

Abstract
Acute pain is protective and a cardinal feature of inflammation. Chronic pain after arthritis, nerve injury, cancer, and chemotherapy is associated with chronic neuroinflammation, a local inflammation in the peripheral or central nervous system. Accumulating evidence suggests that non-neuronal cells such as immune cells, glial cells, keratinocytes, cancer cells, and stem cells, play active roles in the pathogenesis and resolution of pain. We review how non-neuronal cells interact with nociceptive neurons by secreting neuroactive signaling molecules that modulate pain. Recent studies also suggest bacterial infections regulate pain through direct actions on sensory neurons, and specific receptors are present in nociceptors to detect danger signals from infections. We also discuss new therapeutic strategies to control neuroinflammation for the prevention and treatment of chronic pain.
Introduction
Clinically, inflammation is characterized by five cardinal signs: rubor (redness), calor (increased heat), tumor (swelling), dolor (pain), and functio laesa (loss of function). Acute inflammation is a protective response involving immune cells, blood vessels, and molecular mediators (inflammatory mediators). The function of inflammation is to eliminate the initial cause of cell injury and initiate tissue repair. Acute pain, also known as nociceptive pain, is a cardinal feature of inflammation. The majority of known inflammatory mediators cause pain by binding to their receptors on nociceptive primary sensory neurons in the peripheral nervous system (nociceptors) that innervate injured skin, muscle, and joint tissues(1–3) (Fig. 1). Once thought to be a passive process, the resolution of acute inflammation is now recognized as a distinct, active process involving specialized pro-resolution mediators (SPM) such as resolvins, protectins, and maresins, derived from omega-3 unsaturated fatty acids(2, 4), as well as other pro-resolution mechanisms(5). Resolvins not only regulate the resolution of acute inflammation but also inhibit inflammatory pain through direct actions on nociceptors through specific receptors; activation of ChemR23 by resolvin E1 potently inhibits transient receptor potential ion channel V1 (TRPV1), a key ion channel for pain transduction(4).
Fig. 1.
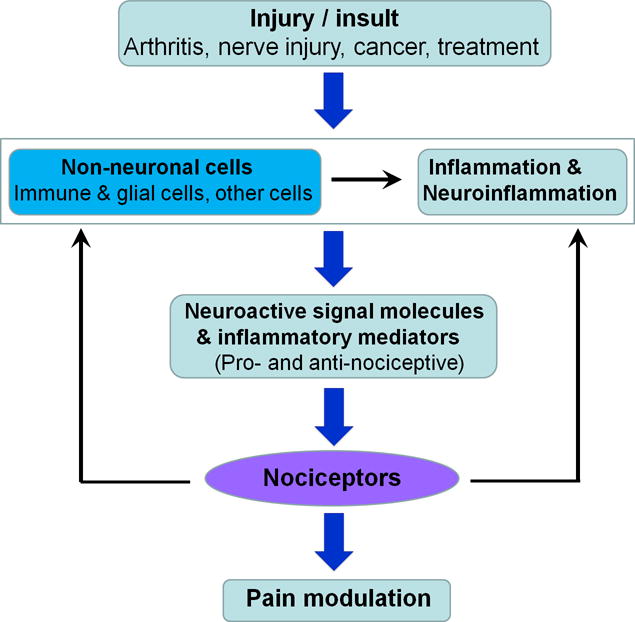
Interactions between non-neuronal cells, neurons, and inflammation/neuroinflammation in different pain conditions after injury and insult. Note that non-neuronal cells can modulate pain in different directions by producing either pro- or anti-nociceptive mediators.
In contrast to acute inflammation, chronic inflammation, is often detrimental, leading to a host of diseases, such as periodontitis, atherosclerosis, rheumatoid arthritis, and even cancer(2). It is unclear if chronic inflammation is also critical for driving chronic pain as acute inflammation is for acute pain. Pain research in the last several decades has established that neuronal plasticity is a key mechanism for the development and maintenance of chronic pain(1, 6). Peripheral sensitization in nociceptors is essential for the development of chronic pain(3) and transition from acute pain to chronic pain(7). Central sensitization (i.e. enhanced responses of pain circuits in the spinal cord and brain) regulates the chronicity of pain, causes the spread of pain beyond the site of injury, and influences the emotional and affective aspects of pain(8).
Neuroinflammation is a localized inflammation occurring in the PNS and CNS, in response to trauma, neurodegeneration, bacterial/viral infection, autoimmunity, and toxin (2, 9). The hallmarkers of neuroinflammation are activation and infiltration of leukocytes, activation of glial cells, and increased production of inflammatory mediators. Neuroinflammation is also associated with changes of vascular cells that facilitate leukocyte infiltration (2, 9). Compared to inflammation, neuroinflammation is more persistent in chronic pain conditions, and therefore, plays a more important role in chronic pain maintenance (2). For example, fibromyalgia, a wide-spread chronic pain syndrome, is associated with small fiber neuropathy and neuroinflammation, although its correlation with systemic inflammation is unclear (10).
The interactions between inflammation and pain are bidirectional (Fig. 1). Nociceptive sensory neurons not only respond to immune signals, but also directly modulate inflammation. For example, nociceptors express receptors for and respond to cytokines and chemokines and also produce these inflammatory mediators (11, 12). In a process called neurogenic inflammation, noxious stimulation causes nociceptors to release neuropeptides such as Substance P (SP) and calcitonin gene-related peptide (CGRP), leading to the extravasation of fluid and cells from the blood. Consistently, silencing nociceptors reduces allergic airway inflammation(13). Nociceptors also serve to dampen and constrain the immune response: ablation of nociceptors abrogated pain during bacterial infection but concurrently worsened inflammation via CGRP(14). Activation of pain circuits also regulates neuroinflammation in the CNS, referred as neurogenic neuroinflammation in chronic pain and neurodegenerative diseases(9).
