

Abstract
Background
Research involving the cold shock gene cspA of the medically important bacterium Staphylococcus aureus is steadily increasing as the relationships between the activity of this gene at 37 °C and a spectrum of virulence factors (e.g., biofilm formation, capsule production) as well as stress-related genes (e.g., alkaline shock protein, asp-23 and the alternative sigma factor, sigB) are distinguished. Fundamental to each of these discoveries is defining the regulation of cspA and the production of its protein product CspA.
Results
In this paper, primer extension analysis was used to identify a transcriptional start point at 112 bp upstream of the initiation codon of the cspA coding sequence from S. aureus Newman RNA collected at 37 °C. Based on the location of the putative −10 and −35 sites as well as putative cold shock protein binding sites, a 192 bp sequence containing an 80 bp promoter + a 112 bp 5′ UTR was generated by polymerase chain reaction. The activity of this 192 bp sequence was confirmed in a pLL38 promoter::xylE reporter gene construct. In addition, Western blots were used to confirm the production of CspA at 37 °C and demonstrated that production of the protein was not constitutive but showed growth-dependent production with a significant increase at the 6 h time point.
Conclusions
The results presented identify another regulatory region for the cold shock gene cspA of S. aureus and show growth-dependent activity of both this cspA regulatory sequence, presented as a 192 bp sequence of promoter + 5′ UTR and the production of the CspA protein at 37 °C. The presence of two active transcription start points, a −112 bp sequence defined in this work and a second previously defined at −514 bp upstream of the cspA initiation codon, suggests the possibility of interactions between these two regions in the regulation of cspA. The growth-dependent production of the cold shock protein CspA supports the availability of this protein to be a modulator of virulence and stress factor genes at 37 °C.
Keywords: Staphylococcus aureus, Cold shock, cspA regulation, CspA protein, xylE activity, Western blot, Total protein normalization
Background
Staphylococcus aureus is a pathogen that effects populations of companion and food animals as well as humans. The treatment options for this bacterium have become more complicated due to both community-acquired and hospital acquired MRSA, methicillin resistant S. aureus strains. These multidrug-resistant strains can be identified in each of these populations with the potential of anthropozoonosis [1]. As the availability of effective antibiotic treatment becomes limited, basic molecular research has become a source of identifying targets for the development of antimicrobial agents. A key component for the future treatment of bacterial diseases is the identification and disruption of the regulatory molecules responsible for modulating the genes coding for these virulence factors [2]. One such regulatory molecule and potential target for disruption in S. aureus is the cold shock protein, CspA.
The cold shock gene cspA and protein of S. aureus have a sequence and structure similar to that of both Escherichia coli cspA and Bacillus subtilis cspB cold shock genes and protein products [3–8]. This gene was not initially recognized in cold shock induction experiments, but identified in testing the susceptibility of S. aureus 8325-4 Tn551 insertional mutants to the antimicrobial peptide CG117-136 at 37 °C [9, 10]. The loss of cspA function resulted in an S. aureus strain with decreased susceptibility to this 20-mer peptide. Because the S. aureus 8325-4 strain carries a regulator of sigma B, rsbU mutation, the Tn551 insertional mutation was transduced into a rsbU repaired strain, S. aureus SH1000 [11]. The susceptibility remained but a new phenotype of carotenoid pigment reduction was identified. This pigment reduction was confirmed by a cspA::kanamycin cassette knockout and trans complementation in a highly pigmented strain of S. aureus COL. This was an indication that the cold shock protein CspA may have specific regulatory functions and was the first cold shock protein in S. aureus to be linked to virulence factors [12].
Subsequent RT-qPCR, real time-quantitative polymerase chain reaction, experiments in a cspA mutant of S. aureus COL resulted in a decrease in transcripts from crtN, one gene in the staphyloxanthin, a carotenoid pigment, operon, sigB, the virulence and stress alternative sigma factor B and asp-23 (a SigB-regulated gene encoding the alkaline shock protein) [13–16]. These results were not due to the degradation of mRNA but to regulation of each gene by the cold shock protein CspA [12].
Since these initial findings, researchers have identified other distinct regulatory links among the presence of the cspA gene, its protein product, and other virulence factors of S. aureus. Among these are the modulation of a major regulator of virulence, the staphylococcal accessory regulator, sarA, the production of capsule in a nutrient dependent fashion, the cap operon, and the production of biofilm [17–19]. In relation to its regulation of sarA, cspA can now been found in the literature as the modulator of sarA, msaB, and has been reported as part of a four gene operon, msaABCR [20]. As part of the investigation into the regulation of the component genes of the msaABCR operon in S. aureus USA300_LAC, an identified 324 bp putative promoter for msaB (cspA) was reported as inactive and the primary promoter for the regulation of msaB (cspA) was identified as that for the msaA gene with a transcriptional start point (TSP) 514 bp upstream of the msaB (cspA) coding sequence [20]. In this current work, we propose a second regulatory region consisting of a 192 bp sequence and composed of a promoter + 5′ untranslated region (UTR) with a TSP −112 bp immediately upstream of the coding sequence for cspA. The growth-dependent activity of this sequence demonstrated by a cspA promoter + 5′ UTR::xylE reporter gene construct in S. aureus Newman [21] and the growth-dependent production of the protein CspA are presented.
