

Abstract
The Mycobacterium tuberculosis early secreted Ag of 6 kDa (ESAT-6) is a potent Ag for human T cells and is a putative vaccine candidate. However, ESAT-6 also contributes to virulence in animal models, mediates cellular cytolysis, and inhibits IL-12 production by mononuclear phagocytes. We evaluated the effects of ESAT-6 and its molecular chaperone, culture filtrate protein of 10 kDa (CFP10), on the capacity of human T cells to produce IFN-γ and proliferate in response to TCR activation. Recombinant ESAT-6, but not CFP10, markedly inhibited IFN-γ production by T cells stimulated with M. tuberculosis or with the combination of anti-CD3 and anti-CD28, in a dose-dependent manner. ESAT-6 also inhibited T cell production of IL-17 and TNF-α but not IL-2. Preincubation of ESAT-6 with CFP10 under conditions that favor dimer formation did not affect inhibition of IFN-γ. ESAT-6 decreased IFN-γ transcription and reduced expression of the transcription factors, ATF-2 and c-Jun, which normally bind to the IFN-γ proximal promoter and stimulate mRNA expression. ESAT-6 inhibited T cell IFN-γ secretion through mechanisms that did not involve cellular cytotoxicity or apoptosis. ESAT-6, but not CFP10, bound to T cells and inhibited expression of early activation markers without reducing activation of ZAP70. We conclude that ESAT-6 directly inhibits human T cell responses to mycobacterial Ags by affecting TCR signaling pathways downstream of ZAP70.
Despite our attempts to understand and control disease due to Mycobacterium tuberculosis for more than a century, this pathogen remains a pressing public health threat, infecting more than one-third of the world’s population, and causing almost two million deaths yearly worldwide (1). The burgeoning epidemic of HIV infection in developing nations and the emergence of multidrug-resistant and extensively drug-resistant strains of M. tuberculosis mandates the development of more effective preventive and therapeutic strategies, such as improved vaccines. Vaccine development hinges on understanding the molecular mechanisms of the host-pathogen interaction, particularly those that affect T cell responses, because this is believed to be pivotal for controlling M. tuberculosis infection.
Early secreted antigenic target of 6 kDa (ESAT-6)3 was originally identified as a potent T cell Ag in the short-term culture filtrate of M. tuberculosis (2, 3). ESAT-6 subunit vaccines induce protection against challenge with M. tuberculosis in mice (4, 5), and a vaccine construct that includes ESAT-6 and Ag85 reduces the bacillary burden after challenge with M. tuberculosis in guinea pigs and nonhuman primates (6, 7).
Although the studies above suggest that ESAT-6 is a potential vaccine candidate, substantial evidence indicates that it is also a virulence factor. The gene encoding ESAT-6, Rv3875 (8), is in the region of difference (RD)1, which is present in all pathogenic mycobacteria, including M. tuberculosis and M. bovis, but not in attenuated M. bovis bacillus Calmette-Guérin (BCG) (9). The gene encoding the M. tuberculosis culture filtrate protein of 10 kDa (CFP10) is cotranscribed with esat-6. ESAT-6 and CFP10 are secreted as a 1:1 dimer by a specialized ESAT-6 secretion system, ESX-1 (10–12). ESAT-6 is produced in the lungs of mice infected with RD1-complemented M. bovis BCG (13), and gene deletion studies of M. tuberculosis, combined with reintroduction of RD1 into M. bovis BCG, demonstrated that secretion of ESAT-6 and CFP10 is required for virulence and pathogenicity of M. tuberculosis, both for growth in macrophages and in mice (13–15). Furthermore, orthologs of ESAT-6 and CFP10 are present in M. leprae, suggesting that they are essential for pathogenic mycobacteria (16).
The mechanisms by which ESAT-6 and CFP10 contribute to virulence are not fully defined, but they are believed to include cytolysis of alveolar epithelial cells and macrophages (14, 17), favoring intercellular spread of M. tuberculosis (17, 18). ESAT-6 dissociates from CFP10 under acidic conditions in the phagosome and can destabilize liposomes, perhaps allowing M. tuberculosis and its products to eventually escape the phagosome (19).
It has been shown that M. tuberculosis inhibits IL-12 p40 gene transcription in infected macrophages (20), and this may affect the T cell responses to M. tuberculosis infection. Recent studies with mutant M. tuberculosis strains with defects in proteins associated with secretion of ESAT-6 and CFP10 suggest that ESAT-6 inhibits macrophage IL-12 p40 expression and secretion (21). Further studies demonstrated that recombinant ESAT-6 inhibits secretion of IL-12 induced by multiple TLR agonists via binding to TLR2 and interfering with the assembly of TLR signaling molecules (22). Therefore, ESAT-6 may indirectly inhibit the capacity of T cell IFN-γ production through reduced IL-12 secretion by APC. IFN-γ is critical for immunity to tuberculosis. Animals with deleted IFN-γ genes have marked susceptibility to tuberculosis (23, 24), and persons with genetic defects in the IFN-γ receptor or in the IL-12 pathway that leads to IFN-γ production develop severe disease due to M. tuberculosis and other less virulent mycobacteria (25). Production of M. tuberculosis-induced IFN-γ by PBMC from tuberculosis patients is decreased, compared with findings in healthy tuberculin reactors (26, 27), and patients with severe disease have the most marked defects in IFN-γ production (28).
Studies on the immunologic effects of ESAT-6 have focused on mice and mononuclear phagocytes, but little is known about the effects of this protein on human T cells. In this report, we demonstrate that ESAT-6 can directly inhibit human T cell responses, reducing IFN-γ secretion, proliferation and expression of CD25 and CD69 without affecting TCR-induced ZAP70 phosphorylation.
Materials and Methods
Human subjects
Blood was obtained from 20 healthy tuberculin reactors and nine tuberculin-negative individuals. All participants provided informed consent. This investigation was approved by the Institutional Review Board of the University of Texas Health Science Center (Tyler, TX).
M. tuberculosis and recombinant mycobacterial proteins
Heat-killed M. tuberculosis Erdman (provided by Dr. Patrick Brennan, Colorado State University, Fort Collins, CO) was used as Ag to stimulate PBMC from healthy tuberculin reactors. Recombinant plasmids containing Rv3874 and Rv3875, encoding CFP10 and ESAT-6, respectively, were obtained through the TB Vaccine Testing and Research Materials Contract (Colorado State University). Escherichia coli BL21 (DE3) cells were transformed with these plasmids, and induced by the addition of isopropyl β-D-1-thiogalactopyranoside to express histidine-tagged CFP10 and ESAT-6. The recombinant proteins were purified by using a His-Bind purification kit (EMD Biosciences), and endotoxin was removed following standard protocols provided by the TB Vaccine Testing and Research Materials Contract. Endotoxin levels in the recombinant proteins were measured by the QCL-1000 Limulus amebocyte assay (Cambrex). Purity of the recombinant proteins were analyzed by performing SDS-PAGE, followed by Coomassie staining and Western blotting using anti-ESAT-6 mAb, HYB 76-8 (provided by Dr. Peter Andersen, Statens Seruminstitut, Copenhagen, Denmark), and anti-ESAT-6 or anti-CFP10 polyclonal IgG (TB Vaccine Testing and Research Materials Contract). Purified proteins were resuspended in HBSS (1 mg/ml), aliquoted, and stored at −70°C before use.
Cell preparation, culture, and detection of cytokines in culture supernatants
PBMC were isolated by differential centrifugation over Ficoll-Paque (GE Healthcare Life Science), and CD3+ T cells were purified from PBMC by immunomagnetic negative selection, using a Human Pan T cell isolation kit (Miltenyi Biotec). The purity of CD3+ cells was >98%, as measured by staining with anti-CD3 and flow cytometry with a FACSCalibur (BD Biosciences). PBMC from healthy tuberculin reactors or CD3+ cells from healthy tuberculin skin test-negative subjects were suspended at 2 × 106/ml in RPMI 1640 (Invitrogen), supplemented with 10% heat-inactivated pooled human serum (Atlanta Biologicals), 100 units or 100 μg/ml penicillin and streptomycin, 1 mM sodium pyruvate and 0.1 mM MEM nonessential amino acids (all from Invitrogen). The cells were then incubated with recombinant CFP10 or ESAT-6 at final concentrations of 0.8 μM, 1.6 μM, or 3.3 μM at 37°C, 5% CO2. Some cells were also treated with a 1:1 molar ratio of CFP10 and ESAT-6 that had been previously mixed and incubated under conditions that favor dimerization of these proteins (29). After 1 h of incubation, PBMC were stimulated with heat-killed M. tuberculosis Erdman (2 μg/ml), and CD3+ cells were stimulated by culturing in a 96-well plate precoated with 5 μg/ml anti-CD3 (OKT3, eBioscience) plus 1 μg/ml anti-CD28 (CD28.2, BD Biosciences) in PBS at 37°C for 2 h. After 48 h further incubation, cell-free supernatants were collected and cytokine concentrations were measured by quantitative sandwich ELISA for IFN-γ, IL-2, and TNF-α with a pair of capture and detection Abs (all from BD Biosciences), and an ELISA kit for IL-17 (R&D Systems).
