

Abstract
Among the mechanisms implicated in the tumor-promoting effects of obesity, signaling by insulin-like growth factor-I (IGF-I) and insulin has received considerable attention. However, the emerging realization that obesity is associated with chronic inflammation has prompted other consideration of how the IGF-I axis may participate in cancer progression. In the present study, we used two mouse models of chronic (LID) and inducible (iLID) igf-1 gene deficiency in the liver to investigate the role of IGF-I in regulating the host microenvironment and colorectal carcinoma growth and metastasis in obese mice. Obese mice had a heightened inflammatory response in the liver, which was abolished in mice with chronic IGF-I deficiency (LID). In control animals changes to the hepatic microenvironment associated with obesity sustained the presence of tumor cells in the liver and increased the incidence of hepatic metastases after intrasplenic/portal inoculation of colon carcinoma cells. These changes did not occur in LID mice with chlonic IGF-1 deficiency. In contrast, these changes occured in iLID mice with acute IGF-1 deficiency, in the same manner as the control animals, revealing a fundamental difference in the nature of the requirement for IGF-1 on tumor growth and metastasis. In the setting of obesity, our findings imply that IGF-1 is critical to activate and sustain an inflammatory response in the liver that is needed for hepatic metastasis, not only through direct, paracrine effect on tumor cell growth, but also through indirect effects involving the tumor microenvironment.
Introduction
The tissue microenvironment plays an important role in tumor development and progression both at the primary site and at sites of metastases (1). The microenvironment consists of the tumor-associated stroma that includes resident fibroblasts, adipocytes, blood and lymph vessel–lining endothelial cells and their respective extracellular matrix proteins, as well as resident and infiltrating host inflammatory and immune cells. The malignant cells can communicate with the stroma through the release of, and response to, soluble mediators, such as growth factors and cytokines, through cell-cell and cell-matrix adhesion and through the release of proteolytic enzymes that remodel tissue barriers around the tumor cells and contribute to neovascularization and tumor migration and invasion (2, 3).
The host inflammatory response, macrophages in particular, plays an important role in tumor progression (4, 5). Chronic inflammation has been linked to tumor initiation and progression in different cancer types, including gastric (6), hepatocellular (7–10), and colorectal (11) carcinoma, and treatment with anti-inflammatory drugs such as cimetidine or aspirin was shown to reduce cancer risk (12). Moreover, a high density of tumor-associated macrophages was found to correlate with poor prognosis in more than 80% of studies published (13, 14). Animal studies have also shown that the host inflammatory response can be acutely activated during the early stages of metastasis and thereby promote tumor cell arrest and extravasation in the invaded organ (15, 16).
Obesity is a common metabolic disorder, frequently associated with chronic low-grade inflammation (17), increased oxidative stress (18), and lipotoxicity, all of which can alter the tissue microenvironment. Obesity often leads to lipid accumulation in the liver (hepatosteatosis) and chronic liver inflammation. Although it has been shown that obesity (and obesity-associated inflammation) can alter the host antitumor immune response and contribute to tumor cell escape from immune surveillance (19), it is unknown how this state of chronic inflammation affects the ability of metastatic tumor cells to induce the more acute and transient inflammatory reactions that are associated with the early stages of organ colonization and known to promote metastasis (15, 20, 21).
The liver is the main site of insulin-like growth factor-I (IGF-I) production. IGF-I has been identified as a critical regulator of tumor growth and metastasis, but its role in inflammation-associated tumor development or the obesity-associated increase in inflammation, and tumor growth, are presently unknown. Likewise, the role of IGF-I in regulating changes to the tumor-associated stroma that affect tumor cell growth in the liver in normal or obese individuals have not been studied. IGF-I is a known activator of the Janus-activated kinase/signal transducer and activator of transcription (STAT) pathway (22) and has been implicated in NF-κB–mediated transcriptional regulation of inflammatory cytokines and vascular endothelial cell adhesion receptors such as intercellular adhesion molecule-1 (ICAM-1; ref. 23). It may therefore play a role both in the maintenance of a chronic inflammatory state and in the induction of the acute inflammatory response that is triggered by tumor cells during the early stages of liver metastasis (24, 25).
The aim of the present study was 2-fold: (a) to assess host inflammation parameters and liver metastasis in mice with diet-induced obesity and (b) to determine the role of IGF-I in the development of liver metastases in the obese state. We used diet-induced obesity and an experimental liver metastasis assay to investigate the effect of a high-calorie diet on the hepatic microenvironment and the development of liver metastasis. To elucidate the role of serum IGF-I in tumor growth under conditions of obesity, we used mice with a liver-specific IGF-I deficiency (the LID model). These mice have no detectable IGF-I mRNA in the liver and a 75% reduction in serum IGF-I levels (26). Additionally, we generated a second mouse model of inducible liver IGF-I deficiency (iLID). The iLID mice express the Cre recombinase under the control of the anti-trypsin 1α promoter, specifically in liver, and a single tamoxifen injection results in gene recombination to produce a liver-specific IGF-I deficiency (iLID). The LID and iLID models were used here to dissect the effects of acute versus chronic IGF-I depletion on tumor cell growth and metastasis.
Materials and Methods
Mice and diets
Male mice were genotyped as described previously (26). Both LID and iLID mouse models were bred on a C57B6/J background. All chows were purchased from Research Diets6 (D12450B: 10 kcal % fat, control diet; D12492: 60 kcal % fat, high-calorie diet). The diets did not contain phytoestrogens.7 Animal care and maintenance were provided through the Mount Sinai School of Medicine AAA-LAC-Accredited Animal Facility. All procedures were approved by the Animal Care and Use Committee of the Mount Sinai School of Medicine.
Body adiposity
Body fat mass was measured in nonanesthetized mice using a Bruker minispec nuclear magnetic resonance analyzer mq 10 (Bruker Optics).
Cells
The colon adenocarcinoma MC-38 cells were maintained in DMEM (BioSource) supplemented with 10% FCS (Invitrogen) and glutamine (BioSource; ref. 27).