Methods
Bacterial strains and plasmids
The bacteria and plasmids used or constructed for the work completed in this paper are listed in Table 1. For the construction and selection of plasmids as well as the overexpression of the cold shock protein CspA using E. coli, all E. coli strains were grown in Luria–Bertani, (LB) broth and on LB agar. All S. aureus strains were grown in tryptic soy broth (TSB) and on tryptic soy agar (TSA). For the broth cultures used in the XylE assays, S. aureus strains were grown in TSB. When required the antibiotics added to cultures were: for S. aureus, 20 μg/mL chloramphenicol, 25 μg/mL kanamycin, 3 μg/mL tetracycline; for E. coli, 50 μg/mL ampicillin, 50 μg/mL kanamycin, 25 μg/mL chloramphenicol, 50 μg/mL spectinomycin.
Table 1.
Bacterial strains and plasmids
| Escherichia coli | ||
| DH5α | F− Φ80 lacZ ΔM15Δ(lacZA-argF) U169 deoR recA1 endA1 hsdR17 (r− K m− K) phoA supE44 λ− thi-1 gyrA96 relA1 | Invitrogen™ |
| BL21(DE3)pLysS | F− ompT hsdS B(r−B,m−B) gal dcm (DE3) pLysS (CamR) | Invitrogen™ |
| SKEPrtEx | E. coli BL21(DE3)pLysS with plasmid pSKPrtExA | This study |
| Staphylococcus aureus | ||
| RN4220 | res − strain | [23] |
| SKC31 | A Km cassette deletion–insertion of the cspA coding region in S. aureus COL, TetR | [10] |
| Newman | Clinical isolate, strong positive coagulase | [21] |
| SKN23 | A Km cassette deletion–insertion of the cspA coding region in S. aureus Newman | This study |
| Plasmids | ||
| pCR2.1 | TA cloning vector | Invitrogen™ |
| pSKCPriEx | pCR2.1 with 238 bp insert of cspA coding region, promoter + 5′ UTR | This study |
| pLL38 | E. coli-staphylococcal shuttle vector that contains promoterless xylE as a reporter gene for promoter-gene fusion; gram-positive RBS (GGAGG) | [29] |
| pSK51 | pLL38 containing a putative sequence of −35 and −10 sites plus P3, −53 bp putative TSP of cspA, ligated to a xylE reporter gene | This study |
| pSK88 | pLL38 containing a putative sequence of −5 and −10 sites plus P2, −88 bp putative TSP of cspA ligated, to a xylE reporter gene | This study |
| pSK96 | pLL38 containing a putative 96 bp sequence of the cspA promoter: upstream protein binding sites, −35 and −10 sites ending at P1, −112 bp putative TSP of cspA ligated to a xylE reporter gene | This study |
| pSK192 | pLL38 containing the sequence of pSK96 + 5′ UTR of cspA | This study |
| pSK324 | pLL38 containing msaB promoter insert from pMOE 482; 5′ PstI and 3′ EcoRI sites | [20]; This study |
| pRHB152 | pRHB153 with six His-tag replacement of GST-tag for protein overexpression | [25] |
| pSKPrtExpA | pRHB152 with six His-tag 200 bp coding region for CspA overexpression | This study |
Transduction to produce the cspA mutant of S. aureus Newman
Phage 80α propagated on S. aureus COL strain SKC31 [10] was used to transduce a kanamycin (Km) cassette deletion–insertion of the cspA coding region to S. aureus Newman and generate strain SKN23 (CY. Lee, personal communication). In brief, a 3 mL overnight culture of S. aureus Newman grown at 37 °C was diluted 1:100 in 10 mL of TSB and grown at 37 °C for 2 h. The cells were centrifuged and suspended in 0.2 mL of TSB. After the addition of 25 μL of 10 mg/mL CaCl2 and 30 μL of phage 80α propagated on SKC31, the infected cells were incubated at room temperature for 10 min. After a 35 min static incubation of the infected cells at 30 °C, 5 mL of TSB was added, the cells centrifuged, and the pellet suspended in 5 mL of TSB. The infected cells were incubated at 37 °C for 1.5 h. The cell pellet was suspended in 0.2 mL of TSB and plated onto three TSA plates containing 25 µg/mL of kanamycin and incubated at 37 °C for selection of cspA::Km deletion–insertion mutants. The replacement of the cspA coding sequence by the Km cassette in a selected S. aureus Newman colony SKN23 was confirmed by PCR, polymerase chain reaction.
Primer extension analysis of the 5′ UTR of cspA
Primer extension was carried out with RNA collected from S. aureus Newman cells (QIAGEN RNeasy kit) grown at 37 °C, harvested at 0.3–0.4 at OD600, and diluted to a CFU/mL of 1 × 108. From the total RNA isolated from these cells, a 20 μg aliquot was first annealed to the 5′-32P-labeled primer R-CPriEx and then combined with AMV reverse transcriptase (Promega) to generate complementary DNA (cDNA) primer extension fragments (Table 2). The template for a parallel GATC sequencing reaction was provided by pSKCPEx that contains a 238 bp putative cspA promoter region and coding sequence. The 238 bp insert is a PCR, (ARKTIK Thermocycler, Thermo Fisher) product of the primers F-CspA238 and R-CspA238 combined with genomic DNA (gDNA) from S. aureus Newman (Qiagen DNeasy Blood and Tissue kit) (Table 2). The 238 bp product was gel purified (QIAGEN Qiaquick Gel Extraction kit), ligated into a pCR2.1 vector (Invitrogen™) and transformed into E. coli DH5α. The transformants were selected on LB agar containing 40 μg/mL X-Gal and 50 μg/mL kanamycin. The presence of a 238 bp insert in the pSKCPEx plasmid was confirmed by restriction digestion with EcoRI (Promega) and DNA sequencing (DNA Laboratory at Arizona State University). Using a Sequenase 2.0 sequencing kit (Affymetrix), unlabeled primer R-CPriEx annealed to 4 μg of pSKCPEx and then labeled with dATP-α-33P was used to create each of the GATC sequencing reactions. The labeled primer extension cDNA was loaded onto an 8% polyacrylamide gel and run simultaneously with the DNA sequencing reactions of pSKCPEx. Primer extension fragments and the GATC sequence were detected by phosphor screen imaging (GE Healthcare) and analyzed for transcriptional start points (TSP).
