

 This work is licensed under a
This work is licensed under a Abstract
Mutations in hepatocyte nuclear factor 1β gene (HNF1B) are responsible for a multisystemic syndrome where monogenic diabetes (classically known as MODY 5) and renal anomalies, mostly cysts, are the most characteristic findings. Urogenital malformations, altered liver function tests, hypomagnesemia or hyperuricemia and gout are also part of the syndrome. Diabetes in these patients usually requires early insulinization. We present the case of a young non-obese male patient with a personal history of renal multicystic dysplasia and a debut of diabetes during adolescence with simple hyperglycemia, negative pancreatic autoimmunity and detectable C-peptide levels. He also presented epididymal and seminal vesicle cysts, hypertransaminasemia, hyperuricemia and low magnesium levels. In the light of these facts we considered the possibility of a HNF1B mutation. The sequencing study of this gene confirmed a heterozygous mutation leading to a truncated and less functional protein. Genetic studies of his relatives were negative; consequently, it was classified as a de novo mutation. In particular, our patient maintained good control of his diabetes on oral antidiabetic agents for a long period of time. He eventually needed insulinization although oral therapy was continued alongside, allowing reduction of prandial insulin requirements. The real prevalence of mutations in HNF1B is probably underestimated owing to a wide phenotypical variability. As endocrinologists, we should consider this possibility in young non-obese diabetic patients with a history of chronic non-diabetic nephropathy, especially in the presence of some of the other characteristic manifestations.
Learning points:
HNF1B mutations are a rare cause of monogenic diabetes, often being a part of a multisystemic syndrome.
The combination of young-onset diabetes and genitourinary anomalies with slowly progressive nephropathy of non-diabetic origin in non-obese subjects should rise the suspicion of such occurrence. A family history may not be present.
Once diagnosis is made, treatment of diabetes with oral agents is worth trying, since the response can be sustained for a longer period than the one usually described. Oral treatment can help postpone insulinization and, once this is necessary, can help reduce the required doses.
Background
The term MODY (maturity-onset diabetes of the young) has traditionally been used to designate a group of diabetes with the particular facts of young age at diagnosis (less than 25 years old) and absence of insulin dependence. The term monogenic diabetes is preferred now because it is known that it comprises a range of diabetes caused by mutations of autosomal dominant inheritance in different genes involved in insulin secretion by pancreatic beta cells (1). There are fourteen genes identified as responsible, implying different clinical presentations, responses to treatments and risks of chronic complications. Approximately 1–5% of all diabetes cases in the United States and other industrialized countries are thought to be monogenic (2), though the real prevalence is likely to be higher. The most frequent forms are those related to mutations in hepatocyte nuclear factor alfa or HNF1A and in glucokinase, also known as MODY 3 and 2 respectively. The HNF1B gene, also called TCF2, encodes the hepatocyte nuclear factor 1β (HNF1B), which is expressed during embryogenesis in kidney, pancreas, genitourinary epithelium, liver and lung (3). As a result, its heterozygous mutation produces a variable clinical spectrum, where the most frequent facts are kidney involvement and HNF1B-related monogenic diabetes or MODY 5. Genitourinary malformations, hyperuricemia, hypomagnesemia and hypertransaminasemia are also manifestations of the syndrome. All these features may be present without a family history, since half the mutations in HNF1B occur de novo (3).
We present the case of a young male with HNF1B-related disease, including monogenic diabetes, which was successfully managed for years with oral antidiabetic agents before needing insulinization.
Case presentation
A 17-year-old male came to the emergency room for polyuria, polydipsia and weight loss. From his medical history, it was of interest that he had been diagnosed of a renal multicystic dysplasia during the first year of life, with a slowly progressive decrease of glomerular filtration rate. The initial analysis showed glycaemia over 700 mg/dL without ketoacidosis. With the diagnosis of diabetic debut with simple hyperglycemia, he was admitted to our Endocrinology Department. Physical examination did not show any dysmorphic feature or evidence of acanthosis nigricans. The body mass index was 25.2 kg/m2. There was no history of diabetes or renal disease in his family.