Flow cytometry
To identify cell populations in human PBMC that bind to ESAT-6 and CFP10, we first biotinylated ESAT-6 and CFP10 by incubation with a biotinylation reagent, sulfo-NHS-LC-biotin (Thermo Scientific), at a molar ratio of 1:20, according to the manufacturer’s protocol. Biotinylation was confirmed by a dot blot assay that detected biotinylated proteins spotted on a nitrocellulose membrane, through binding to streptavidin-labeled HRP, followed by incubation of the blot with a chromogenic substrate.
PBMC (5 × 105) were incubated with biotinylated CFP10 or ESAT-6 at a final concentration of 3.3 μM and PE-conjugated mAbs to CD3, CD4, CD8, CD14, or CD19 (all from eBiosciences) for 30 min at 4°C in FACS staining buffer (PBS with 2% FBS). After washing, the cells were incubated with FITC-conjugated streptavidin for 30 min at 4°C, and analyzed with a FACSCalibur, using Flowjo software (Tree Star).
Expression of CD69 and CD25 was measured by incubating cells with PE-conjugated anti-CD3 (eBiosciences), in combination with FITC-conjugated anti-CD25 (M-A251) or anti-CD69 (FN50) mAbs (both from BD Biosciences) on ice for 30 min. Flow cytometry analysis was performed with a FACSCalibur after gating on CD3+ cells, using Flowjo software.
ELISPOT
The frequency of IFN-γ producing CD3+ cells was determined by ELISPOT, as described previously (30). Briefly, CD3+ cells were purified from M. tuberculosis-stimulated PBMC from healthy tuberculin reactors, plated in 96-well nitrocellulose-backed plates coated with anti-human IFN-γ capture Ab (1-D1K). After 16 h incubation, the spots were developed using anti-human IFN-γ detection Ab (7-B6-1), according to the manufacturer’s instructions (Mabtech), The spots in air-dried plates were counted under a stereomicroscope.
Cell viability and apoptosis assays
CD3+ cells were stained with trypan blue (Invitrogen), and live cells were counted in a hemocytometer. The MTT cleavage assay (USB Corporation) was performed, according to the manufacturer’s instructions. Apoptosis of CD3+ cells was measured by flow cytometry, using an Annexin V-FITC Apoptosis Detection Kit (BD Biosciences).
Cell proliferation assay
Purified CD3+ cells were labeled with CFSE (Invitrogen) as described (31) and incubated in the absence or presence of recombinant protein, in a 96-well plate precoated with anti-CD3 plus anti-CD28 mAbs. After 72 h, the cells were collected, washed and stained with PE-conjugated anti-CD8 mAb, and proliferation was analyzed by flow cytometry.
Real-time PCR
Total RNA was extracted from 2 × 106 PBMC or CD3+ cells with TRIzol reagent (Invitrogen), following the manufacturer’s instructions. cDNA was synthesized in a 20 μl mixture with 200 ng total RNA, using random primers and Moloney Murine Leukemia Virus reverse transcriptase (both from Invitrogen). IFN-γ cDNA was quantified by real-time PCR on an ABI 7300 Real-Time PCR System (Applied Biosystems) in a 20 μl reaction containing 2 μl cDNA, 1 μl TaqMan Gene Expression Assay for human IFN-γ, and 10 μl TaqMan Universal PCR Master Mix (Applied Biosystems). Amplification was conducted at 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 s, and 60°C for 1 min. For endogenous control, 18S rRNA was used and all reactions were run in duplicate. Relative quantity of IFN-γ mRNA was calculated using the ΔΔCt method, as described previously (31).
Western blot
Total cell protein extracts were prepared by resuspending PBMC or purified CD3+ cells in 1% Nonidet P-40 cell lysis buffer, supplemented with proteinase inhibitors. After three cycles of freeze-thawing in liquid nitrogen, cell suspensions were incubated at 4°C for 30 min with rotation, followed by centrifugation at 18,000 × g at 4°C for 15 min. The supernatants were collected as total cell protein extracts, the amount of protein was quantified by a bicinchoninic acid assay (Thermo Scientific), and the protein extracts were aliquoted, snap frozen in liquid nitrogen and kept at −70°C before use.
Reducing SDS-PAGE (10%) was performed, using 30 μg of total protein extract in each sample. The gel was then electroblotted in Tris glycine buffer containing 20% methanol onto a nitrocellulose membrane (Bio-Rad). After blocking with 5% fat-free milk in TBS [10 mM Tris (pH 8.0), 30 mM NaCl] for 1 h at room temperature, the membrane was incubated with a primary Ab in TBS with 5% BSA overnight at 4°C. Thereafter, the membrane was washed four times with TBST, and incubated with secondary Ab in blocking buffer for 35 min at room temperature. After washing four times in TBST and once in TBS, the membrane was drained briefly and Ab binding was detected by ECL (GE Healthcare Life Science). Each membrane was blotted, stripped, and reblotted with a series of primary Abs (all from Santa Cruz Biotechnology) to ATF2 (c-19), c-Jun (H79), CREB-1 (CX-12), and GAPDH (FL-335).
For detection of phosphorylated ZAP70, CD3+ cells were incubated with or without 3.3 μM CFP10 or ESAT-6 at 37°C for one hour. The cells were then incubated with 10 μg/ml anti-CD3 on ice. Fifteen min later, the cells were washed and incubated with 20 μg/ml goat anti-mouse IgG (Sigma) on ice for another 15 min to cross-link cell bound anti-CD3. The cells were incubated at 37°C for a further 5 min and total cell protein extracts were prepared for evaluation of phosphorylated ZAP70 and total ZAP70 by Western blotting, using mouse anti-human ZAP70pY319/SykpY352 (BD Biosciences) and rabbit anti-human ZAP70 (BioLegend). Secondary Abs were goat anti-mouse (Santa Cruz Biotechnology) and goat-anti-rabbit IgG (Bio-Rad), conjugated with HRP.
Depletion of ESAT-6
ESAT-6 was removed from the recombinant protein preparations by incubating 500 μg of protein with 250 μl of activated nickel resin in a 1.5 ml Eppendorf tube at room temperature. For sham depletion, an equal amount of ESAT-6 was incubated with unactivated nickel resin under the same conditions. After 30 min incubation, samples were centrifuged to pellet the resin, and the resin-free supernatants were collected as the ESAT-6-de- pleted samples. The protein content of the samples was measured by the bicinchoninic acid assay. Samples were also subjected to SDS-PAGE, followed by Coomassie staining and Western blotting with the anti-ESAT-6 mAb, HYB 76-8.
Statistical analysis
Student’s t test was used to evaluate differences between different treatment groups. Values of p < 0.05 was considered to be statistically significant.
Results
Preparation of recombinant ESAT-6 and CFP10
Recombinant ESAT-6 and CFP10 were prepared as outlined in Materials and Methods and subjected to SDS-PAGE. The recombinant proteins formed single bands of the same m.w. as the standards prepared by Colorado State University (Fig. 1A). By the QCL-1000 Limulus amebocyte assay, the LPS content in ESAT-6 and CFP10 preparations was 39 pg/mg (.0039%) and 39–78 pg/mg (.0039–.0078%), respectively. The m.w. of ESAT-6 is 9.9 kDa, and with the extra amino acids and histidine tag introduced by cloning, it is 11.4 kDa. Despite not having any cysteine residues, ESAT-6 spontaneously forms dimers, which are SDS-stable, resulting in the appearance of additional bands at 24 and 35 kDa on both reduced and nonreduced SDS gels (Karen Dobos, unpublished data). CFP10 also runs slightly slower on the gel than expected for its m.w., consistent with previous publications (32, 33). Western blotting analysis with mAb to ESAT-6 (HYB 76-8) or polyclonal Ab to CFP10 visualized the respective protein bands at expected molecular weights (Fig. 1B). There were some additional minor protein bands with higher m.w. that account for 13 and 18% of the total bands in ESAT-6 and CFP10 lanes, respectively, according to the densitometry analysis. These higher m.w. bands likely represent dimers and multimers, as these proteins are known to form aggregates in solution. Together, these results confirmed the identity of the recombinant ESAT-6 and CFP10 that we prepared.