Numerous non-neuronal cell types influence pain sensation, including immune, glial, epithelial, mesenchymal, cancer, and bacterial cells. In this review, we focus on non-neuronal cells that interact with nociceptors in distinct anatomical compartments in the PNS and CNS (glial cells) under normal and pathological conditions (Fig. 2). Despite the diversity of these cells, the ways in which they modulate pain are surprisingly consistent. In response to an injury or insult, non-neuronal cells release neuromodulatory substances in close proximity to nociceptors, which either promote or dampen pain depending on the specific identities of the mediators involved (Fig. 1 and Fig. 2). Although the details will differ in each case, this general model will serve as a useful lens through which to examine specific non-neuronal cell types in pain.
Fig. 2.
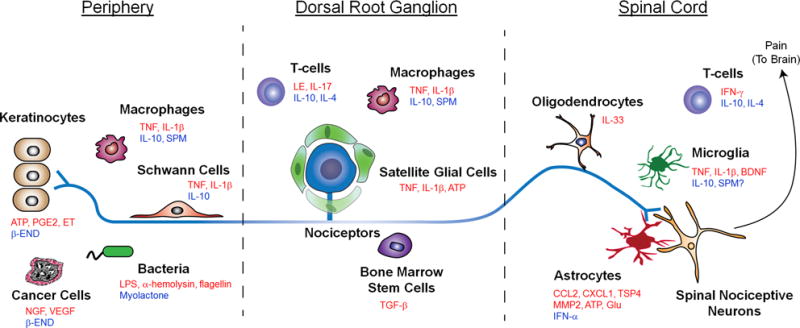
Interactions between distinct parts of a nociceptor with different types of non-neurons cells including keratinocytes, Schwann cells, satellite glial cells, oligodendrocytes, and astrocytes, as well as immune cells (e.g., macrophages and T cells), microglia, cancer cells, and stem cells. These non-neuronal cells produce both pro-nociceptive (highlighted in red) and anti-nociceptive (highlighted in blue) mediators, which can bind their respective receptors on the nociceptor to modulate its sensitivity and excitability. The central terminal of the nociceptor forms a nociceptive synapse with a postsynaptic neuron in the spinal cord dorsal horn to mediate pain transmission in the CNS.
Pain modulation by non-neuronal cells
Monocytes and macrophages
Monocytes and their macrophages serve three main functions in the immune system: phagocytosis, antigen presentation, and cytokine production. Monocytes and macrophages in the periphery play an active role in pain, exhibiting diverse mechanisms that are shaped by the causes and context of pain. In most cases, these cells produce pain through the release of pro-inflammatory mediators such as TNF and IL-1β(11), resulting in enhanced pain transduction and conduction via modulation of ion channels such as TRPA1, TRPV1 and Nav1.7-1.9(1, 2). Cell-specific depletion of proliferating monocytes and macrophages impairs the development of mechanical and thermal hypersensitivity caused by sterile incision and pathogens, in parallel with a decrement in IL-1β and other pro-algesic mediators at the site of inflammation(15). However, in a nerve injury model deletion of peripheral monocytes does not affect neuropathic pain development(16). In a model of chemotherapy-induced neuropathic pain, CX3CR1+ monocytes migrate into peripheral nerves and produce reactive oxygen species, which then elicit pain via the activation of TRPA1(17). After peripheral nerve injury, infiltration of monocytes/macrophages to the spinal cord is limited, arguing against a central role of these cells in neuropathic pain(16, 18). Conversely, monocytes and macrophages can also effect analgesia by releasing anti-inflammatory mediators such as IL-10 and SPMs, together promoting the resolution of the initial insult(2, 4). In support of this view, depletion of monocytes and macrophages delayed the resolution of inflammatory pain(19). Macrophages have different phenotypes related to their functional states, including pro-inflammatory M1-like and anti-inflammatory M2-like phenotypes, which may play distinct roles in the induction and resolution of pain.
T-Lymphocytes
T-cells are critical elements of adaptive immunity that have also been implicated in pain, with the most evidence for a role in neuropathic pain. Following nerve injury, T-cells infiltrate the DRG and release the pro-algesic mediator leukocyte elastase (LE), resulting in mechanical allodynia. Consistently, T-cell-specific ablation of LE or antibody blockade of LE reduces nerve injury-induced allodynia(20). In the spinal cord, T-cell infiltration occurs following nerve injury and is required for the development of mechanical sensitivity, as indicated by a reduction of pain behaviors in T-cell deficient Rag1-null mice(21). A recent report supported the role of spinal T-cells in nerve injury-induced neuropathic pain, but limited this function only to female mice, with male mice instead depending on microglial signaling for pain. This intriguing finding was attributed to the sexually dimorphic expression of peroxisome proliferator activated receptors (PPARs) α and γ in T-cells (22). Of note, different types of T-cells play different roles in chronic pain. For example, adoptive transfer of toxic T cells (CD8+ T cells) via intrathecal injection enhances neuropathic pain, whereas injection of regulatory T cells (Tregs) decreases neuropathic pain after chemotherapy(23).
