

Abstract
Objective
We built India Allele Finder, an online searchable database and command line tool, that gives researchers access to variant frequencies of Indian Telugu individuals, using publicly available fastq data from the 1000 Genomes Project. Access to appropriate population-based genomic variant annotation can accelerate the interpretation of genomic sequencing data. In particular, exome analysis of individuals of Indian descent will identify population variants not reflected in European exomes, complicating genomic analysis for such individuals.
Results
India Allele Finder offers improved ease-of-use to investigators seeking to identify and annotate sequencing data from Indian populations. We describe the use of India Allele Finder to identify common population variants in a disease quartet whole exome dataset, reducing the number of candidate single nucleotide variants from 84 to 7. India Allele Finder is freely available to investigators to annotate genomic sequencing data from Indian populations. Use of India Allele Finder allows efficient identification of population variants in genomic sequencing data, and is an example of a population-specific annotation tool that simplifies analysis and encourages international collaboration in genomics research.
Keywords: Population genomics, India, Variant annotation, Whole exome sequencing
Introduction
Whole exome sequencing (WES) has revolutionized genomic diagnostics and is a key tool in identifying the causal genes underlying rare Mendelian disorders [1–3]. A critical strategy in post-sequencing analysis involves screening a proband’s exome variants against exomes from reference individuals matching the ethnic makeup of the proband. While these data are widely available for individuals from European and African American descent [4, 5], such reference data is less accessible when analyzing exomes from individuals from India. We present India Allele Finder (IAF), an online database table of allele frequencies of individuals from the Indian subcontinent.
The 1000 Genomes web browser (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) effectively presents complete allele frequencies, but rapid queries are more difficult, and annotation of local variant call files (vcfs) is not possible. In contrast, the IAF website and its accompanying command line tool are focused only on the South Indian population, and allow researchers to easily annotate their own exome data sets. Clinicians who want a more ordered method of browsing 1000 Genome data will find the query-based website intuitive to use, while bioinformaticians who work with vcfs will easily adopt the IAF command line tool into their workflow.
Main text
Accessing 1000 Genomes data
Fastq data of individuals specific to Indian populations (flagged with “ITU” indicating Indian Telugu ancestry) available via the 1000 Genomes Project [6] were aggregated via ftp from the 1000 Genomes Project, and combined into two fastq files per individual, one per paired end read. We downloaded 100 fastqs out of 118 available ITU individuals from the 1000 Genomes data set. Automated shell scripts facilitated the downloading of fastq files, while an aggregator written in Python concatenated fastqs of the appropriate paired end such that each individual had two fastq files of equal size.
Data analysis
Fastqs were mapped with the Burrows–Wheeler alignment (BWA) tool 0.7.9a to hg19. The resulting bam files were then analyzed with SAMtools 0.1.19, Picard 1.114, and the Genome Analysis Toolkit (GATK) 3.1.1. Annotation of resulting vcfs was performed with Annovar. A command line Python script, indiaAlleleAnnotator.py, takes as its input a tab delineated vcf and outputs a modified vcf with an additional column representing the allele frequency among the Indian Telugu population.
Database schema
The vcf generated from the analysis was converted into structured query language (SQL) format, and imported into mysql v.14.14 database as one table. The database is accessed on-line via a Perl Catalyst front-end. The files for this implementation, including the raw SQL file, are available at https://github.com/Paciorkowski-Lab/IndiaAlleleFinder.
IAF allows query of variants through its web-based database, as well as providing a command line tool to annotate exome vcfs. Accepted formats for the web-based query include gene symbol, variant genomic location, or rsID number. The command line annotation tool identifies variants that are present in the IAF data set, and therefore likely to be population variants that may be excluded from further analysis in disease gene identification studies. The IAF workflow is represented in Fig. 1.
Fig. 1.
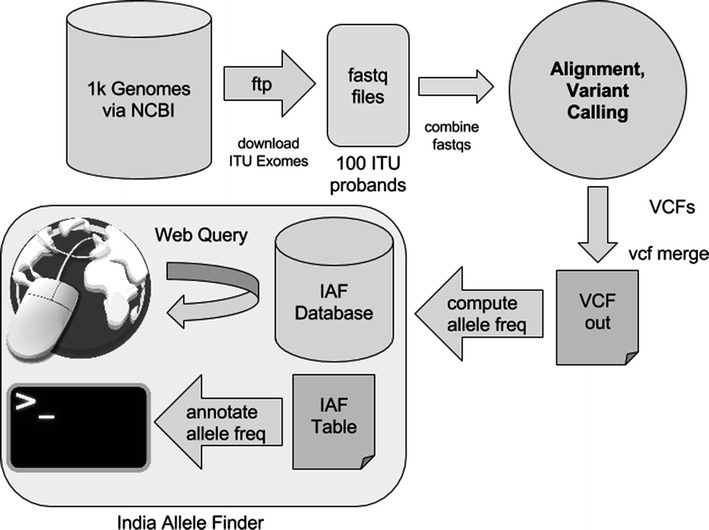
Workflow of analysis of publicly available ITU fastqs from 1000 Genomes used to construct the IAF dataset. Users wishing to annotate exome results with frequency data from IAF may do so using web-based or the command-line interface
IAF use case study
Subjects MP14-001a1, MP14-001a2, two siblings presenting with achalasia–addisonianism–alacrima syndrome (AAAS), as well as the father and mother, were selected for study. Saliva-derived DNA underwent WES using the Agilent Sure-Select 50 Mb whole exome capture kit, and 100 basepair paired-end reads were generated on an Illumina HiSeq 2500 machine at the University of Rochester Genomics Research Center. Sequence was aligned, analyzed as described previously. De novo, autosomal recessive, and X-linked variants were identified and common variants in the database of single nucleotide polymorphisms (dbSNP) version 137 excluded. We then used IAF to identify and exclude variants found in the 100 Telugu Indian individuals from 1000 Genomes. After filtering by pedigree hypothesis, candidate variants were reduced from 84 to seven when using IAF. We found that MP14-001a1 and MP15-001a2 were homozygous for c.43C>A/p.Q15K variant, a known AAAS sequence variation [7]. Their mother and father were both heterozygous for this variant.