FIGURE 1.
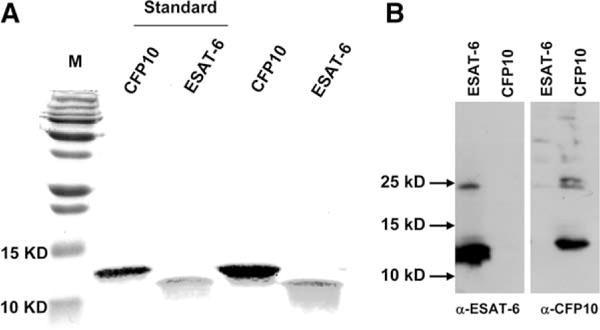
Preparation of recombinant ESAT-6 and CFP10. Plasmids containing inserts encoding histidine-tagged ESAT-6 or CFP10 were expressed in E. coli, and proteins were purified, as described in Materials and Methods. Purity of the recombinant proteins was analyzed by 15% SDS- PAGE, and visualized by staining with Coomassie Blue, followed by destaining the gel (A), or by Western blot (B), after electroblotting to a nitrocellulose membrane and blotting with Abs as indicated (bottom). M: m.w. markers. Standards are recombinant proteins from the TB Vaccine Testing and Research Materials Contract, the two far right lanes show the recombinant proteins we prepared. A representative result from three independent preparations is shown.
ESAT-6, but not CFP10, inhibits M. tuberculosis-induced IFN-γ secretion by PBMC from healthy tuberculin reactors
To study the effect of ESAT-6 and CFP10 on M. tuberculosis-induced IFN-γ secretion, we pretreated PBMC from nine healthy tuberculin reactors with increasing concentrations of recombinant ESAT-6 or CFP10 from three different preparations, then added heat-killed M. tuberculosis Erdman. Recombinant ESAT-6 inhibited IFN-γ secretion by PBMC by up to 90% in a dose-dependent manner (Fig. 2A). In contrast, recombinant CFP10, the molecular partner of ESAT-6, did not affect IFN-γ production. When equimolar concentrations (3.3 μM) of CFP10 and ESAT-6 were added together to generate ESAT-6/CFP10 heterodimers, the effect was similar to that of ESAT-6 alone (Fig. 2A). Addition of polymyxin B (10 μg/ml) did not affect the inhibition of IFN-γ by ESAT-6 (data not shown), indicating that inhibition was not due to LPS in the recombinant proteins.
FIGURE 2.
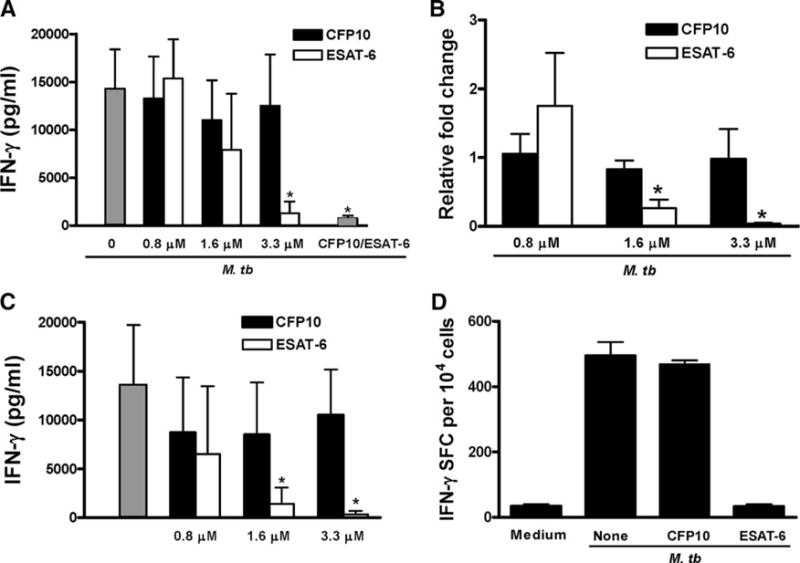
ESAT-6 inhibits M. tuberculosis-induced IFN-γ secretion by PBMC. A, PBMC from nine healthy tuberculin rectors were incubated with different concentrations of ESAT-6, CFP10, or ESAT-6/CFP10 heterodimers (ESAT-6 and CFp10 at 3.3 μM, far right gray bar) for 1 h, before adding 2 μg/ml heat-killed M. tuberculosis. Some wells were cultured with medium alone (far left gray bar). After 48 h of incubation, supernatants were collected and IFN-γ levels were measured by ELISA. For 0.8 μM, ESAT-6 = 5 and CFP10 = 8.3 μg/ml. IFN-γ levels with medium alone were <50 pg/ml. Means and SD are shown. *, p < 0.05, compared with the cells treated with M. tuberculosis and 3.3 μM CFP10. B, PBMC from three healthy tuberculin reactors were cultured, as in A. After 48 h, total RNA from cells was isolated and reverse transcribed to cDNA, and IFN-γ mRNA expression was measured by real-time PCR, after normalization for 18S rRNA content. The fold-change of IFN-γ mRNA in M. tuberculosis-stimulated cells, compared with cells cultured in medium alone, was given an arbitrary value of 1.0. Due to significant person-to-person variability in IFN-γ mRNA level, the expression of IFN-γ mRNA in cells with ESAT-6 or CFP10 was expressed as relative fold change, compared with that expressed in the cells without CFP10 or ESAT-6. *, p < 0.05, compared with the cells treated with M. tuberculosis and equimolar concentrations of CFP10. C, PBMC from six healthy tuberculin reactors were incubated with increasing concentrations of ESAT-6 or CFP10 for 48 h. Stimulation with heat-killed M. tuberculosis Erdman is shown for comparison (far left gray bar). IFN-γ levels in culture supernatants were measured by ELISA. Means and SD are shown. *, p < 0.005, compared with the cells stimulated with M. tuberculosis and equimolar concentrations of CFP10. D, PBMC were stimulated, as in A. After 48 h, CD3+ cells were purified and cultured in a 96-well ELISPOT plate, precoated with anti-IFN-γ capture Ab. After 16 h further incubation, the number of IFN-γ+ cells was determined by incubation with IFN-γ detection Ab, followed by development of the substrate. A representative result from experiments with three donors is shown.
To explore the mechanisms underlying the effect of ESAT-6 on IFN-γ production, we measured IFN-γ gene transcription by real-time PCR. ESAT-6, but not CFP10, reduced IFN-γ mRNA expression induced by M. tuberculosis stimulation of PBMC from three healthy tuberculin reactors by more than 95% (Fig. 2B), suggesting that ESAT-6 inhibits T cell IFN-γ secretion by decreasing transcription of IFN-γ.
These data appear to contradict prior publications demonstrating the immunogenicity of ESAT-6. To investigate this issue further, we stimulated PBMC from healthy tuberculin reactors with increasing concentrations of ESAT-6 and CFP10, in the absence of heat-killed M. tuberculosis. CFP10 induced strong IFN-γ responses at all concentrations. For ESAT-6, IFN-γ concentrations were comparable to those elicited by CFP10 at 0.8 μM, but IFN-γ levels fell markedly as the ESAT-6 concentration increased (Fig. 2C). These results show that, consistent with published data, ESAT-6 can stimulate production of IFN-γ, but that higher concentrations (1.6–3.3 μM) of ESAT-6 fail to induce IFN-γ production.
T cells are the principal cellular source of IFN-γ when PBMC are stimulated with M. tuberculosis. To confirm that ESAT-6 inhibited IFN-γ production by T cells, PBMC were treated with 3.3 μM of CFP10 or ESAT-6, and stimulated with heat-killed M. tuberculosis. After 48 h, CD3+ cells were purified and the frequency of IFN-γ-producing T cells was examined by ELISPOT. The number of IFN-γ+ cells was markedly less in cells treated with ESAT-6 than in those treated with CFP10 (Fig. 2D).