Investigation
A complete biochemistry (Table 1) showed hypomagnesemia, hypertransaminasemia and renal failure (serum creatinine of 2.7 mg/dL, descending to 2.1 mg/dL with intravenous fluid therapy). The study of pancreatic autoimmunity was negative and C-peptide value was 2.17 ng/mL basal and 3.93 ng/mL after glucagon stimulation. The study was completed with an abdominopelvic magnetic resonance, showing the following findings (Figs 1, 2 and 3): kidneys of a reduced size with multiple cysts of a maximum size of 1 cm, renal pelvis ecstasy, reduced pancreatic volume, bilateral epididymal cysts and a mass of 87 × 85 × 75 mm in the seminal vesicle area.
Table 1.
Initial analytic parameters.
| Parameter | Value | Reference interval |
|---|---|---|
| Glycaemia (mg/dL) | 717 | 74–109 |
| HbA1c (%) | 13 | 4–6 |
| pH | 7.42 | 7.34–7.44 |
| Bicarbonate (mEq/L) | 32 | 22–26 |
| pCO2 (mmHg) | 51 | 35–48 |
| Uric acid (mg/dL) | 7.3 | 2.4–5.7 |
| Creatinine (mg/dL) | 2.7 | 0.5–0.9 |
| Urea (mg/dL) | 110 | 10–50 |
| GOT (U/L) | 206 | 5–32 |
| GPT (U/L) | 233 | 5–33 |
| GGT (U/L) | 127 | 5–36 |
| Alkaline phosphatase (U/L) | 184 | 35–104 |
| Magnesium (mg/dL) | 1.1 | 1.58–2.55 |
| Potassium (mmol/L) | 3.3 | 3.5–5.1 |
| Ketonuria | Negative | – |
Figure 1.
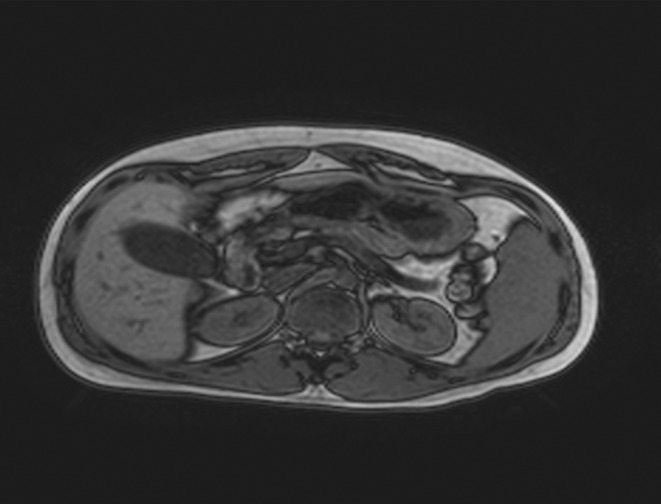
Out-of-phase sequence of abdominal MRI showing pancreatic atrophy and reduced size kidneys with bilateral subcentimetrical cysts and pelvic ecstasy.
Figure 2.
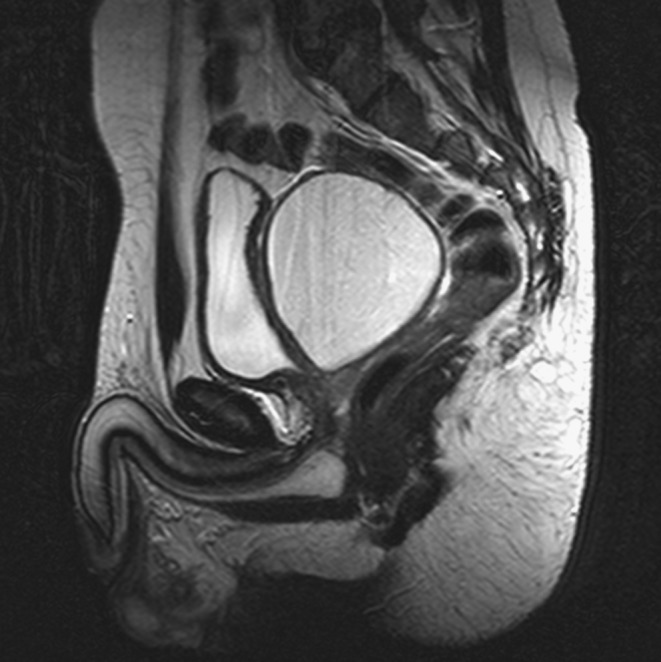
T2-weighted sequence of pelvic MRI showing seminal vesicle tumor measuring 87 × 85 × 75 mm.