Table 2.
Primers
| R-CPriEx | CCATTTAACTGTACCTTGTTTCATAATCTGAAACC |
| F-CspA238 | GTTTCATTGTTTACAAAATAATGAAGTATATT |
| R-CspA238 | CCTTGTTTCATAATCTGAAACCTCC |
| F-P1/96-192 | CAAAATACTGCAGTATATTATAAACTACC |
| F-P2/88 | GTACTGCAGTTTGGTAATAACTGC |
| F-P3/51 | GTTAAGCTGCAGATTATTCCATATTGC |
| R-P3′ | CCTCGAATTCTAAAATTCATTCAATATGC |
| R-P96 | GAATTCTGCTTAACTTGTATTATAGTGC |
| F-P324 | TGCGAAGATCTGCAGGAGGATTACAAATATTTTA |
| R-P324 | ACGTCATTGAATTCTTCAACTTCGATAAAGCC |
Construction of the cspA promoter region ± the 5′ UTR::xylE reporter plasmids
For the construction of each of the putative cspA promoter region or promoter region + 5′ UTR::xylE reporter gene plasmids (Table 1), a standard protocol was used. In brief, a set of 5′ and 3′ primers (Table 2) for each of the five pLL38 xylE inserts, P3 (51 bp), P2 (88 bp), cspA promoter (96 bp), cspA promoter + 5′ UTR (192 bp), and a 324 bp sequence, was combined with gDNA extracted from overnight cultures of S. aureus Newman for a PCR. The primer R-P3′ was paired with each of the 5′ primers for all PCR except the 96 bp cspA promoter which required primer R-P96. The 5′ primer for the 192 and 96 bp inserts is the same, F-P1/96-192. Each PCR fragment was gel-purified and ligated into the cloning vector pCR2.1 and transformed into E. coli DH5α to provide plasmids (Qiagen QIAprep Spin Miniprep Kit) to confirm the sequence of each insert. Each insert was removed by a double-digest, PstI and EcoRI, of the pCR2.1 construct, gel purified and ligated into a similarly digested xylE reporter vector pLL38 [29] and transformed into E. coli DH5α. After confirmation of the sequence and orientation in pLL38, plasmids were first transformed into S. aureus RN4220 by electroporation using a Gene Pulser Xcell™ (Bio-Rad) followed by extraction and then electroporation into the target strain, S. aureus Newman [21–23].
Assays for cspA::xylE reporter gene constructs
The protocol published by Torres et al. [24] was adapted for these assays. In brief, 25 mL of TSB with no antibiotics were inoculated (initial OD600 between 0.074 and 0.080) from an overnight culture of a cspA::xylE reporter construct. The culture was grown at 37 °C. Optical density (OD600) was measured (0–6 h) and samples were taken after 1 h (8 mL), 3 h (4 mL) and 6 h (0.5 mL) of growth. Times are comparable to pre-logarithmic, mid-logarithmic and post-logarithmic growth. Each sample was centrifuged and the pellet suspended in potassium phosphate buffer (20 mM potassium phosphate buffer pH 7.5) and centrifuged. The cell pellets were immediately stored at −80 °C and the XylE assay performed within 24 h after sample collection. The protein from each pellet was extracted with 150 μL of lysis buffer (100 mM potassium phosphate buffer pH 8.0; 10% (v/v) acetone; 25 μg/mL lysostaphin), incubated at 37 °C for 20 min and placed on ice for 5 min before a final centrifugation.
For each assay, a 20 μL volume of the supernatant from each timed sample was combined with 200 μL of the reaction substrate (22.5 mL ddH2O; 2.5 mL 1 M potassium phosphate buffer pH 8.0 and 2.5 μL 0.2 M Catechol) in a 96 flat-bottom well, microtiter plate for each assay (Greiner Bio-One). The specific velocity of 2-hydroxymuconate semialdehyde production was determined on a BioTek Cytation™3 Multi-Mode reader (BioTek Instruments, Inc). Readings of this kinetic reaction were taken at 375 nm for 30 min at 30 °C. The change in concentration of 2-hydroxymuconate semialdehyde was calculated in nmol/min. A bicinchoninic acid (BCA) protein assay (Pierce Protein Research Products) endpoint reaction was used to determine the protein concentration of each sample. Each protein sample (20 μL total volume, 10 μL lysis supernatant + 10 μL XylE lysis buffer) was combined with 200 μL of bicinchoninic acid reaction substrate for this assay. The change in concentration of 2-hydroxymuconate semialdehyde was divided by mg protein to generate units of XylE specific activity (nmol/min/mg protein).