ESAT-6 inhibits expression of transcription factors that regulate IFN-γ gene expression
Transcription of the IFN-γ gene in activated T cells is controlled by essential regulatory regions in the distal (−96 to −80 bp) and proximal (−73 to −48 bp) portions of the IFN-γ promoter (34–36). We focused on the proximal promoter to study T cell production of IFN-γ in response to M. tuberculosis for several reasons. First, in Jurkat T cells, the proximal promoter directs activation- specific IFN-γ gene expression in the same manner as that mediated by the full-length promoter (37). Second, activation of T cells through the TCR and the IL-12 receptor, a process that parallels production of IFN-γ in response to M. tuberculosis, increased activation of the proximal but not the distal IFN-γ promoter element (38). Third, we have shown that the transcription factors, cyclic AMP response element binding protein (CREB), activating transcription factor (ATF)-2 and c-Jun bind to the IFN-γ proximal promoter and positively regulate M. tuberculosis-induced IFN-γ production in human T cells (39–41).
Because ESAT-6 reduced IFN-γ transcription, we studied the effect of ESAT-6 on expression of CREB, ATF-2 and c-Jun in M. tuberculosis-stimulated PBMC. ESAT-6, but not CFP10, inhibited expression of ATF-2, c-Jun, and to lesser extent CREB (Fig. 3A). This was not due to global reduction of proteins, as GAPDH levels were unchanged. Densitometry analysis of three independent experiments (Fig. 3B) demonstrated that ESAT-6, but not CFP10, inhibited expression of c-Jun and ATF-2 by 75 and 49%, respectively.
FIGURE 3.
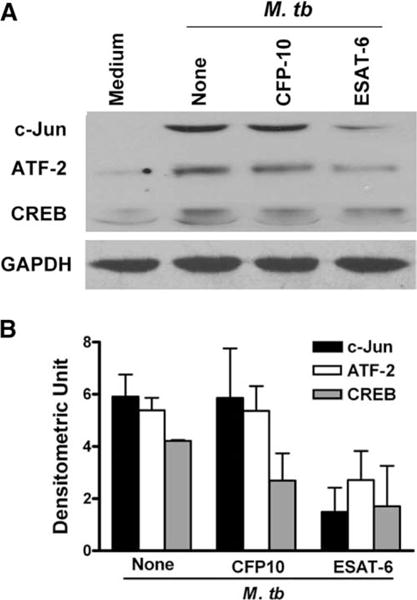
ESAT-6 inhibits expression of transcription factors that positively regulate IFN-γ gene expression in human T cells. PBMC from three healthy tuberculin reactors were treated with medium alone, or with 3.3 μM ESAT-6 or CFP10 for 1 h, before adding heat-killed M. tuberculosis. After 48 h, cells were collected, total protein extracts were prepared, and the proteins were separated by SDS-PAGE (30 μg per lane). A, Western blotting was performed to evaluate the expression of CREB, ATF-2, and c-Jun. GAPDH was used as a loading control. A representative result is shown. B, Means and SD of densitometry analysis from three experiments for each transcription factor is shown.
Effect of ESAT-6 on T cell proliferation and cytokine production in the absence of monocytes
Previous studies have shown that mycobacterial components, such as lipoarabinomannan, can inhibit T cell production of IFN-γ by inducing monocytes to produce immunosuppressive cytokines such as IL-10. To determine whether ESAT-6 inhibits IFN-γ production through effects on monocytes or T cells, we first determined the effect of ESAT-6 on M. tuberculosis-induced IL-10 production by PBMC of three healthy tuberculin reactors. ESAT-6 and CFP10 did not increase IL-10 concentrations (data not shown).
To determine whether ESAT-6 acted directly on T cells, CD3 + cells were purified from PBMC and labeled with CFSE. Stimulation of the labeled cells with anti-CD3 plus anti-CD28 induced strong T cell proliferation after 72 h (Fig. 4A, top right panel). Addition of CFP10 did not affect proliferation (middle panels), but addition of ESAT-6 almost completely abrogated proliferation by both CD4+ and CD8+ cells, as measured by dilution of CFSE (Fig. 4A, lower panels). Furthermore, when purified human CD3 + cells were stimulated with anti-CD3 plus anti-CD28, addition of recombinant ESAT-6, but not CFP10, inhibited IFN-γ secretion in a dose-dependent manner (Fig. 4B). ESAT-6 also reduced expression of IFN-γ mRNA (Fig. 4C) and the transcription factors, ATF-2, c-Jun, and CREB (results not shown). These results indicated that ESAT-6 directly inhibits T cell responses, in the absence of APCs. To determine whether ESAT-6 inhibited production of all T cell cytokines, we evaluated production of IL-17, TNF-α, and IL-2 by T cells stimulated with anti-CD3 plus anti-CD28. ESAT-6 inhibited production of IL-17 (Fig. 4D) and TNF-α (Fig. 4E). In contrast, ESAT-6 slightly increased IL-2 secretion by T cells (Fig. 4F). CFP10 did not affect secretion of these cytokines. These results suggested that inhibition of T cell IFN-γ production by higher ESAT-6 concentrations is not due to a generalized inhibitory effect on T cell cytokine production.
FIGURE 4.
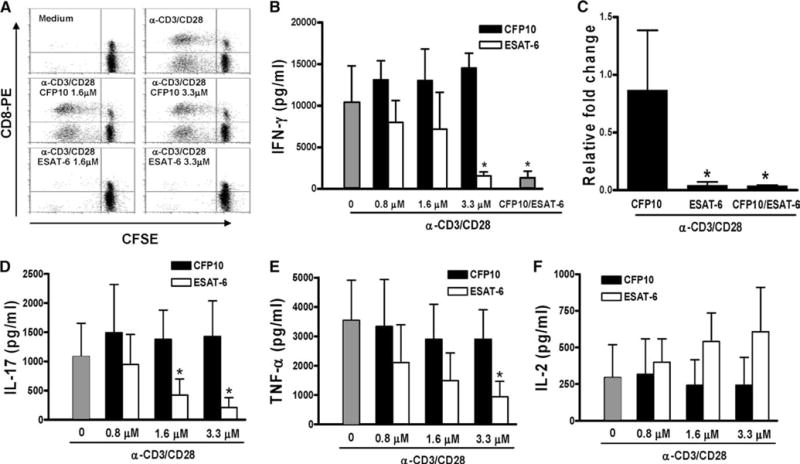
Effect of ESAT-6 on proliferation and cytokine production by CD3+ cells stimulated with anti-CD3 and anti-CD28. A, Purified CD3+ cells were labeled with CFSE and cultured in medium alone (top right panel), CFP10 (middle panels), or ESAT-6 (lower panels) for 1 h, before addition of anti-CD3 (10 μg/ml) and anti-CD28 (1 μg/ml). The top left panel shows cells cultured in medium alone, without anti-CD3 or anti-CD28. After 72 h, the cells were stained with anti-CD8-PE, and CFSE content was analyzed by flow cytometry. One representative result of experiments performed for three donors is shown. B, Purified CD3+ cells from five healthy donors were cultured with medium alone, ESAT-6, CFP10, or ESAT-6/CFP10 heterodimer (3.3 μM each, far right bar) for 1 h, before addition of anti-CD3 and anti-CD28, as in A. After 48 h, IFN-γ levels in the culture supernatants were measured by ELISA. Mean values and SD are shown. *, p < 0.001, compared with the cells treated with anti-CD3 plus anti-CD28 mAbs and equimolar concentration of CFP10. C, Purified CD3+ cells from three healthy donors were cultured as described in B for 1 h, before addition of anti-CD3 and anti-CD28. After 48 h, IFN-γ mRNA expression was measured by real-time PCR. *, p < 0.005, compared with the cells stimulated with anti-CD3 plus anti-CD28 in the presence of CFP10. D–F, Production of IL-17 (D), TNF-α (E) and IL-2 (F) by purified CD3+ cells, cultured as described in B. Mean values and SD from five subjects (IL-17), and eight individuals (TNF-α and IL-2), respectively, are shown. *, p < 0.005 (both D and E), compared with cells stimulated with anti-CD3 plus anti-CD28 in the presence of CFP10.
To exclude the possibility that the inhibitory effects of ESAT-6 on IFN-γ secretion by human T cells is due to contaminants in the recombinant protein preparations, we depleted ESAT-6 from recombinant protein preparations, using a nickel resin, as outlined in Materials and Methods. The depletion process removed 99% of total protein, as measured by the bicinchoninic acid protein assay, and this was further confirmed by Western blot for ESAT-6 (Fig. 5A). Depletion of ESAT-6 completely abrogated the capacity of the recombinant ESAT-6 preparation to inhibit IFN-γ production (Fig. 5B), indicating that inhibition was mediated by ESAT-6 and not by contaminants.