The analysis of exome data from populations other than European and African American can be challenging due to difficulty accessing appropriate normal population data sets. This can result in an excess of candidate variants in disease gene identification studies. We have designed IAF to fit into existing workflows.
There are differences between results reported in 1000 Genomes vs IAF. Overall, the IAF data set reports fewer variants, likely due to our use of the newer version GATK v3.1.1 versus v2.4 [8]. Additionally, we sampled from a smaller group of 100 individuals. 1000 Genomes overall collected data from 2535 individuals from 26 different populations for their phase 3 study. As a result, 1000 Genomes aggregated over 5.2 million entries for chromosome 5 alone. Our data set for chromosome 5 contains 8520 entries aggregated from 100 individuals. We anticipate more variants will be represented in IAF as more exomes from the Indian continental population are added.
Limitations
IAF is a proof of concept implementation of a filtering mechanism based on population-derived variant frequencies. It is a unique tool to further annotate vcfs for the specific purpose of analyzing WES data from individuals of Indian subcontinent descent. We anticipate a proliferation of reference databases for populations that are not of European origin. Additional features are planned for the IAF website, including the ability to input multiple variants, and access a subset of the vcf output corresponding to the genes and/or variants queried. Further exome data sets from individuals of continental Indian ancestry will be added in the future as they become available.
Authors’ contributions
JFZ study design, acquisition, analysis, and interpretation of data, manuscript preparation. FJ analysis of data, manuscript preparation. AS acquisition of data, manuscript preparation. KMG acquisition of data, manuscript preparation. ARP study conception and design, acquisition, analysis, and interpretation of data, manuscript preparation. All authors read and approved the final manuscript.
Acknowledgements
We would like to acknowledge the University of Rochester Genomics Research Center for sequencing support, and the University of Rochester Center for Integrated Research Computing for providing high-performance computing resources.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
IAF is hosted at https://iaf.urmc.rochester.edu. The source code and SQL file are available to download at https://github.com/Paciorkowski-Lab/IndiaAlleleFinder, where instructions are provided to set up a local instance of the database backend to IAF. Vcfs may be annotated with IAF data using a Python script available at https://www.iaf.urmc.rochester.edu/static/assets/commandline_vcf_annotator.tar.gz. This allows for integration of IAF into command-line workflows.
Consent for publication
Individuals in this study underwent informed consent through research protocols approved by the Research Subjects Review Board of the University of Rochester Medical Center and the research ethics board of Manipal University, which included consent to publish.
Ethics
Individuals in this study underwent informed consent through research protocols approved by the Research Subjects Review Board of the University of Rochester Medical Center and the research ethics board of Manipal University.
Funding
Research reported in this work was supported by the National Institutes of Health, National Institute of Neurologic Disorders and Stroke under Award Number K08NS078054 (to A.R.P.).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AAAS
achalasia–addisonianism–alacrima syndrome
- BWA
Burrows–Wheeler alignment tool
- dbSNP
database of single nucleotide polymorphisms
- GATK
Genome Analysis Toolkit
- ITU
Telugu
- SQL
structured query language
- vcf
variant call file
- WES
whole exome sequencing
Contributor Information
Jimmy F. Zhang, Email: jfz8009@rit.edu
Francis James, Email: fjames1@lion.lmu.edu.
Anju Shukla, Email: dranju2003@yahoo.co.in.
Katta M. Girisha, Email: girishkatta@gmail.com
Alex R. Paciorkowski, Phone: (585) 275-2808, Email: Alex_Paciorkowski@urmc.rochester.edu
References
- 1.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu Y-F, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D, et al. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med. 2015;17:774–781. doi: 10.1038/gim.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston JJ, Biesecker LG. Databases of genomic variation and phenotypes: existing resources and future needs. Hum Mol Genet. 2013;22:R27–R31. doi: 10.1093/hmg/ddt384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song W, Gardner SA, Hovhannisyan H, Natalizio A, Weymouth KS, Chen W, Thibodeau I, Bogdanova E, Letovsky S, Willis A, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: insights of relevance to variant classification. Genet Med. 2015;18:850–854. doi: 10.1038/gim.2015.180. [DOI] [PubMed] [Google Scholar]
- 6.1000 Genomes Project Consortium. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papageorgiou L, Mimidis K, Katsani KR, Fakis G. The genetic basis of triple A (Allgrove) syndrome in a Greek family. Gene. 2013;512:505–509. doi: 10.1016/j.gene.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
IAF is hosted at https://iaf.urmc.rochester.edu. The source code and SQL file are available to download at https://github.com/Paciorkowski-Lab/IndiaAlleleFinder, where instructions are provided to set up a local instance of the database backend to IAF. Vcfs may be annotated with IAF data using a Python script available at https://www.iaf.urmc.rochester.edu/static/assets/commandline_vcf_annotator.tar.gz. This allows for integration of IAF into command-line workflows.