CspA overexpression, purification, and antibody production
For the overexpression of CspA using a His-tagged construct, PCR with the primers F-CPrtnNdeI and R-CPrtnXhoI was used to produce the cspA coding region from S. aureus Newman gDNA, The 201 bp PCR product was gel purified, ligated into pCR2.1, and transformed into E. coli DH5α. After selection on LB agar, putative colonies were grown overnight in LB broth at 37 °C. Plasmids were extracted, digested with EcoRI to confirm the presence of an insert, and sequenced to confirm a correct insert. After sequence confirmation, the plasmids were digested with NdeI and XhoI (Promega) and the inserts ligated into an identically restriction digested and gel-purified overexpression vector pRHB152. The construction of the pRHB152 plasmid is identical to the pRHB153 plasmid with the replacement of the GST-tag with a six His-tag sequence [25] (Hernandez, personal communication). The overexpression construct pCProtEx was transformed into E. coli BL21(DE3)pLysS competent cells. The transformants were selected by plating on LB agar plates at 37 °C. The presence of a correct cspA coding sequence, and sequence alignment with the His-tag for read-through transcription were confirmed in selected isolated colonies. For overexpression of CspA, cultures were grown in LB broth with selection to 0.600 at OD600 and protein production was induced by adding 1 mM IPTG. After 3 h, the induced cultures were harvested by centrifugation. Each cell pellet was washed twice with PBS pH 7.3 and centrifuged. The cell pellet for each sample was flash frozen with liquid nitrogen before storing at −80 °C. For purification, 10 mg of frozen cells were suspended in buffer (50 mM Tris–HCl pH 8.0, 50 mM NaCl, 0.5 μg/mL leupeptin, 2 mM PMSF, and 5 μg/mL DNase I) and lysed by sonication on ice (Fisher Scientific™ Sonic Dismembrator Model 500). The crude cell lysate was centrifuged for protein extract and the His-tagged CspA protein purified from the soluble protein using Nickel bound to Chelating Sepharose Fast Flow beads (GE Healthcare) at 4 °C.
Immunoaffinity purification of the rabbit-CspA antibody for Western blots
Polyclonal rabbit antibodies raised against a purified CspA protein [26] were produced by the Proteintech Group Inc. To reduce the amount of secondary binding observed in preliminary Western blots, the rabbit antiserum against purified CspA was further purified following an Affinity-Purification of Antibodies via Protein-Coupled Sepharose beads protocol (Proteintech). In brief, purified CspA was coupled to CNBr-activated Sepharose™ 4B beads (GE Healthcare) in the presence of a coupling buffer (100 mM NaHCO3, 500 mM NaCl pH 8.3) and incubated for 2 h at room temperature. The protein coupled beads were then washed three times with PBS followed by incubation with rabbit anti-CspA antiserum overnight at 4 °C. The protein-bead-antiserum suspension was loaded onto a 10 mL column, and washed with 10 mL PBS pH 7.4, and then 10 mL of 150 mM NaCl–HCl pH 5.0. The protein was eluted from the column with 6, 0.9 mL fractions of 150 mM NaCl–HCl pH 2.75 and the eluent was immediately neutralized with 0.1 mL of saturated PBS pH 7.5.
Western blot analysis and quantitation of CspA production
CspA protein production at different growth phases of S. aureus Newman and SKN23, a cspA::kanamycin cassette mutant, was initiated by growing the two strains overnight in TSB at 37 °C. The overnight cultures were normalized at an OD600 between 0.075 and 0.08 in 125 mL TSB and aliquots were harvested at 1, 3, and 6 h time points post dilution. Each aliquot was centrifuged, washed with 20 mM potassium phosphate buffer pH 7.5 and the pellet frozen at −80 °C until extraction. For the extraction of total cellular protein, each cell pellet was suspended in 500 μL of buffer (25 mM Tris–HCl, 100 mM NaCl, 5 mM EDTA pH 8.0 buffer, 0.5 μg/mL leupeptin, 2 mM PMSF, and 5 μg/mL DNase I). Then 50 μg/mL lysostaphin was added and the cells incubated at 37 °C for 30 min followed by ice incubation for 1 h. Cells were subjected to sonication on ice. Whole cell lysates were centrifuged at 4 °C (Sorval ST16R, Thermo Scientific), to collect supernatants that were centrifuged again at 4 °C. The clear supernatants were collected and analyzed for total protein concentrations using BCA. Equal concentrations of 30 μg of total protein for each sample were heat denatured at 95 °C for 5 min and quick chilled before being loaded onto a 15% SDS-polyacrylamide gel. The electrophoresis resolved proteins were electroblotted onto 0.2 micron PVDF membrane (Bio-Rad Mini Protean 3 gel system, Millipore Immobilon PSQ), air dried, and stained with Ponceau S to confirm total protein transfer. The blot was rinsed with water followed by 1% (v/v), Tris-buffered saline with Tween 20 (TBST) and blocked for 48 h with 5% (w/v) non-fat dry milk in 1% (v/v), TBST. CspA protein was detected with a rabbit-anti-CspA (1:1000 dilution) followed by goat anti-rabbit IgG horseradish peroxidase (HRP) (1:10,000 dilution) incubation. CspA bands were exposed to ECL Prime substrate (GE Amersham) and were detected using a Bio-Rad Chemi-Doc (Universal Hood III) gel imaging system. To measure the relative expression, CspA bands were quantitated using Image lab 5.2.1 software following a total protein normalization method (Bio-Rad).
