa T, et al. Leptin deficiency suppresses progression of atherosclerosis in apoE-deficient mice. Atherosclerosis. 2008;196:68–75. doi: 10.1016/j.atherosclerosis.2007.01.040. [DOI] [PubMed] [Google Scholar]
- 18.Blake GJ, Ridker PM. Novel clinical markers of vascular wall inflammation. Circ Res. 2001;89:763–771. doi: 10.1161/hh2101.099270. [DOI] [PubMed] [Google Scholar]
- 19.Chiba T, Nakazawa T, Yui K, Kaneko E, Shimokado K. VLDL induces adipocyte differentiation in ApoE-dependent manner. Arterioscler Thromb Vasc Biol. 2003;23:1423–1429. doi: 10.1161/01.ATV.0000085040.58340.36. [DOI] [PubMed] [Google Scholar]
- 20.Kim H, Pennisi PA, Gavrilova O, et al. Effect of adipocyte beta3-adrenergic receptor activation on the type 2 diabetic MKR mice. Am J Physiol Endocrinol Metab. 2006;290:E1227–E1236. doi: 10.1152/ajpendo.00344.2005. [DOI] [PubMed] [Google Scholar]
- 21.Kim H, Haluzik M, Gavrilova O, et al. Thiazolidinediones improve insulin sensitivity in adipose tissue and reduce the hyperlipidaemia without affecting the hyperglycaemia in a transgenic model of type 2 diabetes. Diabetologia. 2004;47:2215–2225. doi: 10.1007/s00125-004-1581-6. [DOI] [PubMed] [Google Scholar]
- 22.Hofmann SM, Perez-Tilve D, Greer TM, et al. Defective lipid delivery modulates glucose tolerance and metabolic response to diet in apolipoprotein E-deficient mice. Diabetes. 2008;57:5–12. doi: 10.2337/db07-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 24.Pickersgill L, Litherland GJ, Greenberg AS, Walker M, Yeaman SJ. Key role for ceramides in mediating insulin resistance in human muscle cells. J Biol Chem. 2007;282:12583–12589. doi: 10.1074/jbc.M611157200. [DOI] [PubMed] [Google Scholar]
- 25.Krssak M, Falk Petersen K, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 26.Roden M, Price TB, Perseghin G, et al. Mechanism of non-esterified fatty acids-induced insulin resistance in humans. J Clin Invest. 1996;97:2859–2865. doi: 10.1172/JCI118742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russell AP, Gastaldi G, Bobbioni-Harsch E, et al. Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs bad lipids? FEBS Lett. 2003;551:104–106. doi: 10.1016/s0014-5793(03)00875-5. [DOI] [PubMed] [Google Scholar]
- 28.Yamaguchi K, Yang L, McCall S, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007;45:1366–1374. doi: 10.1002/hep.21655. [DOI] [PubMed] [Google Scholar]
- 29.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neschen S, Morino K, Hammond LE, et al. Prevention of hepatic steatosis and hepatic insulin resistance in mitochondrial acyl-CoA:glycerol-sn-3-phosphate acyltransferase 1 knockout mice. Cell Metab. 2005;2:55–65. doi: 10.1016/j.cmet.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Savage DB, Choi CS, Samuel VT, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mao J, DeMayo FJ, Li H, et al. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci U S A. 2006;103:8552–8557. doi: 10.1073/pnas.0603115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen P, Miyazaki M, Socci ND, et al. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243. doi: 10.1126/science.1071527. [DOI] [PubMed] [Google Scholar]
- 34.Miyazaki M, Flowers MT, Sampath H, et al. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007;6:484–496. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Rask-Madsen C, King GL. Mechanisms of disease: endothelial dysfunction in insulin resistance and diabetes. Nat Clin Pract Endocrinol Metab. 2007;3:46–56. doi: 10.1038/ncpendmet0366. [DOI] [PubMed] [Google Scholar]
- 36.Kim JA, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- 37.Kuhlencordt PJ, Gyurko R, Han F, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 38.Soro-Paavonen A, Watson AM, Li J, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duckworth W, Abraira C, Moritz T, et al. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- 40.ADVANCE Collaborative Group. Patel A, MacMahon S, Chalmers J, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560–2572. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 41.Action to Control Cardiovascular Risk in Diabetes Study Group. Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]