Intrasplenic/portal inoculation of tumor cells
Experimental hepatic metastases were generated using the intrasplenic/portal route of inoculation as described previously (15). Mice were anesthetized using isoflurane. Spleens were exposed through a small abdominal incision; 0.5 × 105 tumor cells in 100 μL of saline or saline alone were inoculated and the mice were splenectomized 1 min later. The livers were removed at the indicated time intervals, snap frozen in liquid N2, and stored at −80°C until further analysis.
Gene expression studies
Total RNA was extracted from snap-frozen livers using a combined Trizol/Qiagen RNeasy Midi kit. One microgram of total RNA was reverse transcribed and real-time PCR was performed using TaqMan reagents and specific primers. Real-time PCR was performed with the QuantiTect SYBR green PCR kit (Qiagen) according to the manufacturer’s instructions in ABI PRISM 7900HT sequence detection systems (Applied Biosystems). Each transcript in each sample was assayed three times and the fold-change ratios between experimental and control samples for each gene used in the analysis were calculated using β-actin or 18S levels as a reference.
Detection of serum hormones
Serum IGF-I and leptin were determined as described in detail previously (27).
Ex vivo experimental metastasis imaging (in vitro vital microscopy)
Cells were fluorescently labeled using 5 mol/L 5-chloromethylfluorescein diacetate (CMFDA; Molecular Probes), according to the manufacturer’s recommendation (28). At 1, 6, 18, and 24 h after the intrasplenic injection of 3 × 105 cells, mice were euthanized by CO2 inhalation. Livers were dissected and then imaged by inverted fluorescent videomicroscopy (Leica DM IRB) at ×100 magnification. Ten random frames were captured and the OpenLab software was used to define and count fluorescent (green) CMFDA tumor events larger than 10 pixels. The means of the total number of events per liver were calculated based on five mice per time point.
Histology and immunohistochemistry
Livers were isolated and fixed overnight in 4% paraformaldehyde in PBS. The tissues were then embedded in paraffin and 5-μm sections were stained by H&E. Immunostaining was performed with an anti-F4/80 antibody (Abcam) at a dilution of 1:150 and detected with the ImmunoCruz Staining System (Santa Cruz Biotechnology).
Statistical analysis
Values are presented as mean ± SEM. Statistical significance was determined by one- or two-way ANOVA or by a t test using the SigmaStat software (SPSS, Inc.). Means ± SEM are indicated. P < 0.05 was considered statistically significant.
Results
Chronic IGF-I deficiency in LID mice abolishes the obesity-associated enhancement of tumor growth
In our previous study, we explored the effect of diet-induced obesity on local subcutaneous growth of colon cancer using the murine colorectal carcinoma MC-38 cells (27). Our results revealed that diet-induced obesity increased the tumor incidence and growth rate in both male and female syngeneic mice, suggesting that obesity contributed to enhanced tumor development. To elucidate the role of serum IGF-I in the obesity-associated enhancement of colon carcinoma growth, we therefore used liver IGF-I–deficient (LID) male mice (26). These mice exhibit 75% to 80% reductions in serum IGF-I levels without changes in extrahepatic igf-1 gene expression, and their overall growth is not significantly different from that of control mice (26). When male LID and control mice were maintained on high-calorie diet for 10 to 14 weeks, they exhibited increased body adiposity and serum leptin levels, compared with age- and sex-matched mice that were fed regular chow (lean mice; Fig. 1A and B) while still maintaining low serum IGF-I levels (i.e., 25% of control levels; Fig. 1C). As expected, local tumor growth in obese mice injected s.c. with MC-38 cells was significantly increased relative to lean mice. However, this increase was abolished in obese LID mice where mean tumor weight at 21 days postinjection was essentially identical to that in lean LID mice (Fig. 1D). This identified IGF-I as a causative factor in enhanced tumorigenicity in these mice. It is also important to note that the decreased tumor growth in LID mice was seen despite increased circulating levels of insulin (secondary to elevations in serum GH; ref. 29).
Figure 1.
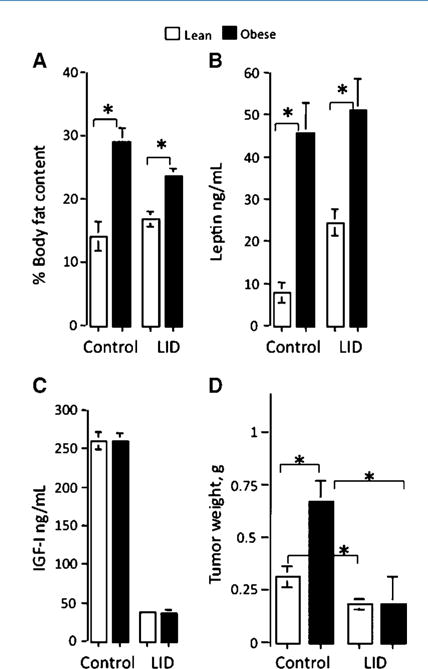
Chronic IGF-I deficiency in LID mice abolishes the obesity-associated enhancement of subcutaneous tumor growth. Mice were maintained on standard or high-calorie diet for 8 to 10 wk. Percent body adiposity (A) was assessed by magnetic resonance imaging. Serum leptin levels (B) and serum IGF-I levels (C) were determined using RIA. Subcutaneous tumor growth in lean and obese LID and control mice was monitored following injection of 5 × 105 MC-38 cells. Columns, mean tumor weights (g; n > 20 mice per group) as measured 21 d after tumor injection; bars, SEM. *, P < 0.05.
Differential effects of acute and chronic serum IGF-I depletion on tumor growth
LID mice are IGF-I deficient throughout their entire life span. Our results indicated that the chronic reduction in IGF-I levels attenuated tumor growth. To distinguish the potential effects that this long-term IGF-I deprivation can have on the host and thereby on tumor growth from the direct paracrine effect that IGF-I has on tumor cell growth in the liver, we developed an inducible, liver IGF-I deficiency model (iLID) whereby the Cre recombinase is expressed under the trypsin-1α promoter and can be induced by a single tamoxifen injection (Fig. 2A). This mouse model allowed us to deplete IGF-I from the liver at different time intervals relative to tumor inoculation, thereby providing the means for a more precise control of IGF-I bioavailability, while also avoiding the long-term effects that serum IGF-I deficiency can have on the host. RNase protection assay (Fig. 2B) was performed on livers dissected from control and iLID mice injected or not with tamoxifen. Dose-response (Fig. 2C) and kinetic (Fig. 2D) analyses revealed that a single i.p. injection of tamoxifen (0.3–0.4 mg) significantly decreased liver IGF-I gene expression 24 hours postinjection (Fig. 2D) and was sufficient to deplete serum IGF-I levels by 60% to 70% for at least 4 weeks postinjection.