Statistical analysis
A t test was used to determine the statistical significance between samples at p ≤ 0.05. All experiments were carried out ≥3 times and all samples were measured in triplicate except for the Western blot data in which duplicate samples were loaded and run on the same or simultaneously on a separate polyacrylamide gel.
Results and discussion
Identification of a TSP for cspA in S. aureus
Based on previous work in defining a 5′ UTR, untranslated region, upstream of a putative coding sequence in the cold shock gene cspA of S. aureus [10], a primer extension protocol was used to distinguish an actual TSP for the promoter of this gene. The primer extension analysis revealed three putative fragments (Fig. 1). These putative transcripts begin at −112 (P1), −88 (P2), and −51 (P3) base pairs upstream of the start site for translation (Fig. 1). Only one TSP had been previously reported in the cold shock genes, cspA of E. coli and cspB of B. subtilis [4, 7]. The presence of one TSP per cold shock gene changed when the production of multiple start sites was described in the primer extension analysis of the cold shock genes cspC and cspP in Lactobacillus plantarum [27]. The smaller primer extension fragments observed for both cspC and cspP arose from specific degradation of the mRNA and the actual transcription initiation sites of these two genes corresponded to the 5′ ends of the longest extension products [28]. This work was followed by the identification of three TSPs in both the cspA and cspB cold shock genes of Caulobacter crescentus [28]. To test the activity of each of these primer extension fragments, promoter-like sequences for each of these fragments were created by PCR through the production of sequences that included putative −10 and −35 RNA polymerase binding sites upstream of each potential start site (Table 2; Fig. 1) and ligating each of these to the xylE reporter gene of plasmid pLL38 [29]. The XylE analyses of these plasmids, pSK51 containing the P3 fragment and pSK88 with the P2 fragment, produced a range of activity (0.00–0.17 units of XylE specific activity) (Table 3). We subsequently classified both pSK51 and pSK88 constructs as inactive.
Fig. 1.
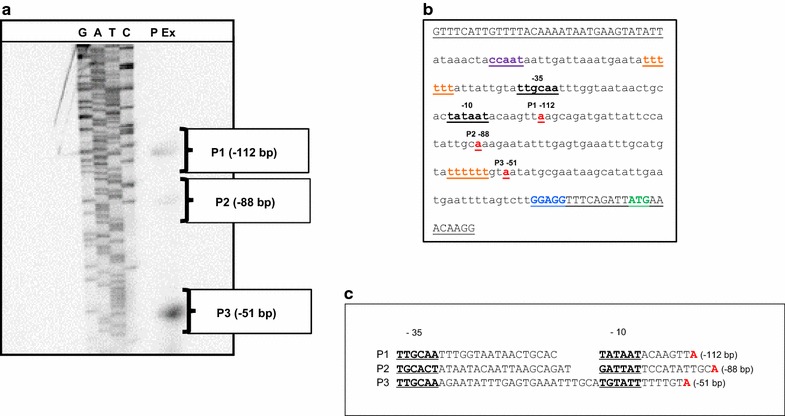
Primer extension to identify a transcription start point for the cold shock gene cspA. a The lanes for each of the specific nucleotide bases of the cspA sequence are labeled G, A, T, C. P Ex identifies the primer extension fragments. The putative transcription start points, P1 (−112 bp), P2 (−88) and P3 (−51) are labeled with bold letters and numbers. Brackets indicate the cDNA primer extension fragments. b The 238 bp sequence used to identify the locations for the P1 (−112 bp), P2 (−88) and P3 (−51) TSP. Each proposed TSP is labeled and the corresponding nucleotide is in bold red and underlined. The 5′ and 3′ primers are in capital letters and underlined. The translation start codon for cspA is in bold green and a ribosomal binding site is in bold blue. The putative −10 and −35 RNA polymerase binding sites are labeled and the sequence is in bold black and underlined. The proposed binding sites, CCAAT and TTTTTT, for a cold shock protein are labeled in bold purple and bold orange, respectively and underlined. c The putative promoter sequences for each of the putative TSPs. Each sequence is identified by P1, P2 and P3 on the left, the putative TSP in bold red at the 3′ end, and sequence location in parentheses at the right. The putative −35 and −10 sequences are labeled and in bold black underlined letters. Space has been added in the P1 and P2 sequences to align the −10 sites
Table 3.