Keratinocytes
Keratinocytes are the primary cells of the epidermis. They reside near the peripheral terminals of nociceptors and produce various neuroactive mediators such as ATP, IL-1β, prostaglandin E2, endothelin, and NGF that are known to elicit pain, suggesting that these cells can directly activate nociceptors. Photostimulation of channelrhodopsin-expressing keratinocytes is sufficient to generate nocifensive behaviors and evoke action potentials in specific subsets of cutaneous nociceptive afferents(24). Keratinocytes are multifunctional, promoting pain or analgesia depending on the condition and context. The familiar experience of sunburn illustrates the dual nature of these cells. Before the pain of sunburn sets in, the feeling of sun on the skin is a pleasurable experience. Here, keratinocytes participate by releasing the endogenous opioid peptide β–endorphin (β-END), which produces both analgesia and reward(25). After light overexposure, when inflammation and injury occur, keratinocytes also elicit pain via the activation of TRPV4 and the subsequent release of endothelin onto cutaneous nociceptors(26). Although keratinocytes are not immune cells, they can release inflammatory mediators including cytokines(27), illustrating how non-immune cells can also regulate inflammation.
Central glial cells: microglia, astrocytes, and oligodendrocytes
Microglia serve as the resident macrophages of the spinal cord and brain, and abundant evidence supports their role in pathological pain(28, 29). A hallmark of microglia is their rapid activation in response to even minor pathological changes in the CNS. Peripheral nerve injury induces a marked proliferation and activation of microglia in parallel with up-regulation of microglial markers such as IBA1 and CD11b in the spinal cord. Indeed, spinal microglia have been strongly implicated in the pathogenesis of neuropathic pain after nerve injury(18, 29–32). The signals that activate these microglia include ATP, colony-stimulating factor-1 (CSF1), chemokines (CCL2, CX3CL1), and proteases, which can originate from injured or activated sensory neurons (Fig. 3). In parallel, expression of the receptors for ATP and CX3CL1 (P2X4, P2X7, P2Y12, CX3CR1) is increased selectively on spinal microglia in response to nerve injury(28, 29). Activation of these receptors typically converges on an intracellular signaling cascade involving the phosphorylation of p38 MAP kinase, which leads to the increased production and release of TNF-α, IL-1β, IL-18, and brain-derived growth factor (BDNF), as well as increased expression of cyclooxygenase (COX) and subsequent synthesis of prostaglandin E2(6). These neuromodulators can then fine tune both excitatory and inhibitory synaptic transmission, which ultimately enhances pain signal transmission to the brain. For example, TNF and IL-1β enhance excitatory synaptic transmission and suppress inhibitory synaptic transmission in spinal cord lamina II neurons(33). BDNF disrupts chloride homeostasis and suppresses GABAergic inhibitory synaptic transmission (disinhibition) in spinal lamina I projection neurons(6, 34). Selective activation of spinal microglia by fractalkine (CX3CL1) is sufficient to rapidly facilitate synaptic strength between primary afferent C-fibers and lamina I projection neurons(35). Caspase-6 is uniquely expressed in axonal terminals in the spinal cord and mediates neuron-microglial interaction. Recombinant caspase-6 triggers marked TNF release from microglia and drives robust acute inflammatory pain behaviors prior to the morphological activation of microglia(36). Interestingly, spinal microglia signaling is sex-dependent, and microglial signaling inhibitors such as minocycline and p38 inhibitor reduce neuropathic pain primarily in male mice, with little to no effect on female mice(22). While microglia are critical to the development of chronic pain, they may function in the maintenance of chronic pain as well. For example, examination of microglial enhancers using ChIP-Seq revealed persistent alterations in close proximity to transcriptionally regulated genes, leading to the hypothesis that these changes may contribute to the “memory” recorded at a molecular level(18). Microglia are also involved in opioid-induced hyperalgesia through microglia-mediated disruption of neuronal chloride homeostasis(34). Morphine paradoxically prolongs neuropathic pain in rats, and selective inhibition of spinal cord microglia in vivo through DREADD (Designer Receptor Exclusively Activated by Designer Drugs) reverses morphine-induced pain sensitization for weeks(37). In the future, it will also be important to determine whether microglia are involved in the resolution of chronic pain, which would be expected based on the dual role of their peripheral macrophage counterparts(19).
Fig. 3.
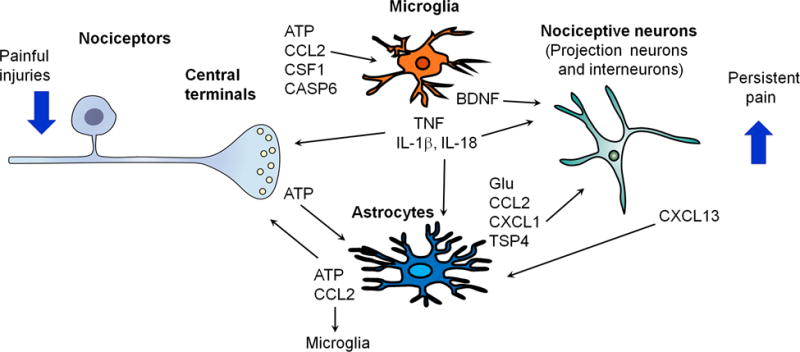
Neuron-glial interactions in the spinal cord for the amplification of chronic pain. Painful injuries such as nerve injury, arthritis, cancer, and treatment (chemotherapy) cause hyperactivity of nociceptors and secretion of glial modulators from their central terminals, leading to the activation of microglia and astrocytes in the spinal cord dorsal horn. Upon activation, microglia and astrocytes secrete neuromodulators to drive chronic pain, by inducing synaptic and neuronal plasticity. Note that pre- and post-synaptic neurons can both “listen” and “talk” to microglia and astrocytes.