FIGURE 5.
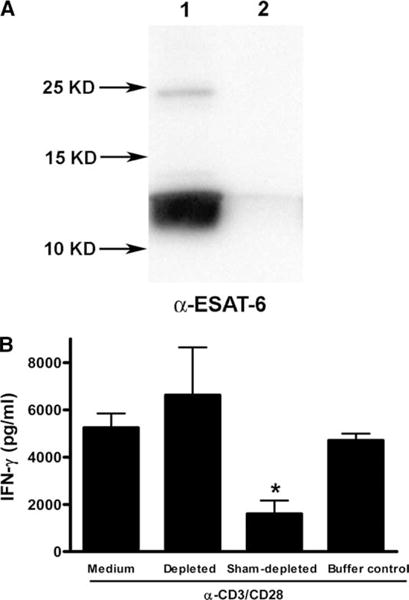
Depletion of ESAT-6 abrogates the inhibition of IFN-γ production by T cells. A, Preparations of recombinant histidine-tagged ESAT-6 were sham-depleted (lane 1) or depleted of ESAT-6 by incubation with a nickel resin (lane 2), and the efficiency of depletion was determined by Western blotting for ESAT-6 with mAb HYB76-8, after resolution by 15% SDS-PAGE and electroblotting to a nitrocellulose membrane. A representative result of two experiments is shown. B, Purified CD3+ cells from three healthy tuberculin-negative individuals were cultured for 1 h in medium alone, 3.3 μM of the sham-depleted recombinant ESAT-6, an equal volume of the ESAT-6-depleted preparation, or the buffer used for the depletion process, before stimulation with anti-CD3 and anti-CD28. After 48 h, IFN-γ levels in the culture supernatants were measured by ELISA. Mean values and SD are shown. *, p < 0.05, compared with the cells treated with the ESAT-6-depleted protein preparation.
The inhibitory effects of ESAT-6 are not mediated by cytolysis or apoptosis
Because ESAT-6 is known to lyse human lung epithelial cells and monocytic cell lines (14), we determined whether ESAT-6 inhibited T cell proliferation and IFN-γ production through cell lysis in our experimental system. When T cells were stimulated with anti- CD3 plus anti-CD28 for 48 h, the percentage of live cells was unaffected by the presence of ESAT-6 or CFP10, as determined by trypan blue exclusion (Fig. 6A) and MTT assay (Fig. 6B).
FIGURE 6.
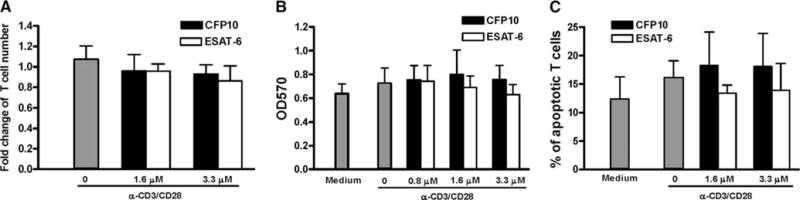
Effect of ESAT-6 and CFP10 on the viability and apoptosis of T cells. CD3+ cells from three healthy donors were cultured with medium alone, ESAT-6, or CFP10 for 1 h, then stimulated with anti-CD3 and anti-CD28. After 48 h, cell viability was analyzed by trypan blue exclusion (A) and MTT assay (B). C, Apoptosis was analyzed by annexin V staining, followed by flow cytometry analysis. In A, the number of viable cells in the wells treated with medium alone was arbitrarily given a value of 1.0. Mean values and SD are shown.
It was previously shown that ESAT-6 induces apoptosis of THP-1 human monocytic cells (42), therefore, we also determined whether the inhibitory effects of ESAT-6 on T cells were mediated through increased apoptosis. When CD3+ cells were stimulated with anti-CD3 plus anti-CD28 for 48 h, ESAT-6 did not increase the percentage of apoptotic, annexin V+ cells (Fig. 6C) or necrotic cells that stained with propidium iodide (data not shown). Taken together with the cytokine studies, these results clearly indicated that the effects of ESAT-6 on human T cells are not due to cytotoxicity or apoptosis.
ESAT-6 down-regulates markers of T cell activation
Our results so far demonstrated that ESAT-6 inhibited TCR signal- induced T cell IFN-γ production by reducing IFN-γ mRNA expression, and IFN-γ gene transcription is known to be initiated by T cell activation. Based on these data, we hypothesized that ESAT-6 inhibited T cell activation. To evaluate this possibility, we studied the effect of ESAT-6 on expression of T cell activation markers, CD25 and CD69, on CD3+ cells after stimulation with anti-CD3 and anti-CD28. ESAT-6 caused a dose-dependent reduction in the percentages of CD25+ and CD69+ cells by up to 30 and 40%, respectively (Fig. 7, A and B). In contrast, CFP10 had no such effect.
FIGURE 7.
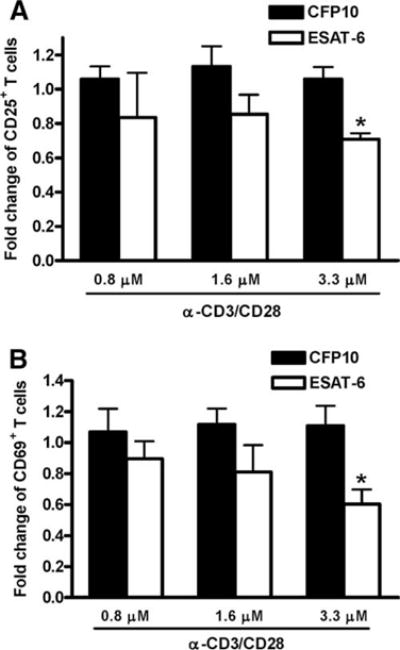
Effect of ESAT-6 and CFP10 on expression of early T cell activation markers. Purified CD3+ cells from PBMC of three healthy donors were incubated with medium alone, ESAT-6, or CFP10 for 1 h, then cultured with anti-CD3 and anti-CD28. After 48 h, the expression levels of CD25 (A) and CD69 (B) on CD3 + cells were analyzed by flow cytometry. Due to significant variability from donor to donor, the percentage of CD25+ or CD69+ cells obtained after stimulation with anti-CD3 and anti- CD28 was arbitrarily given a value of 1.0. Mean values and SD are shown. *, p < 0.05, compared with the cells treated with an equimolar concentration of CFP10, anti-CD3, and anti-CD28.
ESAT-6 binds to human PBMC
Because the results above demonstrated that ESAT-6 directly inhibited T cell activation without affecting cell viability or apoptosis, we hypothesized that ESAT-6 exerts its effects by binding directly to T cells. To test this hypothesis, we stained PBMC from three healthy donors with biotinylated ESAT-6 or CFP10, in combination with PE-conjugated Abs to CD3, CD4, or CD8, followed by incubation with FITC-conjugated streptavidin. Flow cytometry analysis showed that CD3+ T cells stained very strongly with ESAT-6, but not CFP10. Furthermore, ESAT-6 bound to the surface of both CD4+ and CD8+ subpopulations of CD3+ T cells (Fig. 8 and data not shown). In contrast, both ESAT-6 and CFP10 bound to CD14+ monocytes and CD19+ B cells (Fig. 8).
FIGURE 8.
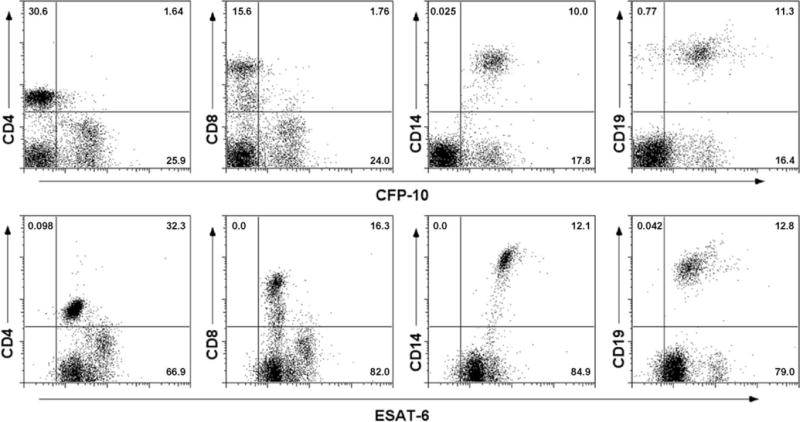
Binding of ESAT-6 to T cells and other cell populations. PBMC from a healthy donor were incubated with 3.3 μM biotinylated ESAT-6 or CFP10 in combination with PE-conjugated mAbs to CD4, CD8, CD14, and CD19. After washing, the cells were incubated with FITC-streptavidin and analyzed by flow cytometry. The numbers indicate the percentage of cells in the corresponding quadrants. One representative result of experiments with cells from three donors is shown.