XylE units of specific activity for the putative P2 and P3 promoters
| 1 HR | 3 HR | 6 HR | Average | |
|---|---|---|---|---|
| P2 (−88 bp)a | 0.13 | 0.17 | 0.00 | 0.10 |
| P3 (−51 bp)a | 0.12 | 0.04 | 0.00 | 0.05 |
Units of specific activity = nmol/min/mg protein
aData normalized by subtraction of promoterless vector pLL38 XylE velocity before calculation of units specific activity
The 112 bp putative P1 promoter containing a 5′ UTR was combined with an additional 80 base pairs upstream from the −112 bp putative TSP. These 80 bp contain a putative consensus TATAAT, −10 and a TTGCAA, −35 RNA polymerase binding site as well as proposed cold shock protein binding sites of a hexathymidine sequence and a pentamer CCAAT (Fig. 1) [5, 34]. This 192 bp putative promoter + 5′ UTR::xylE reporter gene construct pSK192 demonstrated XylE activity in S. aureus Newman (Fig. 3). The change in the level of units of XylE specific activity of pSK192 was not statistically significant when comparing the 1–3 h (p > 0.05) but increased significantly, ~14× at 6 h (p ≤ 0.05) (Fig. 3). In recent work, each of these fragments has been identified in relation to the endonuclease activity of RNase III [30]. The 113 bp (P1 −112 bp) fragment represents a full length 5′ UTR of the cspA gene. The −88 bp (P2 −88 bp) and the −53 bp (P3 −51 bp) are the products of post transcriptional modification of this 5′ UTR by RNase III to increase the stability of the cspA mRNA and increase the binding affinity of the cspA mRNA to a ribosome [30]. This results in an increased production of the protein product CspA. This work adds support to our findings of the P3 and P2 fragments as inactive and not transcriptional regulatory sequences and the P1 fragment as a 5′ UTR of a larger regulatory sequence. In our study, we have identified the promoter component to the 5′ UTR as an 80 bp sequence (Fig. 1). To determine the contribution of the 5′ UTR to the regulation of cspA, we constructed a putative promoter only plasmid pSK96 and tested for XylE activity at the same time points of 1, 3, and 6 h for comparison to the activity of pSK192. When compared to pSK192, the overall results for pSK96 showed a similar pattern of growth-dependent activity with significantly less XylE activity at the 3 h and 6 h time points (p ≤ 0.05) (Fig. 3), This reduction in units of XylE specific activity by pSK96 at 3 and 6 h could be related to the loss of the 5′ UTR that results in the absence of a potential regulatory protein binding site within the 5′ UTR (i.e., the hexathymidine at −54 to −59 bp) (Fig. 1) [31], a change in the secondary structure of the shortened mRNA that limits ribosomal binding, or a decrease in longevity of the mRNA. We feel it is important to note that these experiments demonstrate that the 5′ UTR is a component of regulation and the 80 bp putative promoter sequence also makes a contribution to the activity of these XylE constructs and potentially to cspA. The specifics of these latter contributions will need further work.
Fig. 3.

The activity of cspA sequence::xylE reporter gene constructs in Staphylococcus aureus Newman. Units of XylE specific activity were measured over a 6 h time period with samples collected at 1, 3, and 6 h. The values for the cspA promoter::xylE plasmid pSK96 are represented by black-filled squares joined by red lines and the cspA promoter + 5′ UTR plasmid pSK192 values are open circles linked by blue lines. The 324 bp sequence::xylE reporter construct pSK324 is represented by black-filled diamonds connected by green lines. The XylE velocity for each time point was normalized by subtraction of the velocity of the promoterless xylE reporter plasmid pLL38 before the units specific activity (nmol/min/mg protein) were calculated. Double-ended error bars represent the standard error of the mean for each time point
Comparison of the structure and activity of the 192 bp XylE active sequence to a reported inactive 324 bp sequence
The 192 bp active promoter + 5′ UTR identified in this work is a distinct sequence within the previously identified inactive 324 bp cspA/msaB sequence reported by Sahukhal and Elasri [20] (Fig. 2). This 324 bp sequence contains an additional 29 bp upstream of the 5′ end of the 192 bp construct and extends 103 bp downstream of the 3′ end of the 192 bp cspA promoter + 5′ UTR and 31 amino acids into the coding region of the cspA gene (msaB). Interestingly both sequences extend into the 3′ end of the coding region for the proposed upstream msaA gene, seven amino acids for pSK192 and 20 amino acids for the 324 bp sequence (Fig. 2) [20]. Although the sequence for the 324 bp in the previous work was obtained from S. aureus strain USA300_LAC (NC_ 007793.1) [32], the alignment of the sequence for this chromosomal segment is 100% identical to that found in S. aureus strain Newman (NC_009641.1) [33]. The sequence for the 5′ UTR and the coding sequence for S. aureus cspA have been previously submitted as AF259960 to GenBank [10]. Since the XylE active 192 bp promoter + 5′ UTR sequence is a component of this inactive 324 bp sequence, we decided to test the activity of this 324 bp in a xylE reporter construct pSK324. In the work reported by Sahukhal and Elasri [20], this 324 bp linked to the luxAB reporter gene of pCN58 showed no activity. However in our hands this same 324 bp sequence showed activity in excess of and significantly greater (p ≤ 0.05) than both pSK96 and pSK192 at all 3 collection times (Fig. 3). The pSK324 units of XylE specific activity also demonstrated a similar growth-dependent pattern to pSK96 and pSk192 plasmids. There are possible reasons that could be related to no activity in the pCN58 construct pMOE 482 and activity in the pLL38 construct pSK324. When we generated the 324 bp insert by PCR, we used the same primer sequences [20] (Table 2), except for changes in the sequence for the restriction sites (5′ PstI for BamHI and 3′ EcoRI for KpnI) for ligation to the xylE reporter gene in pLL38. These new restriction sites introduced a total of 10 bp changes, 6 at the 5′ PstI and 4 at the 3′ EcoRI, These changes could effect the confirmation of the DNA or the mRNA and effect the process of transcription and/or translation. The origin of replication (ori) and copy number for each plasmid are different, The ori for pCN58 is from pT181 with a copy number of 22 and the ori for pLL38 is from pC194 with a copy number of 15 [34]. In this case the copy number appears to favor pCN58 in terms of expression. Another source of this measurable activity in pSK324 could be due to the kinetic protocol of XylE expression over 30 min versus a protocol of luciferase expression that requires measurement within a 10 s time frame [20]. The former protocol provides a longer time for the development of measurable end product. Resolution of this discrepancy between these results could occur through the transformation of pSK324 into S. aureus USA300_LAC and test for XylE activity, or the transformation of S. aureus Newman with pMOE 482 and test for luciferase activity. A third possibility is to use another promoter::reporter plasmid such as pGL or pXEN-1 in both S. aureus backgrounds [35, 36].