Astrocytes perform numerous critical functions such as neurotransmitter recycling, formation of the blood-brain barrier, regulation of extracellular ion concentration, and modulation of synaptic transmission, among many others. Nerve injury induces myriad changes in astrocytes that lead to enhanced pain. For example, after nerve injury, astrocytes lose their ability to maintain the homeostatic concentrations of extracellular potassium (K+) and glutamate, leading to neuronal hyperexcitability(28). Astrocytes can also signal directly to neurons through physically coupled networks mediated by gap junctions to facilitate intercellular transmission. Gap junction communication is mediated by connexin-43 (Cx43), the predominant connexin expressed in astrocytes. Nerve injury induces persistent upregulation of Cx43 in astrocytes and switches the function of Cx43 from gap junction communication to paracrine modulation(38). This paracrine regulation leads to the increased release of glutamate, ATP, and chemokines through a paracrine mechanism. The astrocytes-derived chemokines act as neuromodulators and can potentiate excitatory synaptic transmission in the spinal cord pain circuitry(38). Furthermore, nerve injury upregulates CXCL13 in spinal cord neurons, which can activate astrocytes via CCR5 to maintain neuropathic pain(39). Thus, chemokines facilitate neuropathic pain via bi-directional neuron-astrocyte interactions (Fig. 3). Nerve injury also induces spinal cord and cortical astrocytes to up-regulate thrombospondin-4 (TSP4), which promotes neuropathic pain through the formation of new synapses and rewiring of somatosensory cortical circuits(40, 41). Notably, a single human astrocyte may contact more than 1 million synapses, and such complexity points to a more important role of astrocytes in humans that bears further investigation. As compared to microglial activation, astrocyte activation in chronic pain conditions are more persistent, indicating their contribution to the chronicity of pain(28).
Oligodendrocytes create the myelin sheath that provides support and insulation to axons in the CNS (Fig. 2). In spite of their ubiquity and importance, only recently has a role for oligodendrocytes been revealed in pain. In the chronic constriction injury model of nerve injury-induced neuropathic pain, oligodendrocyte-derived IL-33 contributes to pain hypersensitivity via MAP kinases and NF-κB(42). Similarly, in post-mortem spinal cord samples from HIV patients, expression of oligodendrocyte markers, such as NG2, Oligo2 and PDGFRα, increases, reflecting a persistent activation of oligodendrocytes in chronic pain(43). Conversely, toxin-mediated ablation of oligodendrocytes induces neuropathic pain symptoms, suggesting a potential protective role of these cells(44). These divergent findings suggest that, as with other non-neuronal cells, oligodendrocytes play active and context-specific roles in pain.
Peripheral glia: Schwann cells and satellite glial cells
Like their central counterparts, the major glial cells of the PNS, the Schwann cells and the satellite glial cells (SGCs), contribute to pain. In response to painful stimuli, these peripheral glial cell are activated prior to central glia and release various inflammatory mediators, sensitizing nociceptors at axons (Schwann cells) and cell bodies (SGCs). After nerve injury, activated Schwann cells mediate the breakdown of the blood-nerve barrier via the secretion of MMP-9, which promotes the recruitment of immune cells from the vasculature and their subsequent release of more pro-nociceptive mediators(27). SGCs surround the somata of DRG neurons and are directly coupled to them via gap junctions. Following nerve injury, SGCs become activated and proliferate(27). SGCs contribute to chronic pain sensitization by producing cytokines and MMPs that regulate the cleavage and activation of cytokines(28). Nociceptive activity also causes ATP release from neuronal soma to activate P2X7 in SGCs, leading to TNF release from SGCs and subsequent increase in neuronal excitability(45). Activation of P2X7 receptors in SGCs also reduces pain through downregulation of P2X3 receptors in nociceptive neurons(46).
Stem cells
Bone marrow stromal cells or bone marrow stem cells (BMSCs) produce many beneficial effects to promote tissue regeneration and tissue repair by secreting growth factors. These cells can also effectively control inflammation and neuroinflammation by secreting anti-inflammatory mediators. The duration of pain relief by BMSCs is remarkable following systemic or local injection(47, 48). A single intrathecal injection of BMSCs inhibits nerve injury-induced neuropathic pain for many weeks via secretion of transforming growth factor-beta1 (TGF-β1), a potent anti-inflammatory cytokine(48). The analgesic effects of intrathecal BMSCs was abolished by a neutralizing antibody against TGF-β1. Intrathecal BMSCs effectivity suppress nerve injury-induced glial activation and neuroinflammation in DRGs and spinal cord. After intrathecal injection, BMSC migrate to DRG via a chemotaxic signal (CXCL12) triggered by nerve injury(48). This paracrine modulation of pain by BMSCs is very different from other stem cell strategies for chronic pain management. For example, implantation of forebrain GABAergic precursor cells into the spinal cord reduced nerve injury-induced neuropathic pain, because these precursor cells can differentiate into functional GABAergic neurons in the spinal cord(49).