ESAT-6 inhibits human T cells without affecting early TCR signaling
ESAT-6 may bind to target cells by interacting with cell membranes (19, 43) and we found that ESAT-6 binds to human T cells (Fig. 7). This suggested that ESAT-6 may inhibit T cell activation by binding to the cell membrane and perturbing the early TCR signaling. ZAP70 is a key tyrosine kinase in TCR signaling and phosphorylation of ZAP70 is the limiting factor before it is recruited to the phosphorylated ζ-chain of TCR to relay T cell activation signaling (44), hence we evaluated the effect of ESAT-6 on phosphorylation of ZAP70. CD3+ cells from five individuals were pretreated with or without ESAT-6 and CFP10 and stimulated with cross-linked anti-CD3. Anti-CD3-stimulated T cells showed a significant increase in phosphorylation of ZAP70, compared with unstimulated cells and pretreatment of CD3+ cells with either ESAT-6 or CFP10 did not affect ZAP70 phosphorylation (Fig. 9).
FIGURE 9.
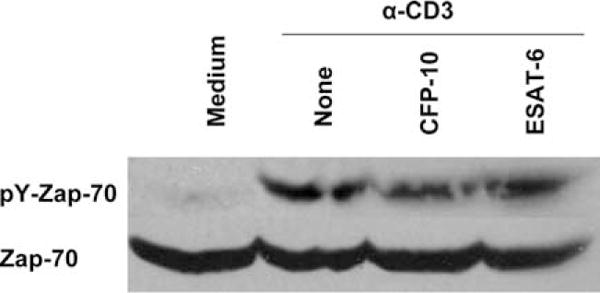
Effect of ESAT-6 on phosphorylation of ZAP70. CD3+ cells purified by negative selection from four healthy donors were incubated on ice with medium alone, or with 3.3 μM of CFP10 or ESAT-6 for one hour, and the cells were treated with anti-CD3 mAb and cross-linked with goat anti-mouse IgG as described in methods. Total cell protein extracts of cells were prepared and the level of phosphorylated and total ZAP70 was evaluated by Western blotting. A representative result is shown.
Discussion
A large body of literature indicates that ESAT-6 and CFP10 induce potent immune responses to protect against M. tuberculosis infection in animal models. In contrast, secretion of these proteins by the ESX-1 specialized secretion system is associated with mycobacterial virulence and pathogenicity. Because these proteins mediate critical host-pathogen interactions, it is important to understand the full range of their biological effects.
In this study, we made the surprising finding that higher concentrations (1.6–3.3 μM) of ESAT-6 inhibited IFN-γ production by M. tuberculosis-responsive T cells in PBMC, and suppressed both proliferation and IFN-γ production by purified CD3+ T cells stimulated with anti-CD3 and anti-CD28, indicating that these effects were independent of APCs. A direct effect of ESAT-6 on human T cells was also suggested by flow cytometry studies that demonstrated binding of ESAT-6 but not CFP10 to CD4+ and CD8+ T cells. Further, ESAT-6 inhibited expression of T cell activation markers and reduced expression of the transcription factors, ATF-2 and c-Jun, which normally bind to the IFN-γ proximal promoter and stimulate mRNA expression. However, ESAT-6 did not affect ZAP70 phosphorylation, which transmits intracellular signals after TCR activation, suggesting that ESAT-6 acts at down- stream TCR-induced signaling cascade. These findings provide the first evidence that ESAT-6 directly inhibits the capacity of human T cells to respond to mycobacterial Ags.
Because ESAT-6 is a putative vaccine Ag that is being evaluated in human trials (45), it is critical to understand its effects on T cells and mononuclear phagocytes that mediate immunity to M. tuberculosis. The ESAT-6/CFP10 complex has been reported to bind to monocytic but not fibroblast cell lines through the C-terminal flexible arm of CFP10 (43). We found that ESAT-6 binds to T cells, monocytes, and B-cells, whereas CFP10 only binds to monocytes and B-cells (Fig. 8). When ESAT-6 binds to cells, the effects on cell viability are controversial. Some studies have suggested that ESAT-6 induces apoptosis in human THP-1 cells (42) and lyses artificial lipid bilayers (23). However, the solution structure of the ESAT-6/CFP10 complex suggests that these proteins do not have pore-forming activity and the complex did not lyse MonoMac6 cells (43). In the current study, ESAT-6 did not increase apoptosis or reduce viability of T cells, as measured by the trypan blue assay, which evaluates cell membrane integrity, or the MTT assay, which measures cellular metabolic activity. Furthermore, ESAT-6 did not inhibit IL-2 production, indicating that it did not act as a toxin that inhibits production of all T cell cytokines. Thus, general cellular toxicity and apoptosis induced by ESAT-6 binding to human T cell membrane are unlikely to mediate its inhibition of T cell IFN-γ production and proliferation.
We demonstrated that ESAT-6 directly inhibits T cell proliferation and production of IFN-γ in response to TCR stimulation by mycobacterial Ags and anti-CD3 and anti-CD28. This effect was independent of APCs, and was not due to contamination of LPS and production of IL-10. Other bacterial products have similar effects to those observed with ESAT-6. Bacillus anthracis lethal toxin inhibits proliferation and IL-2 production by murine and human CD4+ T cells, without increasing apoptosis or cell death (46, 47). Lethal toxin inhibits T cell function by cleaving MEKs of MAPKs and inhibiting activation of many intracellular signaling molecules in T cells, including ERK, ATF-2, Akt, NFAT, and AP-1 (46). Helicobacter pylori vacA inhibits T cell proliferation by inhibiting cell cycle progression (48), preventing calcium influx and translocation of NFAT to the nucleus, as well as inhibiting phosphorylation of p38 MAPK (49). Bordetella pertussis adenylate cyclase toxin inhibits T cell activation and production of IL-12 by dendritic cells through increasing intracellular cAMP (50, 51). Thus, ESAT-6 is but one example of several bacterial products that inhibit immune responses by both T cells and mononuclear phagocytes with similar mechanisms.
Like the toxins produced by B. anthracis and B. pertussis, ESAT-6 affects both T cells and mononuclear phagocytes. In monocytic cells, ESAT-6 inhibits LPS-induced activation of ERKs (52), and binds to TLR2 and inhibits Akt and NF-κB activation (22). However, neutralizing anti-TLR2 did not abrogate inhibition of T cell IFN-γ production by ESAT-6 (Buka Samten, unpublished data), suggesting that the effects we observed in T cells were not mediated through this interaction. ESAT-6 inhibited IFN-γ production and reduced expression of ATF-2 and c-Jun (Fig. 3), transcription factors that up-regulate IFN-γ transcription by binding to the proximal promoter (31). However, ESAT-6 did not affect phosphorylation of ZAP70 (Fig. 9) or intracellular calcium levels (data not shown), both of which are part of the proximal signaling cascade induced by TCR signaling. We speculate that ESAT-6 inhibits T cell immune responses by affecting downstream signaling molecules, such as kinases that activate transcription factors, as is the case for anthrax lethal toxin (46). Future studies are needed to address this issue.
Although ESAT-6 is composed of only 95 amino acids, it is intriguing that different portions of the molecule appear to have specific effects on different cell types. The amino terminal portion is highly immunogenic for T cells (53, 54), whereas the C-terminal portion bound to TLR2 on murine macrophages (22) and contributed to virulence in animals (55). We speculate that certain portion of ESAT-6 would mediate its capacity to inhibit IFN-γ production by T cells, and work is ongoing to identify the structural basis for this effect. These studies would allow us to design improved ESAT-6-based subunit vaccines by removing domains that inhibit T cell immune responses.