Fig. 2.
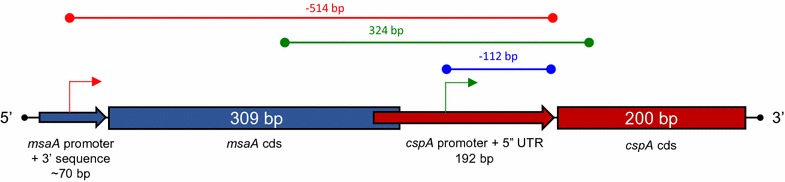
A chromosomal arrangement of regulatory sequences and two proposed cspA transcriptional start points in Staphylococcus aureus. The location and length in base pairs (bp) for the promoter + 3′ sequence (arrow) and coding sequence (cds) (rectangle) for msaA in light blue are shown in relation to the length and location of the promoter + 5′ UTR (arrow) and cds (rectangle) for cspA in red. The TSP for cspA identified by primer extension is identified by a green bent arrow and located by a 112 bp dark blue overline upstream of the 200 bp coding sequence for the cspA gene in red. The initial promoter for cspA/msaB identified by Sahukhal and Elasri [20] is identified by a red bent arrow at the 5′ end of the proposed promoter for msaA in blue and located by a 514 bp red overline upstream of the cds for cspA [20]. The red 192 bp promoter + 5′ UTR of cspA designed for the XylE assays is labeled. The location of the 324 bp regulatory sequence used for XylE assays is marked by a green overline [20]. The relative locations of these features are based on sequences from S. aureus Newman (NC_009641.1) [33]
Production of the CspA protein over a time course of 6 h
In using the vector pRHB152, a six His-tag CspA expression construct, pSKPrtExpA was created and used to produce a soluble form of the protein. The CspA protein was purified from the total soluble component of sonicated E. coli BL21(DE3)pLysS cells after 3 h of induction. The CspA protein was used to generate and to affinity purify rabbit anti-CspA antiserum. Based on the results of Western blots, over the time course of 6 h, the CspA protein is present in the wild type culture from samples taken at 1, 3, and 6 h. Using a total protein normalization method (BioRad), the calculated amount of CspA production at 6 h was significantly greater (p ≤ 0.05) than the other two timed collections (Fig. 4). These data support the production of CspA in a growth-dependent fashion and not constitutively. These results also add support to the regulation of the msaABCR operon by an anti-sense RNA of msaR in a growth phase dependent fashion proposed by Sahukhal and Elasri [20, 30]. These investigators demonstrated that the anti-sense RNA of msaR is present in both the pre-logarithmic and mid-logarithmic phase of growth yet absent in the post-logarithmic phase of growth. If the production of CspA seen in this study is superimposed over the production of the anti-sense RNA of msaR, it is possible to see a lower production of CspA at 1 h (pre-logarithmic) and 3 h (mid-logarithmic) growth in the presence of the msaR anti-sense RNA and the increase production of CspA at 6 h (post-logarithmic) growth in the absence of this RNA. These findings also appear to align with the production of a shorter and more active transcript of cspA by RNase III degradation of a longer and less active transcript through increased access to cspA mRNA in the absence of the anti-sense RNA of msaR [30]. Verification of this same data, production of the anti-sense RNA msaR in S. aureus Newman and CspA production in S. aureus USA300_LAC, seems highly likely based on the 100% identity in the alignment of these two sequences for the cspA 5′ UTR and coding sequences.
Fig. 4.
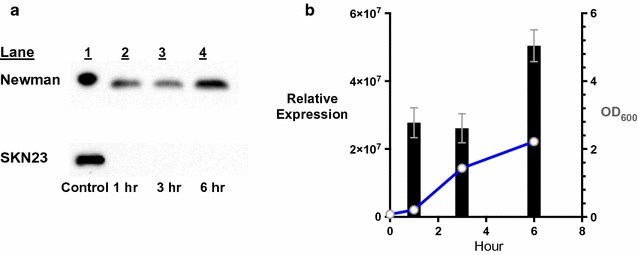
CspA protein production and growth of Staphylococcus aureus Newman. a The Western blot of CspA production by the wild type S. aureus Newman is the upper band. The lower band is the Western blot of a negative control S. aureus cspA mutant SKN23. Lane 1 contains 35 ng of purified His-tag CspA control. In both the wild type and mutant Western blots, Lanes 2, 3, and 4 contain 30 µg of total soluble protein from aliquots of cells taken at 1, 3, and 6 h. The Lane, Control, and h for each sample are labeled. b The relative expression values for CspA bands were calculated using Image Lab 5.2.1 software following a total protein normalization method. The relative expression of CspA for each time point, 1, 3, and 6 h, is represented by black columns with double-ended bars for the standard error of the mean (SEM) (left Y axis). The OD600 for each sample is represented by a gray circle and time points connected by blue lines with doubled-ended error bars for the SEM (right Y axis). The double-ended error bars are not shown if the SEM is less than the width of symbol. The OD600 sample for 6 h was diluted 1:2 before measurement
Conclusions
In this paper, we present results that show a growth-dependent relationship between the production of the cold shock protein CspA (Western blot) and the activity of a cspA promoter + 5′ UTR (units of XylE specific activity) adjacent to the cspA coding region. CspA is produced in a growth-dependent fashion that significantly increases at the post-logarithmic time point of 6 h. This production of CspA at 37 °C demonstrates the availability of this protein to modulate virulence factors (pigment and biofilm production, antimicrobial peptide susceptibility, capsule) and the major regulatory molecule SarA. The growth phase dependent increase in the production of CspA also supports the association between the transcription of cspA and regulation by a proposed anti-sense RNA identified as msaR [20, 30]. The identification of this cspA promoter + 5′ UTR adds both definition and potential complexity to the regulation of the cspA gene. The results presented define a new cspA regulatory region, designed as a 192 bp sequence composed of a promoter region + a 5′ UTR upstream of the CspA translation start site. The presence of two active promoters, one initiating transcription at 112 bp upstream of the cold shock initiation codon defined in this work and the second initiating transcription from the promoter of the proposed upstream gene msaA [20] at 514 bp upstream of the cspA initiation codon, suggests the possibility of interactions in the regulation of cspA. We are currently pursuing the study of this interaction with recombinant chromosomal constructs that will allow us to identify the activity of and report the contribution of each of these promoters to the regulation of cspA transcription and CspA protein production both in vitro and in vivo.