Figure 2.
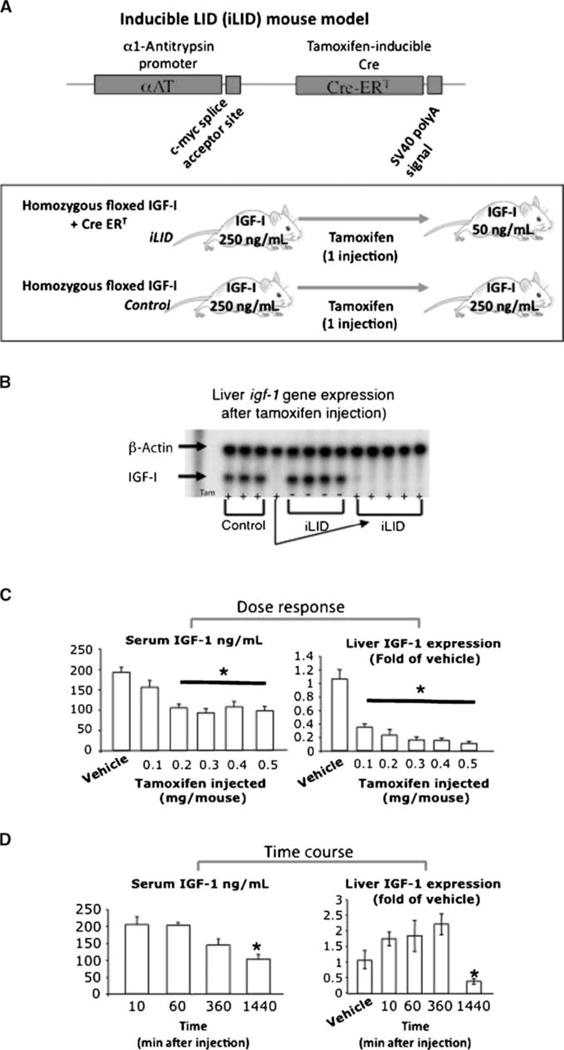
Tamoxifen-inducible liver IGF-I depletion in the iLID mouse model. In the iLID mice (A), the Cre recombinase is expressed under the α1-antitrypsin promoter and can be induced by a single tamoxifen injection (0.4 mg per mouse). RNase protection assay (B) was performed on livers from mice injected with tamoxifen. Dose-response analysis (C) was performed following a single i.p. tamoxifen injection at the indicated dose. Serum IGF-I levels were determined 4 wk later by RIA. Liver IGF-I gene expression was determined 4 wk postinjection by real-time PCR. Time course analysis (D) was performed following a single i.p. tamoxifen injection (0.4 mg). Serum IGF-I levels were determined at 10 min, 1 h, 8 h, and 18 h postinjection by RIA. Liver IGF-I gene expression was determined at the same time points by real-time PCR. Columns, mean (n > 5 mice per group); bars, SEM; *, P < 0.05.
To assess the effects that an acute IGF-I depletion had on the metabolic parameters characteristic of obesity, mice were fed a high-calorie (or regular) diet for 8 to 10 weeks, injected with 0.4 mg tamoxifen, and the serum samples were collected and analyzed 2 weeks later. Similarly to LID mice, we found that serum IGF-I levels in iLID lean or obese animals were decreased relative to tamoxifen-treated controls (Supplementary Fig. S1). Serum leptin and insulin levels increased in obese compared with lean iLID mice. Blood glucose and serum adiponectin levels were comparable with those in lean mice. These data suggested that both LID and iLID mice had increased body adiposity in response to a high-calorie diet and developed a state of insulin resistance.
We next asked whether this acute IGF-I depletion could affect tumor cell growth in the subcutaneous site. IGF-I depletion was induced in iLID mice by a tamoxifen injection 4 days before or after s.c. inoculation of MC-38 cells. We found that an acute IGF-I depletion before or after tumor inoculation failed to significantly reduce the growth of local subcutaneous tumors in lean mice and did not suppress the increase in tumor growth seen in obese mice (Fig. 3). This indicated that the effects of chronic and acute IGF-I depletion on tumor growth were fundamentally different and suggested that the tumor-promoting effects of IGF-I in vivo were not restricted to a direct paracrine, growth-stimulating effect on the tumor cells and entailed other IGF-I–mediated mechanisms, likely involving the host.
Figure 3.
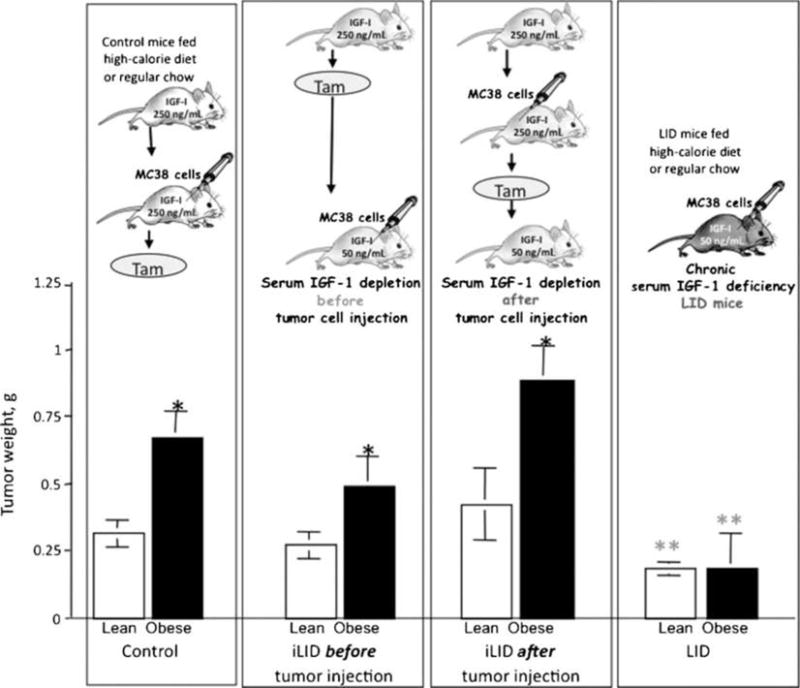
Acute depletion of hepatic IGF-I does not affect subcutaneous tumor growth in obese mice. Mice were maintained on regular chow or high-calorie diet for 8 to 10 wk. A single injection of tamoxifen (0.4 mg) was administered to both control and iLID mice 4 d before or after the subcutaneous inoculation of 5 × 105 MC-38 cells per mouse. Tumors were removed and weighted. Columns, mean weights (g; n > 15 mice per group); bars, SEM. *, P < 0.05 as compared to mice fed regular chow within the same group; **, P < 0.05 as compared to control group fed the same diet.