Recent studies with mice transgenic for TCRs that recognize specific mycobacterial peptides demonstrated that T cell-mediated adaptive immune responses in the lungs during M. tuberculosis infection are delayed, despite substantial bacterial burdens and high local concentrations of T cells (56–58). ESAT-6 is produced in M. tuberculosis culture filtrate predominantly during the early phase of growth in vitro (3, 59) and is present in infected lung tissue (13). Therefore, it is intriguing to speculate that, in the initial stages of pulmonary infection by M. tuberculosis, ESAT-6 may delay the onset of T cell adaptive immunity by inhibiting T cell IFN-γ responses. As the infection progresses, other authors have recently suggested that the transcription factor, EspR (Rv3849) acts through a negative feedback loop to reduce secretion of ESAT-6 and other proteins of the ESX-1 system (60). The immune system then has the opportunity to respond to other dominant Ags of M. tuberculosis, such as Ag85, which elicit robust T cell responses (56, 61, 62). This mechanism may also play a role, at least in part, in reduced IFN-γ secretion by T cells in human tuberculosis.
In conclusion, we found that higher concentrations of ESAT-6 markedly inhibit IFN-γ production by T cells in response to M. tuberculosis or TCR activation by reducing activation of T cells without affecting upstream TCR signaling events. Further work to delineate the molecular mechanisms underlying these effects are critical to better understand interactions between M. tuberculosis and the human immune system, and to facilitate development of ESAT-6-based vaccines.
Acknowledgments
We thank Dr. Peter Andersen for provision of HYB 76-8 and Dr. Patrick Brennan for provision of heat-killed M. tuberculosis Erdman.
Footnotes
This work was supported by grants from the National Institutes of Health (A1063514), the James Byers Cain Research Endowment, and the Center for Pulmo-nary and Infectious Disease Control. P.F.B. holds the Margaret E. Byers Cain Chair for Tuberculosis Research.
Abbreviations used in this paper: ESAT-6, early secreted Ag of 6 kDa; ATF, activating transcription factor; BCG, bacillus Calmette-Guérin; CFP10, culture filtrate protein of 10 kDa; CREB, cyclic AMP response element binding protein; RD, region of difference.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Raviglione MC, Snider DE, Jr, Kochi A. Global epidemiology of tuberculosis. Morbidity and mortality of a worldwide epidemic. J Am Med Assoc. 1995;273:220–226. [PubMed] [Google Scholar]
- 2.Andersen P, Andersen AB, Sorensen AL, Nagai S. Recall of long-lived immunity to Mycobacterium tuberculosis infection in mice. J Immunol. 1995;154:3359–3372. [PubMed] [Google Scholar]
- 3.Sorensen AL, Nagai S, Houen G, Andersen P, Andersen AB. Purification and characterization of a low-molecular-mass T-cell antigen secreted by Mycobacterium tuberculosis. Infect Immun. 1995;63:1710–1717. doi: 10.1128/iai.63.5.1710-1717.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietrich J, Billeskov R, Doherty TM, Andersen P. Synergistic effect of bacillus calmette guerin and a tuberculosis subunit vaccine in cationic liposomes: increased immunogenicity and protection. J Immunol. 2007;178:3721–3730. doi: 10.4049/jimmunol.178.6.3721. [DOI] [PubMed] [Google Scholar]
- 5.Pym AS, Brodin P, Majlessi L, Brosch R, Demangel C, Williams A, Griffiths KE, Marchal G, Leclerc C, Cole ST. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat Med. 2003;9:533–539. doi: 10.1038/nm859. [DOI] [PubMed] [Google Scholar]
- 6.Olsen AW, Williams A, Okkels LM, Hatch G, Andersen P. Protective effect of a tuberculosis subunit vaccine based on a fusion of antigen 85B and ESAT-6 in the aerosol guinea pig model. Infect Immun. 2004;72:6148–6150. doi: 10.1128/IAI.72.10.6148-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langermans JA, Doherty TM, Vervenne RA, van der Laan T, Lyashchenko K, Greenwald R, Agger EM, Aagaard C, Weiler H, van Soolingen D, et al. Protection of macaques against Mycobacterium tuberculosis infection by a subunit vaccine based on a fusion protein of antigen 85B and ESAT-6. Vaccine. 2005;23:2740–2750. doi: 10.1016/j.vaccine.2004.11.051. [DOI] [PubMed] [Google Scholar]
- 8.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 9.Harboe M, Oettinger T, Wiker HG, Rosenkrands I, Andersen P. Evidence for occurrence of the ESAT-6 protein in Mycobacterium tuberculosis and virulent Mycobacterium bovis and for its absence in Mycobacterium bovis BCG. Infect Immun. 1996;64:16–22. doi: 10.1128/iai.64.1.16-22.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiGiuseppe Champion PA, Cox JS. Protein secretion systems in mycobacteria. Cell Microbiol. 2007;9:1376–1384. doi: 10.1111/j.1462-5822.2007.00943.x. [DOI] [PubMed] [Google Scholar]
- 11.Behr MA, Sherman DR. Mycobacterial virulence and specialized secretion: same story, different ending. Nat Med. 2007;13:286–287. doi: 10.1038/nm0307-286. [DOI] [PubMed] [Google Scholar]
- 12.Abdallah AM, Gey van Pittius NC, Champion PA, Cox J, Luirink J, Vandenbroucke-Grauls CM, Appelmelk BJ, Bitter W. Type VII secretion-mycobacteria show the way. Nat Rev Microbiol. 2007;5:883–891. doi: 10.1038/nrmicro1773. [DOI] [PubMed] [Google Scholar]
- 13.Majlessi L, Brodin P, Brosch R, Rojas MJ, Khun H, Huerre M, Cole ST, Leclerc C. Influence of ESAT-6 secretion system 1 (RD1) of Mycobacterium tuberculosis on the interaction between mycobacteria and the host immune system. J Immunol. 2005;174:3570–3579. doi: 10.4049/jimmunol.174.6.3570. [DOI] [PubMed] [Google Scholar]
- 14.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 16.Geluk A, van Meijgaarden KE, Franken KL, Subronto YW, Wieles B, Arend SM, Sampaio EP, de Boer T, Faber WR, Naafs B, Ottenhoff TH. Identification and characterization of the ESAT-6 homo-logue of Mycobacterium leprae and T-cell cross-reactivity with Mycobacterium tuberculosis. Infect Immun. 2002;70:2544–2548. doi: 10.1128/IAI.70.5.2544-2548.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol. 2004;53:1677–1693. doi: 10.1111/j.1365-2958.2004.04261.x. [DOI] [PubMed] [Google Scholar]
- 18.van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 19.de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, Honore N, Marchal G, Jiskoot W, England P, Cole ST, Brosch R. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol. 2007;189:6028–6034. doi: 10.1128/JB.00469-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA. Human macrophage activation programs induced by bacterial pathogens. Proc Natl Acad Sci USA. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA. 2003;100:13001–13006. doi: 10.1073/pnas.2235593100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pathak SK, Basu S, Basu KK, Banerjee A, Pathak S, Bhattacharyya A, Kaisho T, Kundu M, Basu J. Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat Immunol. 2007;8:610–618. doi: 10.1038/ni1468. [DOI] [PubMed] [Google Scholar]
- 23.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon γ in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon γ gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ottenhoff TH, Kumararatne D, Casanova JL. Novel human immunodeficiencies reveal the essential role of type-I cytokines in immunity to intracellular bacteria. Immunol Today. 1998;19:491–494. doi: 10.1016/s0167-5699(98)01321-8. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Lin Y, Iyer DV, Gong J, Abrams JS, Barnes PF. T-cell cytokine responses in human infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:3231–3234. doi: 10.1128/iai.63.8.3231-3234.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirsch CS, Toossi Z, Othieno C, Johnson JL, Schwander SK, Robertson S, Wallis RS, Edmonds K, Okwera A, Mugerwa R, Peters P, Ellner JJ. Depressed T-cell interferon-γ responses in pulmonary tuberculosis: analysis of underlying mechanisms and modulation with therapy. J In-fect Dis. 1999;180:2069–2073. doi: 10.1086/315114. [DOI] [PubMed] [Google Scholar]
- 28.Sodhi A, Gong J, Silva C, Qian D, Barnes PF. Clinical correlates of interferon γ production in patients with tuberculosis. Clin Infect Dis. 1997;25:617–620. doi: 10.1086/513769. [DOI] [PubMed] [Google Scholar]
- 29.Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, Carr MD. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex: implications for pathogenesis and virulence. J Biol Chem. 2002;277:21598–21603. doi: 10.1074/jbc.M201625200. [DOI] [PubMed] [Google Scholar]