Figure 3.
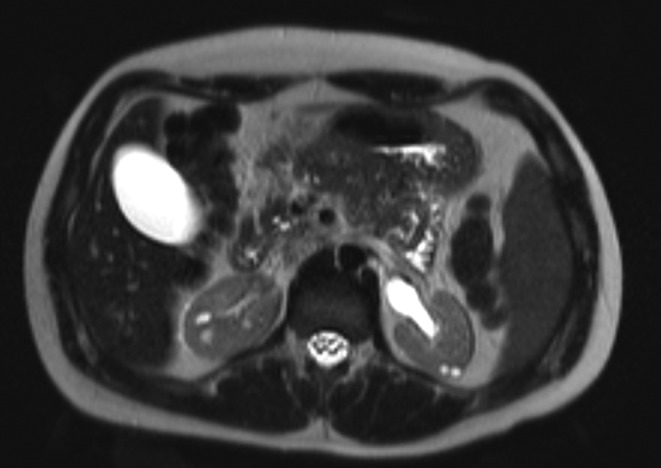
T2-weighted sequence of abdominal MRI where small bilateral renal cyst and pelvic ecstasy are observed.
Treatment
Insulin therapy in basal-bolus regime was started with a basal analogue and three doses of ultra-rapid-acting insulin (0.5 U/kg of weight/day). In subsequent visits, the patient showed excellent metabolic control (glycated hemoglobin of 6.3–6.5%). After initial hospitalization, resection of the paravesical tumor was scheduled. It proved to be dependent on the right seminal vesicle and histopathology was consistent with a benign cyst.
Regarding this phenotype, the sequencing study of the HNF1B gene was requested. The result was a heterozygous mutation involving the deletion of five nucleotides in exon 1 (c207_211delCGCCA). This alteration caused a change in the reading frame resulting in a truncated protein with loss of function. The study was negative for the patient’s parents and sister; therefore, diagnosis of monogenic diabetes derived from de novo mutation in HNF1B was established.
Considering this as well as the excellent glycemic control achieved by lowering insulin doses, it was decided to switch to oral antidiabetic agents. Glimepiride in monotherapy was initially prescribed, associating sitagliptin some time later. Following this treatment, the patient maintained his glycated hemoglobin around 6.5% for six years. After this period, there was a gradual deterioration of metabolic control despite intensifying oral therapy, with glycated hemoglobin up to 8.8%, so we proceeded to insulinization again. In his last visits, he has followed a basal-plus regime with a total daily dose of 0.3 U/kg, including a basal analogue and ultra-rapid-acting insulin at breakfast time. Treatment with sitagliptin was maintained and repaglinide was associated in the remaining meals.
Outcome and follow-up
With this combination, the glycemic control improved again, glycated hemoglobin being in the range of 6.5% eight years after initial diagnosis. Periodical screening of retinopathy and albuminuria has resulted negative so far. The patient is asymptomatic and keeps stable renal function. Biochemical initial anomalies consisting of hyperuricemia, hypomagnesemia and altered liver function tests have remained stable all this time with little fluctuations and without any clinical repercussion.
Discussion
HNF1B gene is located on chromosome 17q12. The encoded protein, a transcription factor with 80% homology with HNF1A, binds DNA either as a homodimer or a heterodimer with HNF1A (3). Genetic alterations can consist of complete gene deletions, little fragment deletions or insertions. Although minor changes in this HNF1B gene have proved to confer a slight predisposition to type 2 diabetes mellitus (4), mutations responsible for the HNFB1-related syndrome are pathogenic by generating a protein that either cannot bind DNA (as in our patient’s case) or fails at transactivation after binding DNA. Though HNF1B mutations have autosomal dominant inheritance, it is not unusual to find de novo mutations as it is our patient’s case. There is no evidence of a genotype–phenotype relationship and clinical manifestations are heterogeneous even among individuals with the same mutation. Since diabetes secondary to HNF1B mutation was first reported in a Japanese family in 1997 (5), the description of the phenotype related to mutations in this gene has been amplified.