Obesity increases the growth of hepatic metastases in an IGF-I–dependent manner
The liver is the primary site for colorectal carcinoma metastasis, and the IGF-I axis was identified as a positive regulator of liver metastasis. It was therefore of interest to analyze the effect of obesity on colon carcinoma liver metastasis. Mice were inoculated with 5 × 104 MC-38 cells through the intrasplenic/portal route to generate experimental hepatic metastases. Livers were dissected 2 weeks later, the incidence of hepatic metastases was recorded (Fig. 4A), and the increase in liver weight was measured to determine the metastatic burden (Fig. 4B). We found that the incidence of metastases and the liver weights dramatically increased in obese compared with lean mice (Supplementary Figs. S2 and S3; Fig. 4). The incidence of liver metastases was markedly reduced and the difference between lean and obese mice was abolished in the LID mice, where only 1 of 10 lean and 2 of 10 obese mice had visible metastases 2 weeks after tumor cell injection.
Figure 4.
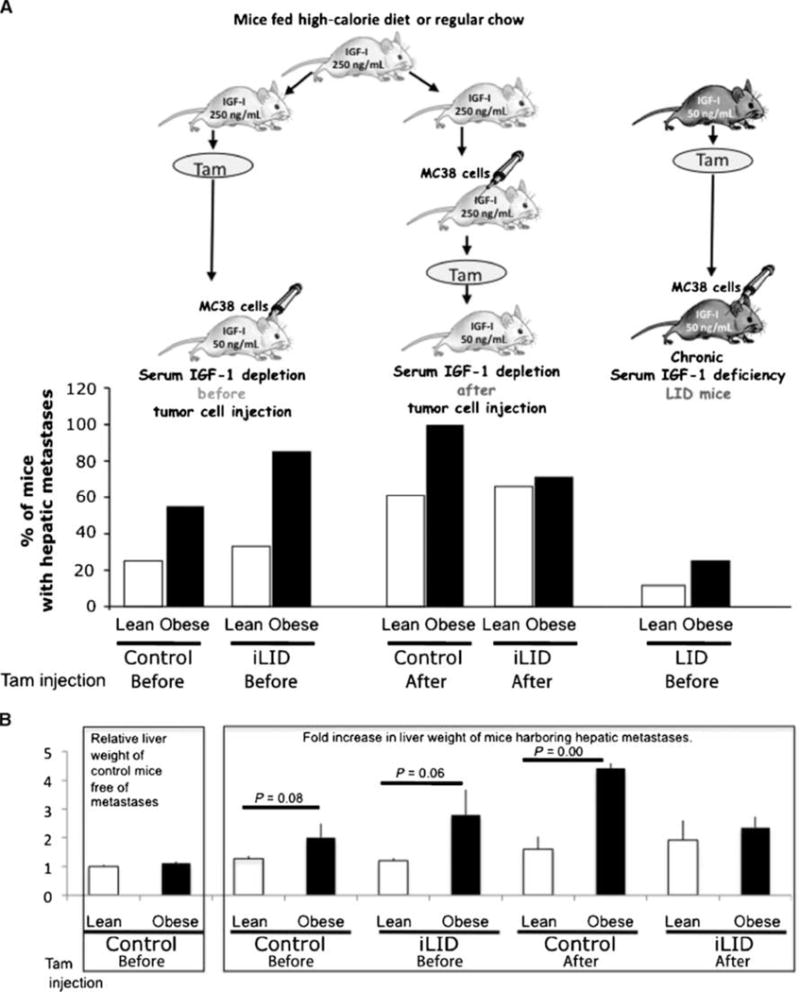
Chronic but not acute IGF-I depletion decreases the growth of hepatic metastases in obese and lean mice. Mice were maintained on regular chow or high-calorie diet for 8 to 10 wk. A single injection of tamoxifen (Tam, 0.4 mg) was administered to mice 4 d before or after the intrasplenic/portal inoculation of 5 × 104 MC-38 cells, mice were splenectomized 1 min later, and livers were harvested 2 wk following tumor cell injection. A, incidence of hepatic metastases is expressed as percent mice with visible metastases. B, metastatic burden was calculated as fold increase in liver weight as compared to livers of tumor-free lean mice. Left, the mean weights (±SEM) of livers obtained from control lean mice noninjected with tumor cells were set as 1 (n = 8). The mean weights (±SEM) of livers obtained from control obese mice noninjected with tumor cells (n = 14), also shown on the left, did not differ significantly from lean controls (P = 0.2), indicating that the baseline mean liver weight in the two groups was comparable. Right, fold increase in liver weight of mice harboring hepatic metastases, injected with tamoxifen before or after tumor cell inoculation.
To assess the effects of acute serum IGF-I depletion on hepatic metastasis, tamoxifen was injected into lean and obese control and iLID animals 4 days before or after intrasplenic/portal tumor cell inoculation, as depicted in Fig. 4A. As expected, the incidence of hepatic metastases increased in obese compared with lean mice (Fig. 4A). This increase was not diminished by a reduction in serum IGF-I levels that was induced 4 days before tumor inoculation (Fig. 4A and B). Tamoxifen-induced reduction in serum IGF-I 4 days after tumor cell inoculation reduced the incidence of metastases in obese iLID mice to the level seen in lean iLID mice. In contrast to the LID mice, the incidence of metastases in the obese iLID mice remained high and this was reflected in increased relative liver weights (Fig. 4B). Histologic examination of the livers 2 weeks after tumor cell inoculation revealed no morphologic differences between tumors that developed in LID, iLID, or control obese mice (Supplementary Figs. S2 and S3).