Cancer cells
Cancer pain is a complex pain state, involving inflammatory, neuropathic, compressive and ischemic mechanisms. Cancer pain is commonly caused by tumors that metastasize from distant sites to the bone and by tumors infiltrating the nerve. Cancers generate and secrete algogenic mediators (e.g. protons, bradykinin, endothelins, prostaglandins, proteases) that sensitize and activate nociceptors in the cancer microenvironment(50). In particular, cancer cells secrete NGF and vascular endothelial growth factor (VEGF) that induce hyper-innervation of pain-mediating nerve fibers in cancer tissues(50, 51). VEGF promotes cancer pain through a non-vascular regulation. VEGF receptor 1 (VEGFR1) is expressed nociceptors, and tumor-derived VEGF family members increase nociceptor excitability and produce pain hypersensitivity through selective activation of VEGFR1(51). Certain cancer cells, such as Walker-256 rat mammary gland carcinoma cells also secret Transforming growth factor-beta (TGF-β). In early stage of cancer, TGF-β acts as a tumor suppressor through anti-proliferative and pro-apoptotic actions. During tumor progression TGF-β becomes an oncogenic factor and causes angiogenesis, immunosuppression, and proliferation, invasion and metastasis of cancers. TGF-β1 also promotes bone cancer pain(52). Thus, this anti-inflammatory cytokine might have different roles in different pain conditions via different signaling mechanisms. Microglia also play an active and distinct role in bone cancer pain. Spinal microglia contribute to the maintenance of bone cancer pain in female rats(53). Bone cancer causes increased ATP secretion in CSF and upregulation of P2X7 receptor in spinal microglia. Activation of P2X7 results in increased synthesis and release of IL-18 via p38 MAP kinase. Spinal inhibition of the P2X7/p-38/IL-18 pathway reduces the advanced-phase of bone cancer pain and suppresses hyperactivity of spinal wide dynamic range neurons(53). This result suggests that spinal cord microglia have an active role in late-phase of bone cancer pain in female rats. Further investigation is warranted to test sex-dependent regulation of microglial signaling in different animal models and also in different phases of chronic pain. Some cancers such as melanoma are not painful in the majority of patients. It is possible that melanoma may produce some analgesic molecules such as β-endorphin to suppress pain (Fig. 2), which can promote the optimal growth of cancers, dampening the warning function of pain.
Pain modulation by bacterial infections through direct interactions with sensory neurons
Infections caused by bacterial, fungal, and viral pathogens are commonly associated with pain. Until recently, it was generally believed that infections cause pain in an indirect manner through the intermediating effects of immune cells and the inflammatory substances they secrete (Fig. 4). However, emerging evidence suggests that pathogens can also directly activate nociceptors and elicit pain using a suite of novel and intriguing mechanisms(14). For example, the bacterium Staphylococcus aureus (S.aureus) possesses a pore-forming toxin called α-hemolysin, which enables it to destroy host cells and also to cause pain by directly forming pores in nociceptors, permitting cation influx and action potential firing through metallopeptidase ADAM10 (14). Lipopolysaccharide (LPS) is the best studied element derived from bacterial cell walls and is also known to produce pain hypersensitivity by sensitizing TRPV1 in nociceptors(54). Bacteria also produce pain by secreting substances that are ligands for receptors on nociceptors, as is the case with N-formyl peptides, which are metabolic byproducts of bacteria that activate nociceptors and elicit pain by binding to the Formyl Peptide Receptor 1 (FPR1), a G-protein coupled receptor. The pain produced through these mechanisms is independent of the immune response(14). Bacterial infection is not always painful, however. Mycobacterium ulcerans, the etiological agent of Buruli ulcer, causes extensive skin lesions but not pain. Mycobacterium ulcerans produces remarkable analgesia, through a specific activation of sensory neurons triggered by the secreted mycobacterial polyketide mycolactone. Mycolactone elicits analgesia through activation of type 2 angiotensin II receptors (AT2Rs), leading to the activation of K2P potassium channels TRAKK (KCNK4) and hyperpolarization of nociceptors(55).
Fig. 4.
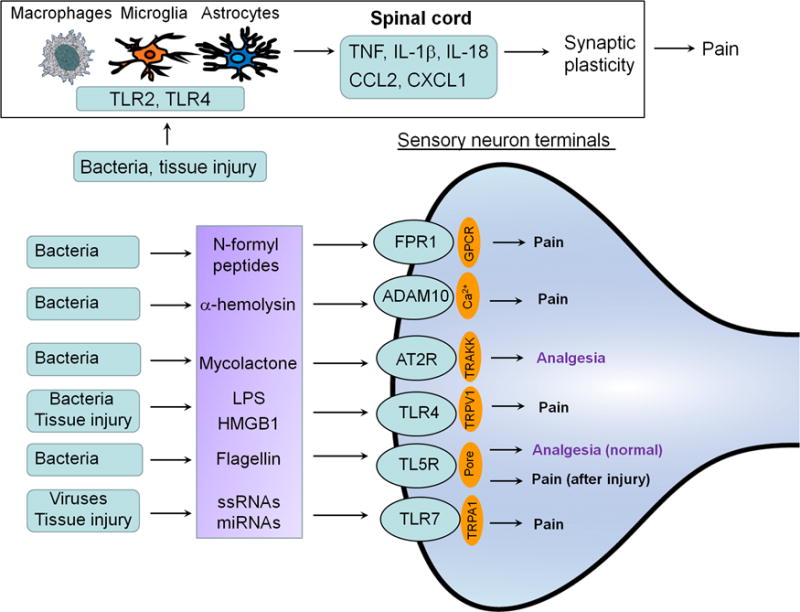
Infections and tissue injury regulate pain via both neuronal and non-neuronal mechanisms through TLRs. Bacterial infections can modulate pain through direct interactions with specific receptors such as FPR and TLR on primary sensory neurons including nociceptors and mechanoceptors, leading to increased and decreased pain sensitivity. TLRs are expressed by neurons, glial cells, and immune cells and are activated by PAMP (infections), as well as DAMP, endogenous TLR ligands (e.g., HMGB1, miRNAs) released after tissue injury.