Renal involvement can occur at any moment from the first year of life, as in our patient, to adult age. Dysplastic kidneys in fetus have also been described. The most common manifestation is renal cystic disease, including simple cysts, glomerulocystic hypoplasia or cystic dysplasia. Less frequently, we can find solitary functioning kidney, horseshoe kidney, oligomeganephronia or pielocalycial anomalies. Glomerular filtrate usually experiments a slowly progressive decline, with a mean annual loss of 2 mL/min. Nevertheless, severe impairment requiring early dialysis and kidney transplant has also been reported (6).
Diabetes associated to HNF1B mutations is usually manifested as the incidental finding of hyperglycemia in asymptomatic patients but symptomatic hyperglycemia with or without ketosis is also frequent. Although the median age at diagnosis of diabetes is 21 years, it can be found at any age (7). There are even cases of neonatal diabetes of this kind, either transitory or permanent. Reduced pancreatic volume, as our patient’s abdominal magnetic resonance showed, is a very typical radiologic finding. This is probably related to hypoplasia more than atrophy, considering the role of HNF1B in embryogenesis. It has been suggested that, in addition to a primary defect in insulin secretion, a component of insulin resistance is also a pathogenic mechanism in this type of diabetes. A model of hyperglycemic hyperinsulinemic clamp suggested reduced insulin sensitivity in these subjects at hepatic level, since glucose intravenous infusion failed to suppress gluconeogenesis. By contrast, peripheral sensitivity is preserved in this model (8). Chronic complications in HNF1B-related diabetes are not frequently reported in the literature. Anyway, there is a lack of long-term data. Our patient showed no chronic complications after an eight-year follow-up. Regarding therapeutic options, it is known that diabetes secondary to HNF1A mutations is often well responsive to oral agents during long periods of time, sulfonylureas being the first-choice treatment (1). Conversely, clinical results with oral therapy in diabetes related to HNF1B mutations are generally poor and the main treatment for these patients is insulin, in most of the cases initiated within the first three years from the diagnosis (6). Our patient experienced satisfying results with oral treatment, initially in monotherapy with sulfonylureas and later with a combination of agents, making it possible to use exclusive oral therapy for six years. Besides, DDP-4 inhibitors and repaglinide proved to be useful choices both before and after insulinization.
Probably related to pancreatic hypoplasia, pancreatic exocrine insufficiency has also been described (3), being generally subclinical and demonstrated by reduced levels of fecal elastase. Considering that our patient was asymptomatic to this respect, fecal elastase was not evaluated.
Our patient has always presented low magnesium levels, always near 1 mg/dL and without any clinical repercussion. This is another characteristic finding in HNF1B-related syndrome. It seems to be related to renal loss and is typically not corrected by oral supplementation. Although this is usually a mild asymptomatic alteration, one case of tetany has also been reported (9). The pathogenic mechanism seems to be a defective transactivation of FXYD2 gene, which encodes the gamma subunit of the Na-K-ATPase responsible for the renal management of electrolytes.
Hypertransaminasemia is another constant finding of this entity, mostly without any clinical repercussion (3). Some patients present a cholestatic pattern that is present at birth and improves spontaneously during the first year of life, reappearing later to become permanent with fluctuations. One single case of neonatal cholestasis requiring portoenterostomy at 32 days of life has been described (10). In other cases, liver involvement is manifested as an adult-onset cholestasis or as a cytolysis pattern at any age. In the present case, abnormal liver function tests with a mixed pattern were found, without any clinical implication.
Urogenital tract anomalies like epididymal and seminal vesicle cysts found in our patient can also be part of this multi-organic syndrome. Wolffian and Müllerian ducts are affected. Deferent ducts atresia, hypospadias, varicocele, solitary ovary, vaginal aplasia and rudimentary or bicorn uterus are the most frequently reported anomalies.
Amplifying the phenotypic spectrum of the entity even further, relationship with hyperuricemia and juvenile-onset gout has been established. The mechanism is not clear but renal underexcretion of uric acid seems to play a role (3). Asymptomatic hyperuricemia is also present in our patient.