Sustained tumor cell presence in the livers of obese mice
To begin to understand how obesity affects the hepatic microenvironment and what role, if any, IGF-I played in regulating these effects, we first measured tumor cell arrest following entry into the hepatic microcirculation, using in vitro vital microscopy. MC-38 cells were labeled with a fluorescent dye (CMFDA), injected through the intrasplenic/portal route, and their presence in the liver was monitored at different time intervals thereafter. When analyzed 1 hour after inoculation, no significant difference could be observed between the number of tumor cells per field detectable in lean and obese mice (Fig. 5), suggesting that similar number of cells reached the livers in both groups. However, although the number of tumor cells detectable in the livers declined steadily in lean mice reaching 10% of the initial number at 24 hours postinjection, the number of tumor cells present in livers of obese mice decreased more slowly with up to 70% of the initial number still detectable 24 hours postinjection (Fig. 5). These results suggested that the liver microenvironment in obese mice was more permissive for the sustained presence of metastatic cells during the first 24 hours after inoculation.
Figure 5.
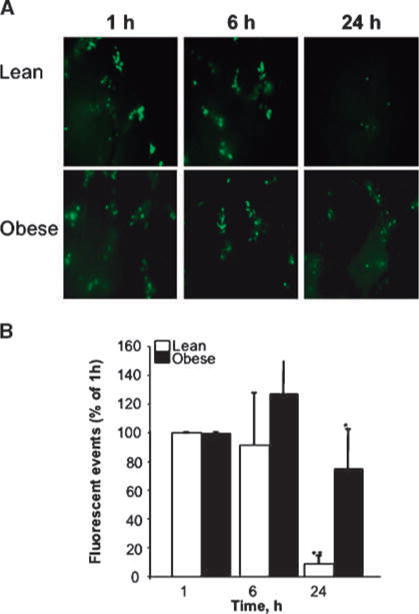
Persistent detection of tumor cells in livers of obese mice. MC-38 cells were labeled with a fluorescent dye (CMFDA), 3 × 105 cells were injected per mouse through the intrasplenic/portal route, and mice were splenectomized 1 min later. Livers were dissected at the indicated time points after tumor cell injection and imaged by inverted fluorescent videomicroscopy (Leica DM IRB) at ×100 magnification (A). Ten random frames were captured and analyzed using OpenLab software to define and count fluorescent (green) CMFDA tumor events larger than 10 pixels. Columns, mean total number of events per liver per mouse (n = 5 mice per group) per time point; bars, SEM. *, P < 0.05 as compared to lean animals at the same time point; **, P < 0.05 as compared to lean animals at 1 h after cell injection.
Chronic, but not acute, IGF-I depletion alters macrophage content and function in the liver
Tumor cell arrest in the liver microcirculation was previously linked to an upregulation of cytokine-induced expression of vascular endothelial cell adhesion molecules such as E-selectin and VCAM-1 (15, 21). A quantitative, real-time PCR analysis revealed that all indicators of an activated inflammatory response, such as the expression of the cytokines tumor necrosis factor α (TNF-α), interleukin (IL)-1β and IL-18, and the levels of MCP-1 (Fig. 6A), were elevated in obese relative to lean mice and that a chronic IGF-I depletion (as present in LID mice) caused a reduction in these indicators in both obese and lean animals. Analysis of the macrophage-specific marker F4/80 revealed a decrease in macrophage numbers and activity in the livers of these mice, and, in parallel, markers of cytokine signaling such as STAT-3 were also reduced (data not shown). In sharp contrast, an acute igf-1 gene deletion, as seen in iLID mice, did not significantly alter F4/80 or inflammatory cytokine mRNA expression in lean mice and actually increased the expression of some cytokines (e.g., TNF-α and IL-1β) in obese mice (Fig. 6B). These data were also confirmed by immunostaining with the macrophage-specific antibody (to F4/80), which showed an increase in macrophage numbers in livers of control and iLID obese mice and a decrease in their numbers in LID mice (Supplementary Fig. S4). Additionally, we observed microvesicular and macrovesicular steatosis (abnormal retention of lipids within hepatocytes) in livers of control and acutely depleted IGF-I (iLID) mice, whereas LID mice developed only a mild steatosis that was mainly microvesicular (Supplementary Fig. S4).
Figure 6.
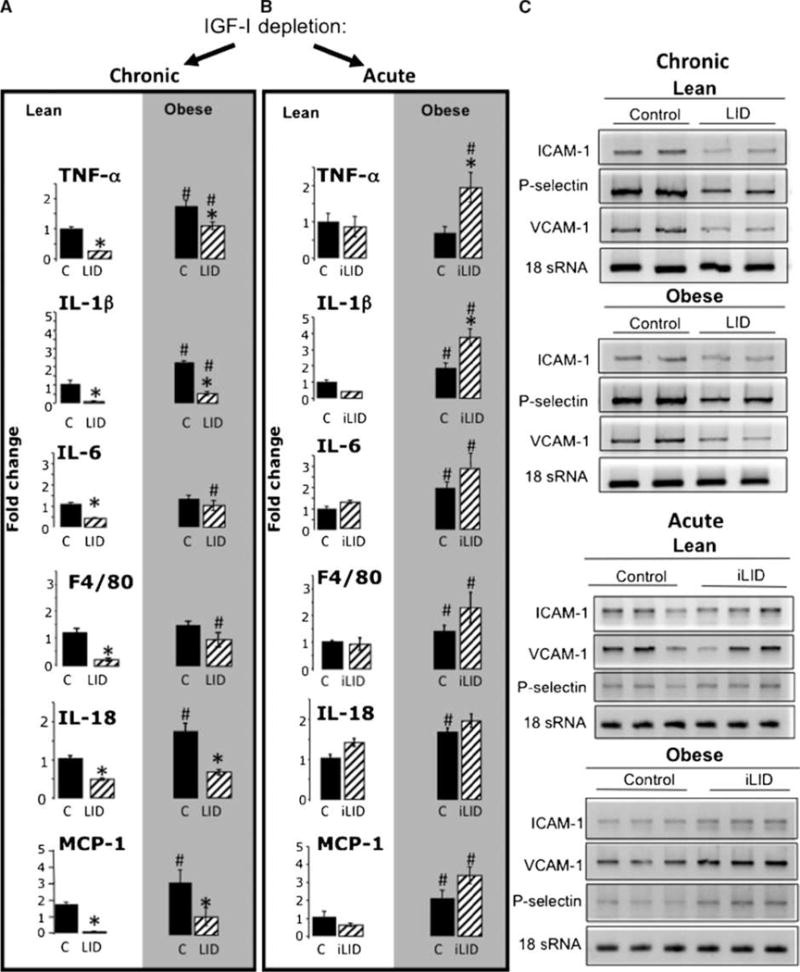
Chronic, but not acute, IGF-I depletion alters macrophage content and function in the liver. Mice were maintained on regular chow (lean) or high-calorie (obese) diets for 8 to 10 wk. RNA was extracted from the livers using Trizol and analyzed by real-time PCR. Columns, mean mRNA expression levels relative to control lean mice that were assigned a value of 1 and are based on ≥6 mice per group; bars, SEM. *, P < 0.05 relative to the respective control group that was fed the same diet. #, P < 0.05 relative to lean animals of the same genetic group. Chronic, but not acute, IGF-I depletion results in reduced expression of vascular cell adhesion molecules in the liver (C). RNA was extracted from livers of lean or obese LID, iLID, and control mice using Trizol, and expression levels of ICAM-1, VCAM-1, and P-selectin were analyzed by reverse transcription-PCR.