Although viruses are not cells, viral infections are frequently associated with inflammation and pain, such as in sore throat. Several types of viruses, such as HSV-1, HSV-2, and VZV (varicella zoster virus) can infect sensory neurons to produce painful syndromes. Shingles, also called herpes zoster, is a painful skin rash and usually appears in a defined region of face or body that corresponds to dermatomes. It is most common in older adults and people who have weak immune systems. Viral infections may activate Toll-like receptors (TLRs) in nociceptors to modulate pain and neuroinflammation.
Toll-like receptors mediate bilateral neuron-immune and neuron-glial interactions
Mounting evidence suggests that TLRs are instrumental in bilateral interactions between neurons and non-neuronal cells (Fig. 4). TLRs are typically expressed by immune and glial cells to regulate innate immunity through the activation of the NF-κB and MAP kinase signaling pathways. TLRs are activated not only by PAMP (pathogen-activated molecular patterns) but also by DAMP (danger-activated molecular patterns), the endogenous ligands released after tissue injury. Activation of TLR4 in spinal microglia and astrocytes critically contributes to the development and maintenance of inflammatory and neuropathic pain, as well as opioid-induced hyperalgesia, by eliciting the synthesis of TNF-α and IL-1β (37, 56, 57). Secretion of these cytokines interact with nociceptive neurons in the spinal cord pain circuitry (Fig. 4).
Notably, primary sensory neurons in the DRG and TG also express TLRs(54, 58–60). TLRs are differentially expressed in primary sensory neurons and modulate different sensory functions. TLR4 appears to be functionally coupled with TRPV1 in nociceptors, such that LPS binding to TLR4 increased Ca2+ influx in sensory neurons(54, 60). In contrast to its intracellular localization in immune cells, TLR7 expresses on the cell surface as well as in axons of DRG nociceptors. Strikingly, activation of TLR7 by synthetic ligands causes immediate activation and excitation of nociceptors(61). miRNA let-7b is an endogenous ligand of TLR7 and activates TLR7 through the GUUGUGU motif. Let-7b causes rapid activation and excitation of nociceptors through the coupling of TLR7 and TRPA1. Consistently, intraplantar injection of let-7b elicits rapid spontaneous pain through TLR7 and TRPA1(58).
TLR5 is expressed by large-sized A-fibers, especially Aβ fibers that normally mediate touch sensation. The canonical ligand for TLR5 is flagellin, a globular protein that forms the filament in bacterial flagella. In large A-fibers, activation of TLR5 by flagellin permits the rapid transport of certain small molecules across the neuronal membrane through a to-be-determined mechanism. This Aβ-fiber-specific transport process was exploited to deliver the membrane-impermeable anesthetic QX-314, resulting in selective blockade of A-fibers. Silencing A-fibers in this manner revealed that these neurons inhibit pain under normal conditions but give rise to mechanical allodynia in chemotherapy-induced neuropathy(59), in support of the “Gate Control Theory”. Further studies are warranted to investigate the ion channels and signaling molecules that give rise to this novel function of TLR5.
While coupling of TLRs with ion channels in nociceptors is important for the induction of acute pain, activation of conventional TLR signaling in nociceptors through myeloid differentiation factor 88 (MyD88), a key downstream signaling molecule for TLRs, is required for the development of chronic pain following painful insults(23, 58). Deletion of MyD88 in nociceptors results in deficits in neuropathic pain as well as in innate and adaptive immunity in the PNS following chemotherapy-induced neuropathy(23). Thus, nociceptors share similar features as immune cells by expressing TLRs, cytokines, chemokines, and inflammatory cascades(62). Through the coupling with ion channels, nociceptor TLRs act as a unique class of “danger receptors” for the rapid detection of PAMP and DAMP. Thus, primary sensory neurons not only sense pain, itch, temperature, and touch(63) but also sense “danger” (DAMP and PMAP, which are partially overlapped with algogenic and prurigogenic agents), and this danger sensation will trigger protective responses such as a rapid withdrawal or scratching response(61), as well as delayed innate and adaptive immunity responses through MyD88 signaling in sensory neurons(23). This evolutionarily conserved signaling pathway persists because of its critical biological function but excessive activity of the same pathway can also lead to autoimmune diseases if left unchecked.
Concluding remarks and future directions
Chronic pain is a rising health concern worldwide, affecting up to 30% of adults. Despite this enormous burden, the current treatments for chronic pain are inadequate. Chronic pain typically includes inflammatory pain following tissue injury, cancer pain, and neuropathic pain following nerve injury as a result of diabetic neuropathy, viral infection, major surgeries. Common treatments, such as chemotherapy and chronic opioid treatment, also cause chronic pain. In the past decade great progress has been made to demonstrate the critical roles of glial cells and immune cells, as well as keratinocytes, cancer cells, and stem cells, in the pathogenesis and resolution of chronic pain. These non-neuronal cells can communicate with nociceptive neurons by “listening” and “talking” to neurons, modifying their activities by secreting neuroactive signaling molecules that can be both pro- and anti-nociceptive (Figs 1–3). There is increasing appreciation that many inflammatory mediators such as cytokines and chemokines are indeed neuromodulators in the PNS and CNS(2). Thus, chronic pain could be caused by both “neuropathy” and “gliopathy”(28), as well as dysregulations of other non-neuronal cells (Fig. 2). We extend the boundary of non-neuronal cells to include immune cells, cancer cells, and stem cells, because they can infiltrate damaged or normal tissues to interact with nociceptors and also produce neuroactive modulators to alter pain states. Given the close link between inflammation and pain, we discussed non-neuronal cells in the context of inflammation and especially neuroinflammation(2) that is tightly associated with chronic pain. Inflammation and neuroinflammation can be modulated not only by immune cells, but also by other non-neuronal cells such as keratinocytes, stem cells, and cancer cells, and even by nociceptive neurons in the PNS and CNS. Infections by bacteria and other pathogens not only affect immune and glial cells but also have a direct impact on sensory neurons, eliciting rapid nociceptive responses through activation of neuronal receptors such as FRP1 and TLRs. In particular, TLRs in immune/glial cells and sensory neurons not only sense the danger signals from tissue injury and infections but also regulate bilateral interactions between neurons and neuron-neuronal cells, and these interactions are important for the induction and chronicity of inflammation and pain.