- 30.Shams H, Klucar P, Weis SE, Lalvani A, Moonan PK, Safi H, Wizel B, Ewer K, Nepom GT, Lewinsohn DM, Andersen P, Barnes PF. Characterization of a Mycobacterium tuberculosis peptide that is recognized by human CD4+ and CD8+ T cells in the context of multiple HLA alleles. J Immunol. 2004;173:1966–1977. doi: 10.4049/jimmunol.173.3.1966. [DOI] [PubMed] [Google Scholar]
- 31.Samten B, Townsend JC, Sever-Chroneos Z, Pasquinelli V, Barnes PF, Chroneos ZC. An antibody against the surfactant protein A (SP-A)-binding domain of the SP-A receptor inhibits T cell-mediated immune responses to Mycobacterium tuberculosis. J Leukocyte Biol. 2008;84:115–123. doi: 10.1189/jlb.1207835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dillon DC, Alderson MR, Day CH, Bement T, Campos-Neto A, Skeiky YA, Vedvick T, Badaro R, Reed SG, Houghton R. Molecular and immunological characterization of Mycobacterium tuberculosis CFP-10, an immunodiagnostic antigen missing in Mycobacterium bovis BCG. J Clin Microbiol. 2000;38:3285–3290. doi: 10.1128/jcm.38.9.3285-3290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okkels LM, Andersen P. Protein-protein interactions of proteins from the ESAT-6 family of Mycobacterium tuberculosis. J Bacteriol. 2004;186:2487–2491. doi: 10.1128/JB.186.8.2487-2491.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penix L, Weaver WM, Pang Y, Young HA, Wilson CB. Two essential regulatory elements in the human interferon γ promoter confer activation specific expression in T cells. J Exp Med. 1993;178:1483–1496. doi: 10.1084/jem.178.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cippitelli M, Sica A, Viggiano V, Ye J, Ghosh P, Birrer MJ, Young HA. Negative transcriptional regulation of the interferon-γ promoter by glucocorticoids and dominant negative mutants of c-Jun. J Biol Chem. 1995;270:12548–12556. doi: 10.1074/jbc.270.21.12548. [DOI] [PubMed] [Google Scholar]
- 36.Penix LA, Sweetser MT, Weaver WM, Hoeffler JP, Kerppola TK, Wilson CB. The proximal regulatory element of the interferon-γ promoter mediates selective expression in T cells. J Biol Chem. 1996;271:31964–31972. doi: 10.1074/jbc.271.50.31964. [DOI] [PubMed] [Google Scholar]
- 37.Young HA, Ghosh P, Ye J, Lederer J, Lichtman A, Gerard JR, Penix L, Wilson CB, Melvin AJ, McGurn ME. Differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-γ gene. J Immunol. 1994;153:3603–3610. [PubMed] [Google Scholar]
- 38.Zhang F, Wang DZ, Boothby M, Penix L, Flavell RA, Aune TM. Regulation of the activity of IFN-γ promoter elements during Th cell differentiation. J Immunol. 1998;161:6105–6112. [PubMed] [Google Scholar]
- 39.Samten B, Ghosh P, Yi AK, Weis SE, Lakey DL, Gonsky R, Pendurthi U, Wizel B, Zhang Y, Zhang M, et al. Reduced expression of nuclear cyclic adenosine 5′-monophosphate response element-binding proteins and IFN-γ promoter function in disease due to an intracellular pathogen. J Immunol. 2002;168:3520–3526. doi: 10.4049/jimmunol.168.7.3520. [DOI] [PubMed] [Google Scholar]
- 40.Samten B, Howard ST, Weis SE, Wu S, Shams H, Townsend JC, Safi H, Barnes PF. Cyclic AMP response element-binding protein positively regulates production of IFN-γ by T cells in response to a microbial pathogen. J Immunol. 2005;174:6357–6363. doi: 10.4049/jimmunol.174.10.6357. [DOI] [PubMed] [Google Scholar]
- 41.Samten B, Townsend JC, Weis SE, Bhoumik A, Klucar P, Shams H, Barnes PF. CREB, ATF, and AP-1 transcription factors regulate IFN-γ secretion by human T cells in response to mycobacterial antigen. J Immunol. 2008;181:2056–2064. doi: 10.4049/jimmunol.181.3.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 2007;9:1547–1555. doi: 10.1111/j.1462-5822.2007.00892.x. [DOI] [PubMed] [Google Scholar]
- 43.Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, Gordon SV, Hewinson RG, Burke B, Norman J, Williamson RA, Carr MD. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 2005;24:2491–2498. doi: 10.1038/sj.emboj.7600732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wange RL, Samelson LE. Complex complexes: signaling at the TCR. Immunity. 1996;5:197–205. doi: 10.1016/s1074-7613(00)80315-5. [DOI] [PubMed] [Google Scholar]
- 45.Reece ST, Kaufmann SH. Rational design of vaccines against tuberculosis directed by basic immunology. Int J Med Microbiol. 2008;298:143–150. doi: 10.1016/j.ijmm.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 46.Fang H, Cordoba-Rodriguez R, Lankford CS, Frucht DM. Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4+ T cells. J Immunol. 2005;174:4966–4971. doi: 10.4049/jimmunol.174.8.4966. [DOI] [PubMed] [Google Scholar]
- 47.Comer JE, Chopra AK, Peterson JW, Konig R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect Immun. 2005;73:8275–8281. doi: 10.1128/IAI.73.12.8275-8281.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci USA. 2004;101:7727–7732. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D’Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–1897. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paccani SR, Dal Molin F, Benagiano M, Ladant D, D’Elios MM, Montecucco C, Baldari CT. Suppression of T-lymphocyte activation and chemotaxis by the adenylate cyclase toxin of Bordetella pertussis. Infect Immun. 2008;76:2822–2832. doi: 10.1128/IAI.00200-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spensieri F, Fedele G, Fazio C, Nasso M, Stefanelli P, Mastrantonio P, Ausiello CM. Bordetella pertussis inhibition of interleukin-12 (IL-12) p70 in human monocyte-derived dendritic cells blocks IL-12 p35 through ade-nylate cyclase toxin-dependent cyclic AMP induction. Infect Immun. 2006;74:2831–2838. doi: 10.1128/IAI.74.5.2831-2838.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ganguly N, Giang PH, Basu SK, Mir FA, Siddiqui I, Sharma P. Mycobacterium tuberculosis 6-kDa early secreted antigenic target (ESAT-6) protein downregulates lipopolysaccharide induced c-myc expression by modulating the extracellular signal regulated kinases 1/2. BMC Immunol. 2007;8:24. doi: 10.1186/1471-2172-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brandt L, Oettinger T, Holm A, Andersen AB, Andersen P. Key epitopes on the ESAT-6 antigen recognized in mice during the recall of protective immunity to Mycobacterium tuberculosis. J Immunol. 1996;157:3527–3533. [PubMed] [Google Scholar]
- 54.Ulrichs T, Munk ME, Mollenkopf H, Behr-Perst S, Colangeli R, Gennaro ML, Kaufmann SH. Differential T cell responses to Mycobacterium tuberculosis ESAT6 in tuberculosis patients and healthy donors. Eur J Immunol. 1998;28:3949–3958. doi: 10.1002/(SICI)1521-4141(199812)28:12<3949::AID-IMMU3949>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 55.Brodin P, de Jonge MI, Majlessi L, Leclerc C, Nilges M, Cole ST, Brosch R. Functional analysis of early secreted antigenic target-6, the dominant T-cell antigen of Mycobacterium tuberculosis, reveals key residues involved in secretion, complex formation, virulence, and immunogenicity. J Biol Chem. 2005;280:33953–33959. doi: 10.1074/jbc.M503515200. [DOI] [PubMed] [Google Scholar]
- 56.Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J Exp Med. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 58.Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J Exp Med. 2008;205:2359–2368. doi: 10.1084/jem.20080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andersen P, Askgaard D, Ljungqvist L, Bennedsen J, Heron I. Proteins released from Mycobacterium tuberculosis during growth. Infect Immun. 1991;59:1905–1910. doi: 10.1128/iai.59.6.1905-1910.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raghavan S, Manzanillo P, Chan K, Dovey C, Cox JS. Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature. 2008;454:717–721. doi: 10.1038/nature07219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harth G, Lee BY, Wang J, Clemens DL, Horwitz MA. Novel insights into the genetics, biochemistry, and immunocytochemistry of the 30-kilodalton major extracellular protein of Mycobacterium tuberculosis. Infect Immun. 1996;64:3038–3047. doi: 10.1128/iai.64.8.3038-3047.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Havlir DV, Wallis RS, Boom WH, Daniel TM, Chervenak K, Ellner JJ. Human immune response to Mycobacterium tuberculosis antigens. Infect Immun. 1991;59:665–670. doi: 10.1128/iai.59.2.665-670.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]