IGF-I promotes endothelial cell activation during obesity-induced inflammatory response
Vascular endothelial cell adhesion molecules such as the selectins, ICAM-1, and VCAM-1 are cytokine inducible, and their expression is upregulated in response to an activated inflammatory response (30). We found that P-selectin, VCAM-1, and ICAM-1 mRNA levels decreased in both lean and obese LID compared with control mice. In contrast, the expression levels of these CAMs in lean or obese iLID mice did not differ significantly from tamoxifen-treated controls (Fig. 6C). Taken together, these data indicate that chronic but not short-term (acute) liver IGF-I depletion alters the hepatic microenvironment.
Discussion
The roles of IGF-I– and insulin-induced signaling in the tumor-promoting effects of obesity have received considerable attention. However, the emerging realization that obesity is associated with chronic inflammation has provided new insight into the possible role that obesity and the IGF-I axis in particular may play in cancer progression. Adhesion and growth of tumor cells often occur at sites of inflammation (31), tissue injury (32), and repair (33). Therefore, sites of inflammation are potentially “fertile ground” for the development of metastases. The present study was undertaken to determine whether enhanced tumor growth and progression in an obese state (where systemic inflammation occurs) is also regulated by IGF-I.
Our study shows the following: (a) the liver microenvironment in obese mice is more permissive for metastatic cells than in lean mice; (b) IGF-I plays a critical role in the induction of a chronic inflammatory state in the liver of obese animals and can thereby indirectly affect tumor growth in this site; and (c) there is a fundamental difference between the effects of chronic and acute liver IGF-I depletion on tumor growth and metastasis, implying that IGF-I promotes liver metastasis not only through a direct paracrine effect on tumor cell survival and proliferation but also through indirect effects, likely involving the host microenvironment and proinflammatory responses.
Hepatic tissue is a primary target for metastases by most disseminating malignant cancers (34, 35). Kupffer and perisinusoidal hepatic stellate cells release inflammatory cytokines, eicosanoids, plasminogen activator inhibitor, and reactive oxygen or nitrogen intermediates (36) that can lead to increased cell proliferation, expression of adhesion molecules, extracellular matrix production, cytotoxicity, changes in coagulation, and altered sinusoidal flow. Furthermore, inflammatory cytokines such as TNF-α and IL-1 (constitutively upregulated in a state of obesity) can stimulate the endothelium (37, 38) and lead to an increased expression of cell surface adhesion molecules. In line with previous studies, we show here that in mice with diet-induced obesity, the basal expression levels of inflammatory cytokines and cell adhesion molecules were upregulated. This increased expression was dependent on hepatic IGF-I, as it was abolished in LID mice. This effect was only seen in mice with a chronic reduction in hepatic IGF-I because acute depletion of liver IGF-I, as induced in obese iLID mice, was not associated with reduced expression of inflammatory cytokines and vascular adhesion molecules and in fact caused an increase in the expression of some cytokines, such as TNF-α and IL-1β, by a mechanism yet to be identified. Taken together, our data identify a new indirect role for IGF-I in promoting tumor growth, at least in the liver, namely, the regulation of the host innate immunity and tumor-promoting inflammation. Studies have shown that GH and IGF-I can each stimulate the expansion of primitive multilineage hematopoietic progenitor cells and increase several hematopoietic subpopulations, including lymphoid progenitor cells (39). In LID animals, we observed significant and consistent reductions (between 4 and 52 weeks of age) in the LeuCam surface protein, CD11b (macrophage marker) in the bone marrow (40), and a markedly reduced expression of F4/80 in the liver. F4/80 is a Kupffer cell marker that was accompanied by a reduction in mRNA levels for several macrophage-derived cytokines, including TNF-α, IL-1β, and IL-18. Together, these results suggest that cells committed to the myeloid/monocytic differentiation pathway may be particularly susceptible to a sustained (but not a short term) reduction in hepatic IGF-I levels and that the altered cytokine profile seen in the liver of LID mice was due, at least in part, to a depletion of hepatic Kupffer cells and/or their impaired function.