Although several types of non-neuronal cells contribute to the pathogenesis of pain, deletion of a cell type entirely could be detrimental. We should bear in mind that these non-neuronal cells, except for cancer cells, are protective in the normal conditions but dysregulated in pathological conditions. While blocking their function may help pain relief for a short period, restoring their normal function through pro-resolution approaches and is probably the best way. Based on this principle, we proposed the following therapeutic strategies as future directions. First, different pharmaceutical strategies should be considered at different development stages of pain and inflammation. Anti-inflammatory drugs such as inhibitors of TNF are more effective in the early-stage of pain and inflammation. Resolution of inflammation is a new therapeutic frontier(5), and SPMs such as resolvins and protectins may promote the resolution of inflammation and pain, which can be partially achieved via dietary control(2). In particular, neuroprotectin D1 is effective in controlling neuroinflammation and neuropathic pain(64). Second, cell therapies such as implantation of bone marrow stem cells are promising, demonstrating long-term pain relief and even final resolution of pain(47, 48). However, only a very small percentage of cells in bone marrow are stem cells for autologous transplantation. Instead, macrophages are an abundant cell source in blood, and implantation of resident and M2-like macrophages could be an alternative to promote the resolution of inflammation and pain. Alternatively, inducible pluripotent stem cells (iPSC) from abundant cells (e.g. skin, blood) could be transformed into the desired cell type and then transplanted. Moreover, TALEN- or CRISPR-mediated gene editing ex vivo could first be used to enhance the production of pro-resolution mediators and the migration capability of the cells to be transplanted so that they can target the damaged tissue more accurately. Finally, neuromodulation, such as vagus nerve stimulation and acupuncture has been used for inflammation control and pain treatment(65, 66). In addition to local acute analgesia via adenosine from non-neuronal cells(67), acupuncture and sciatic nerve activation also regulates inflammation through vagus nerve activation and dopamine release(68). It will be of great interest to test whether neuromodulation can also help to restore the normal function of non-neuronal cells in chronic pain as a non-pharmaceutical approach.
Acknowledgments
Our research is supported by NIH (R01 grants DE17794, DE22743, NS87988 to R.R.J) and National Natural Science Foundation of China (grant 31420103903 to Y.Q.Z and R.R.J). All the figures have not been published before.
Note 69: Abbreviations in figures and figure legends
- ADAM10
A Disintegrin and metalloproteinase domain-containing protein 10
- CASP6
caspase-6
- CSF1
colony-stimulating factor-1
- DAMP
danger-associated molecular patterns
- END
endorphin
- ET
endothelin
- FPR1
formyl Peptide Receptor 1
- Glu
glutamate
- HMGB1
high mobility group box 1 protein
- IFN-α
interferon-α
- LE
leukocyte elastase
- miRNA
microRNA
- MMP-2
matrix metalloprotease-2
- NGF
nerve growth factor
- PAMP
pathogen-associated molecular patterns
- SPM
specialized pro-resolution mediator
- ssRNA
single-strand RNA
- TSP4
thrombospondin-4
- VEGF
vascular endothelial growth factor
REFERENCES AND NOTES
- 1.Basbaum AI, Bautista DM, Scherrer G, Julius D. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ji RR, Xu ZZ, Gao YJ. Nat Rev Drug Discov. 2014;13:533–548. doi: 10.1038/nrd4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gold MS, Gebhart GF. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu ZZ, et al. Nat Med. 2010;16:592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fullerton JN, Gilroy DW. Nat Rev Drug Discov. 2016;15:551–567. doi: 10.1038/nrd.2016.39. [DOI] [PubMed] [Google Scholar]
- 6.Coull JA, et al. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- 7.Reichling DB, Levine JD. Trends Neurosci. 2009;32:611–618. doi: 10.1016/j.tins.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woolf CJ, Salter MW. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 9.Xanthos DN, Sandkuhler J. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 10.Uceyler N, et al. Brain. 2013;136:1857–1867. doi: 10.1093/brain/awt053. [DOI] [PubMed] [Google Scholar]
- 11.Zelenka M, Schafers M, Sommer C. Pain. 2005;116:257–263. doi: 10.1016/j.pain.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 12.White FA, et al. M Proc Natl Acad Sci USA. 2005;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talbot S, et al. Neuron. 2015;87:341–354. doi: 10.1016/j.neuron.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chiu IM, et al. Nature. 2013;501:52–57. doi: 10.1038/nature12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghasemlou N, Chiu IM, Julien JP, Woolf CJ. Proc Natl Acad Sci USA. 2015;112:E6808–6817. doi: 10.1073/pnas.1501372112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng J, et al. Nat Commun. 2016;7:12029. doi: 10.1038/ncomms12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Old EA, Nadkarni S, Grist J, Gentry C, Bevan S. J Clin Invest. 2014;124:2023–2036. doi: 10.1172/JCI71389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denk F, Crow M, Didangelos A, Lopes DM, McMahon SB. Cell reports. 2016;15:1771–1781. doi: 10.1016/j.celrep.2016.04.063. [DOI] [PubMed] [Google Scholar]