Diagnosis of the syndrome is usually made in the context of studying renal malformations without an identified cause or juvenile diabetes with some of the other aforementioned manifestations. A clinical score has been recently proposed in order to select candidates for the genetic study (11).
In conclusion, we present a young male with a de novo mutation in HNF1B gene and several clinical characteristics of the related syndrome, who experienced favorable clinical results with oral therapy both as initial treatment and subsequent to insulinization.
HNF1B mutations are probably underdiagnosed because of their wide phenotypical variability. In general, we should suspect them both in young non-obese diabetic subjects with slowly progressive nephropathy not suspicious of diabetic etiology and in patients with unexplained renal anomalies, present at any age. Features like urogenital tract malformations, pancreatic hypoplasia, altered liver function tests, hypomagnesemia or hyperuricemia reinforce the probability of such diagnosis. Although these patients have been mostly treated with insulin, oral antidiabetic agents in monotherapy or in combination may also play a role at different stages of their treatment.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent was obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
Dr C Lamas is the patient’s main physician and is a co-author.
References
- 1.Murphy R, Ellard S, Hattersley T. 2008. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nature Clinical Practice Endocrinology and Metabolism 4 200–213. ( 10.1038/ncpendmebib778) [DOI] [PubMed] [Google Scholar]
- 2.Fajans SS, Bell GI, Polonsky KS. 2001. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. New England Journal of Medicine 345 971–980. ( 10.1056/NEJMra002168) [DOI] [PubMed] [Google Scholar]
- 3.Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. 2014. HNF1B-associated renal and extra-renal disease-an expanding clinical spectrum. Nature Reviews Nephrology 11 102–112. ( 10.1038/nrneph.2014.232) [DOI] [PubMed] [Google Scholar]
- 4.Imamura M, Takahashi A, Yamauchi T, Hara K, Yasuda K, Grarup N, Kadowaki T. 2016. Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nature Communications 7 10531 ( 10.1038/ncomms10531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horikawa Y Iwasaki N Hara M Furuta H Hinokio Y Cockburn BN Lindner T, Yamagata K Ogata M, Tomonaga O. 1997. Mutation in hepatocyte nuclear factor-1 beta gene (TCF 2) associated with MODY. Nature Genetics 17 384–385. ( 10.1038/ng1297-384) [DOI] [PubMed] [Google Scholar]
- 6.Farguer S Decramer S Chassaing N Bellanné-Chantelot C Calvas P Beaufils S Bessenay L, Lengelé JP Dahan K, Ronco P et al. 2011. Diagnosis, management, and prognosis of HNF1β nephropathy in adulthood. Kidney International Journal 80 768–776. ( 10.1038/ki.2011.225) [DOI] [PubMed] [Google Scholar]
- 7.Chen YZ, Gao Q, Zhao XZ, Chen YZ, Bennett CL, Xiong XS, Chen XM. 2010. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chinese Medical Journal 123 3326–3333. ( 10.3760/cma.j.issn.0366-6999.2010.22.029) [DOI] [PubMed] [Google Scholar]
- 8.Brackenridge A, Pearson ER, Shojaee-Moradie F, Hattersley AT, Russell-Jones D, Umpleby AM. 2006. Contrasting insulin sensitivity of endogenous glucose production rate in subjects with hepatocyte nuclear factor-1B and -1A mutations. Diabetes 55 405–411. ( 10.2337/diabetes.55.02.06.db05-1019) [DOI] [PubMed] [Google Scholar]
- 9.Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Bockenhauer D. 2009. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. Journal of the American Society of Nephrology 20 1123–1131. ( 10.1681/ASN.2008060633) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kotalova R, Dusatkova P, Cinek O, Dusatkova L, Dedic T, Seeman T, Pruhova S. 2015. Hepatic phenotypes of HNF1B gene mutations: a case of neonatal cholestasis requiring portoenterostomy and literature review. World Journal of Gastroenterology 21 2550–2557. ( 10.3748/wjg.v21.i8.2550) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Faguer S, Chassaing N, Bandin F, Prouheze C, Garnier A, Casemayou A, Chauveau D. 2014. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney International 86 1007–1011. ( 10.1038/ki.2014.202) [DOI] [PubMed] [Google Scholar]