In contrast to the marked reductions in cytokine expression in LID mice, we found that an acute depletion of IGF-I did not change basal cytokine production in lean mice, whereas in obese animals, the levels of these cytokines increased relative to wild-type mice. This could most likely reflect different biological effects of sustained (chronic) or acute IGF-I deletion on the number and function of hepatic Kupffer cells. Although the number of these cells in livers of LID mice was markedly reduced, the acute depletion of hepatic IGF-I did not alter the macrophage content in the liver and, as a consequence, basal cytokine production remained relatively normal. In obese mice, cytokine expression was actually augmented in response to acute IGF-I depletion, suggesting that the combined effect of “obesity” and an acute IGF-I depletion may cause a rapid activation of the hepatic macrophages resulting in increased cytokine production. Consistent with these findings, a phenotypic switch from the low cytokine–producing M2 to the “classically activated” M1 macrophages was observed in adipose tissue–associated macrophages of obese mice (41), and Kupffer cell dysfunction was reported in obese animals such as the leptin-deficient ob/ob mice (42). It is possible that in the present study, phenotypic/functional changes in the Kupffer cells in obese mice heightened their sensitivity to an acute drop in hepatic IGF-I levels and, as a consequence, cytokine expression was further activated.
In addition to the differences in the effects of chronic and acute IGF-I depletion on the macrophage population of the liver, differences in the metabolic effects caused by IGF-I depletion may have also contributed to the differential effects on hepatic cytokine production in these mice. For example, chronic depletion of IGF-I (as seen in the LID model) can lead to compensatory increases in serum GH levels, which persist even to an older age (up to 24 weeks).8 GH is one of the regulators of body adiposity and was shown to induce lipolysis (43). It is conceivable that the elevation in GH levels in LID mice improves their lipolytic response to the high-calorie diet in the liver or in the white adipose tissue, thereby leading to alterations in the liver microenvironment. On the other hand, acute depletion of IGF-I (as seen in the iLID model), although leading to a slight increase in GH secretion (∼4–7 days after induction), is unlikely to cause rapid and significant alterations in the liver microenvironment. Our finding that the incidence of hepatic metastases in obese iLID mice was not reduced and even somewhat increased relative to controls is consistent with this lack of effect on hepatic cytokine production and with the increase observed in TNF-α and IL-1β production. This finding also lends further support to the critical role that inflammatory cytokines play in the regulation of hepatic metastasis.
Taken together, our results identify hepatic IGF-I as a critical regulator of basal inflammatory responses in the liver of normal and obese mice. Furthermore, they implicate IGF-I in the obesity-induced increase in tumor cell growth and hepatic metastasis and show that its effect is mediated through a complex mechanism involving both direct effects on the tumor cells and the conditioning of the hepatic microenvironment.
Acknowledgments
We thank Drs. Derek LeRoith and Scott Friedman (Mount Sinai School of Medicine) for their help and advice on this project.
Grant Support
Canadian Institute for Health Research grant MOP-81201.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Unpublished observation.
References
- 1.Stewart TJ, Abrams SI. How tumours escape mass destruction. Oncogene. 2008;27:5894–903. doi: 10.1038/onc.2008.268. [DOI] [PubMed] [Google Scholar]
- 2.Rudmik LR, Magliocco AM. Molecular mechanisms of hepatic metastasis in colorectal cancer. J Surg Oncol. 2005;92:347–59. doi: 10.1002/jso.20393. [DOI] [PubMed] [Google Scholar]
- 3.Wang HH, Qiu H, Qi K, Orr FW. Current views concerning the influences of murine hepatic endothelial adhesive and cytotoxic properties on interactions between metastatic tumor cells and the liver. Comp Hepatol. 2005;4:8. doi: 10.1186/1476-5926-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 5.Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci. 2008;99:1501–6. doi: 10.1111/j.1349-7006.2008.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farinati F, Cardin R, Cassaro M, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: a morphological, biological and molecular pathway. Eur J Cancer Prev. 2008;17:195–200. doi: 10.1097/CEJ.0b013e3282f0bff5. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez-Irigoyen J, Santamaria M, Sanchez-Quiles V, et al. Redox regulation of methylthioadenosine phosphorylase in liver cells: molecular mechanism and functional implications. Biochem J. 2008;411:457–65. doi: 10.1042/BJ20071569. [DOI] [PubMed] [Google Scholar]
- 8.Lukasiak S, Schiller C, Oehlschlaeger P, et al. Proinflammatory cytokines cause FAT10 upregulation in cancers of liver and colon. Oncogene. 2008;27:6068–74. doi: 10.1038/onc.2008.201. [DOI] [PubMed] [Google Scholar]
- 9.Muriel P. NF-κB in liver diseases: a target for drug therapy. J Appl Toxicol. 2008;27:203–10. doi: 10.1002/jat.1393. [DOI] [PubMed] [Google Scholar]
- 10.Wu XZ. New strategy of antiangiogenic therapy for hepatocellular carcinoma. Neoplasma. 2008;55:472–81. [PubMed] [Google Scholar]
- 11.Wang D, DuBois RN. Pro-inflammatory prostaglandins and progression of colorectal cancer. Cancer Lett. 2008;267:197–203. doi: 10.1016/j.canlet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi K, Matsumoto S, Morishima T, Kawabe T, Okamoto T. Cimetidine inhibits cancer cell adhesion to endothelial cells and prevents metastasis by blocking E-selectin expression. Cancer Res. 2000;60:3978–84. [PubMed] [Google Scholar]
- 13.Boring CC, Squires TS, Tong T, Montgomery S. Cancer statistics, 1994. CA Cancer J Clin. 1994;44:7–26. doi: 10.3322/canjclin.44.1.7. [DOI] [PubMed] [Google Scholar]
- 14.Fahy B, Bold RJ. Epidemiology and molecular genetics of colorectal cancer. Surg Oncol. 1998;7:115–23. doi: 10.1016/s0960-7404(99)00021-3. [DOI] [PubMed] [Google Scholar]