- 19.Willemen HL, et al. J Pain. 2014;15:496–506. doi: 10.1016/j.jpain.2014.01.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vicuna L, et al. Nat Med. 2015;21:518–523. doi: 10.1038/nm.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costigan M, et al. J Neurosci. 2009;29:14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sorge RE, et al. Nat Neurosci. 2015;18:1081–1083. doi: 10.1038/nn.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu XJ. Cell Res. 2014;24:1374–1377. doi: 10.1038/cr.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumbauer KM, et al. eLife. 2015;4 doi: 10.7554/eLife.09674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fell GL, Robinson KC, Mao J, Woolf CJ, Fisher DE. Cell. 2014;157:1527–1534. doi: 10.1016/j.cell.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore C, et al. Proc Natl Acad Sci USA. 2013;110:E3225–3234. doi: 10.1073/pnas.1312933110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calvo M, Dawes JM, Bennett DL. Lancet Neurol. 2012;11:629–642. doi: 10.1016/S1474-4422(12)70134-5. [DOI] [PubMed] [Google Scholar]
- 28.Ji RR, Berta T, Nedergaard M. Pain. 2013;154(Suppl 1):S10–28. doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grace PM, Hutchinson MR, Maier SF, Watkins LR. Nat Rev Immunol. 2014;14:217–31. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsuda M, et al. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- 31.Clark AK, Yip PK, Grist J, Gentry C, Staniland F AA, et al. Proc Natl Acad Sci USA. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan Z, et al. Nat Neurosci. 2016;19:94–101. doi: 10.1038/nn.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawasaki Y, Zhang L, Cheng JK, Ji RR. J Neurosci. 2008;28:5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferrini F, et al. Nat Neurosci. 2013;16:183–92. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clark AK, et al. J Neurosci. 2015;35:4552–4570. doi: 10.1523/JNEUROSCI.2061-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berta T, et al. J Clin Invest. 2014;124:1173–1186. doi: 10.1172/JCI72230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grace PM, et al. Proc Natl Acad Sci USA. 2016;113:E3441–50. doi: 10.1073/pnas.1602070113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen G, et al. Brain. 2014;137:2193–2209. doi: 10.1093/brain/awu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang BC, et al. J Clin Invest. 2016;126:745–761. doi: 10.1172/JCI81950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim DS, et al. J Neurosci. 2012;32:8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim SK, et al. J Clin Invest. 2016;126:1983–1997. doi: 10.1172/JCI82859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zarpelon AC, et al. FASEB J. 2016;30:54–65. doi: 10.1096/fj.14-267146. [DOI] [PubMed] [Google Scholar]
- 43.Shi Y, Shu J, Liang Z, Yuan S, Tang SJ. Molecular pain. 2016;12 doi: 10.1177/1744806916656845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gritsch S, et al. Nat Commun. 2014;5:5472. doi: 10.1038/ncomms6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Chen Y, Wang C, Huang LY. Proc Natl Acad Sci USA. 2007;104:9864–9869. doi: 10.1073/pnas.0611048104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y, et al. Proc Natl Acad Sci USA. 2008;105:16773–16778. doi: 10.1073/pnas.0801793105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo W, et al. Stem Cells. 2011;29:1294–1303. doi: 10.1002/stem.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen G, Park CK, Xie RG, Ji RR. J Clin Invest. 2015;125:3226–3240. doi: 10.1172/JCI80883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Braz JM, et al. Neuron. 2012;74:663–675. doi: 10.1016/j.neuron.2012.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mantyh P. Pain. 2013;154(Suppl 1):S54–S62. doi: 10.1016/j.pain.2013.07.044. [DOI] [PubMed] [Google Scholar]
- 51.Selvaraj V D, et al. Cancer Cell. 2015;27:780–796. doi: 10.1016/j.ccell.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Q, et al. J Neurosci. 2013;33:19099–19111. doi: 10.1523/JNEUROSCI.4852-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang Y, et al. J Neurosci. 2015;35:7950–7963. doi: 10.1523/JNEUROSCI.5250-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. J Dent Res. 2011;90:759–764. doi: 10.1177/0022034511400225. [DOI] [PubMed] [Google Scholar]
- 55.Marion E, et al. Cell. 2014;157:1565–1576. doi: 10.1016/j.cell.2014.04.040. [DOI] [PubMed] [Google Scholar]
- 56.Tanga FY, Nutile-McMenemy N, DeLeo JA. Proc Natl Acad Sci USA. 2005;102:5856–61. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christianson CA, et al. Pain. 2011;152:2881–2891. doi: 10.1016/j.pain.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Park CK, et al. Neuron. 2014;82:47–54. doi: 10.1016/j.neuron.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu ZZ, et al. Nat Med. 2015;21:1326–31. doi: 10.1038/nm.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, et al. J Neurosci. 2015;35:13487–13500. doi: 10.1523/JNEUROSCI.1956-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Nat Neurosci. 2010;13:1460–1462. doi: 10.1038/nn.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiu IM, von Hehn CA, Woolf CJ. Nat Neurosci. 2012;15:1063–1067. doi: 10.1038/nn.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zimmerman A, Bai L, Ginty DD. Science. 2014;346:950–954. doi: 10.1126/science.1254229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu ZZ, et al. Ann Neurol. 2013;74(3):490–5. doi: 10.1002/ana.23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borovikova LV, et al. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 66.da Silva MD, et al. Mol Neurobio. 2015;51:19–31. doi: 10.1007/s12035-014-8790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goldman N, et al. Nat Neurosci. 2010;13:883–888. doi: 10.1038/nn.2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Torres-Rosas R, et al. Nat Med. 2014;20:291–295. doi: 10.1038/nm.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]