- 15.Auguste P, Fallavollita L, Wang N, Burnier J, Bikfalvi A, Brodt P. The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. Am J Pathol. 2007;170:1781–92. doi: 10.2353/ajpath.2007.060886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang N, Thuraisingam T, Fallavollita L, Ding A, Radzioch D, Brodt P. The secretory leukocyte protease inhibitor is a type 1 insulin-like growth factor receptor-regulated protein that protects against liver metastasis by attenuating the host proinflammatory response. Cancer Res. 2006;66:3062–70. doi: 10.1158/0008-5472.CAN-05-2638. [DOI] [PubMed] [Google Scholar]
- 17.Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eriksson JW. Metabolic stress in insulin’s target cells leads to ROS accumulation—a hypothetical common pathway causing insulin resistance. FEBS Lett. 2007;581:3734–42. doi: 10.1016/j.febslet.2007.06.044. [DOI] [PubMed] [Google Scholar]
- 19.Nave H, Mueller G, Siegmund B, et al. Resistance of Janus kinase-2 dependent leptin signaling in natural killer (NK) cells: a novel mechanism of NK cell dysfunction in diet-induced obesity. Endocrinology. 2008;149:3370–8. doi: 10.1210/en.2007-1516. [DOI] [PubMed] [Google Scholar]
- 20.Orr FW, Wang HH, Lafrenie RM, Scherbarth S, Nance DM. Interactions between cancer cells and the endothelium in metastasis. J Pathol. 2000;190:310–29. doi: 10.1002/(SICI)1096-9896(200002)190:3<310::AID-PATH525>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 21.Gout S, Tremblay PL, Huot J. Selectins and selectin ligands in extravasation of cancer cells and organ selectivity of metastasis. Clin Exp Metastasis. 2008;25:335–44. doi: 10.1007/s10585-007-9096-4. [DOI] [PubMed] [Google Scholar]
- 22.LeRoith D. Insulin-like growth factor I receptor signaling-overlapping or redundant pathways? Endocrinology. 2000;141:1287–8. doi: 10.1210/endo.141.4.7475. [DOI] [PubMed] [Google Scholar]
- 23.Knittel T, Dinter C, Kobold D, et al. Expression and regulation of cell adhesion molecules by hepatic stellate cells (HSC) of rat liver: involvement of HSC in recruitment of inflammatory cells during hepatic tissue repair. Am J Pathol. 1999;154:153–67. doi: 10.1016/s0002-9440(10)65262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev. 2007;28:20–47. doi: 10.1210/er.2006-0001. [DOI] [PubMed] [Google Scholar]
- 25.Yakar S, Leroith D, Brodt P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: lessons from animal models. Cytokine Growth Factor Rev. 2005;16:407–20. doi: 10.1016/j.cytogfr.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 26.Yakar S, Liu JL, Stannard B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324–9. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yakar S, Nunez NP, Pennisi P, et al. Increased tumor growth in mice with diet-induced obesity: impact of ovarian hormones. Endocrinology. 2006;147:5826–34. doi: 10.1210/en.2006-0311. [DOI] [PubMed] [Google Scholar]
- 28.Khanna C, Wan X, Bose S, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nat Med. 2004;10:182–6. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- 29.Yakar S, Setser J, Zhao H, et al. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest. 2004;113:96–105. doi: 10.1172/JCI200417763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi H, Boelte KC, Lin PC. Endothelial cell adhesion molecules and cancer progression. Curr Med Chem. 2007;14:377–86. doi: 10.2174/092986707779941032. [DOI] [PubMed] [Google Scholar]
- 31.Orr FW, Adamson IY, Young L. Pulmonary inflammation generates chemotactic activity for tumor cells and promotes lung metastasis. Am Rev Respir Dis. 1985;131:607–11. doi: 10.1164/arrd.1985.131.4.607. [DOI] [PubMed] [Google Scholar]
- 32.Lafrenie R, Shaughnessy SG, Orr FW. Cancer cell interactions with injured or activated endothelium. Cancer Metastasis Rev. 1992;11:377–88. doi: 10.1007/BF01307188. [DOI] [PubMed] [Google Scholar]
- 33.Murphy P, Alexander P, Senior PV, Fleming J, Kirkham N, Taylor I. Mechanisms of organ selective tumour growth by bloodborne cancer cells. Br J Cancer. 1988;57:19–31. doi: 10.1038/bjc.1988.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Antin JH, Weinstein HJ, Guinan EC, et al. Recombinant human interleukin-1 receptor antagonist in the treatment of steroid-resistant graft-versus-host disease. Blood. 1994;84:1342–8. [PubMed] [Google Scholar]
- 35.Fisher CJ, Jr, Slotman GJ, Opal SM, et al. Initial evaluation of human recombinant interleukin-1 receptor antagonist in the treatment of sepsis syndrome: a randomized, open-label, placebo-controlled multicenter trial. Crit Care Med. 1994;22:12–21. doi: 10.1097/00003246-199401000-00008. [DOI] [PubMed] [Google Scholar]
- 36.McCloskey TW, Todaro JA, Laskin DL. Lipopolysaccharide treatment of rats alters antigen expression and oxidative metabolism in hepatic macrophages and endothelial cells. Hepatology. 1992;16:191–203. doi: 10.1002/hep.1840160130. [DOI] [PubMed] [Google Scholar]
- 37.Lauri D, Bertomeu MC, Orr FW, Bastida E, Sauder D, Buchanan MR. Interleukin-1 increases tumor cell adhesion to endothelial cells through an RGD dependent mechanism: in vitro and in vivo studies. Clin Exp Metastasis. 1990;8:27–32. doi: 10.1007/BF00155590. [DOI] [PubMed] [Google Scholar]
- 38.Rice GE, Gimbrone MA, Bevilacqua MP. Tumor cell-endothelial interactions. Increased adhesion of human melanoma cells to activated vascular endothelium. Am J Pathol. 1988;133:204–10. [PMC free article] [PubMed] [Google Scholar]
- 39.Hanley MB, Napolitano LA, McCune JM. Growth hormone-induced stimulation of multilineage human hematopoiesis. Stem Cells. 2005;23:1170–9. doi: 10.1634/stemcells.2004-0322. [DOI] [PubMed] [Google Scholar]
- 40.Yakar S, Canalis E, Sun H, et al. Serum IGF-1 determines skeletal strength by regulating sub-periosteal expansion and trait interactions. J Bone Miner Res. 2009;24:1481–92. doi: 10.1359/JBMR.090226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diehl AM. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1–5. doi: 10.1152/ajpgi.00384.2001. [DOI] [PubMed] [Google Scholar]
- 43.Yuen KC, Dunger DB. Therapeutic aspects of growth hormone and insulin-like growth factor-I treatment on visceral fat and insulin sensitivity in adults. Diabetes Obes Metab. 2007;9:11–22. doi: 10.1111/j.1463-1326.2006.00591.x. [DOI] [PubMed] [Google Scholar]