Authors’ contributions
CKU, KDG, GV-B, and SK designed and carried out the experiments. CKU, KDG, GV-B wrote the protocols. SK analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank W. M. Shafer, C. Lee, T. Luong, and J. Hernandez for strains and plasmids essential to this work. The authors would also like to thank D. Stauff, J. Hernandez, B. Wellensiek, and K. Francis for their suggestions and technical expertise. We would also like to thank S. Korch and F. Gonzalez for reading the paper and their constructive comments.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
This work was funded by Midwestern University in the forms of Graduate Research Stipends (KDG. and GV-B) and intramural research funds (SK).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- TSB
tryptic soy broth
- LB
Luria–Bertani medium
- BCA
bicinchoninic acid
- TSP
transcriptional start point
- UTR
untranslated region
- PCR
polymerase chain reaction
- RT-qPCR
real time quantitative polymerase chain reaction
- cDNA
complementary DNA
- gDNA
genomic DNA
- Km
kanamycin
- Tet
tetracycline
Contributor Information
Chandana K. Uppalapati, Email: cuppal@midwestern.edu
Kimberley D. Gutierrez, Email: gutiekd@u.washington.edu
Gina Buss-Valley, Email: ginamvalley@gmail.com.
Sam Katzif, Email: skatzi@midwestern.edu.
References
- 1.Petinaki E, Spiliopoulou I. Methicillin-resistant Staphylococcus aureus among companion and food-chain animals: impact of human contacts. Clin Microbiol Infect. 2011;18(7):626–634. doi: 10.1111/j.1469-0691.2012.03881.x. [DOI] [PubMed] [Google Scholar]
- 2.Committee on New Directions in the Study of Antimicrobial Therapeutics: New Classes of Antimicrobials, Committee on New Directions in the Study of Antimicrobial Therapeutics: Immunomodulation, National Research Council (2006). Treating infectious diseases in a microbial world: report of two workshops on novel antimicrobial therapeutics. Washington, D.C.: The National Academies Press; 2002. http://www.nap.edu/catalog/11471.html. Accessed 13 Oct 2016.
- 3.Goldstein J, Pollitt NS, Inouye M. Biochemistry: major cold shock protein of Escherichia coli (gene regulation/stress response/heat shock response) Proc Natl Acad Sci USA. 1990;87:283–287. doi: 10.1073/pnas.87.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Willimsky G, Bang H, Fischer G, Marahiel MA. Characterization of cspB, a Bacillus subtilis inducible cold shock gene affecting cell viability at low temperatures. J Bacteriol. 1992;174:6326–6335. doi: 10.1128/jb.174.20.6326-6335.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Graumann P, Marahiel MA. The major cold shock protein of Bacillus subtilis CspB binds with high affinity to the ATTGG and CCAAT sequences in single stranded oligonucleotides. FEBS Lett. 1994;338:157–160. doi: 10.1016/0014-5793(94)80355-2. [DOI] [PubMed] [Google Scholar]
- 6.Graumann P, Schroder KS, Schmid R, Marahiel MA. Cold-shock stress proteins in Bacillus subtilis. J Bacteriol. 1996;128:4611–4619. doi: 10.1128/jb.178.15.4611-4619.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamanaka K. Cold shock response in Escherichia coli. J Mol Microbiol Biotechnol. 1999;1:193–202. [PubMed] [Google Scholar]
- 8.Horn G, Hofweber R, Kremer W, Kalbitzer HR. Structure and function of bacterial cold shock proteins. Cell Mol Life Sci. 2007;64:1457–1470. doi: 10.1007/s00018-007-6388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafer WM, Katzif S, Bowers S, Fallon M, Hubalek M, Reed MS, et al. Tailoring an antibacterial peptide of human lysosomal cathepsin G to enhance its broad-spectrum action against antibiotic-resistant bacterial pathogens. Curr Pharm Des. 2002;8:695–702. doi: 10.2174/1381612023395376. [DOI] [PubMed] [Google Scholar]
- 10.Katzif S, Danavall D, Bowers S, Balthazar JT, Shafer WM. The major cold shock gene, cspA, is involved in the susceptibility of Staphylococcus aureus to an antimicrobial peptide of human cathepsin G. Infect Immun. 2003;71:4304–4312. doi: 10.1128/IAI.71.8.4304-4312.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. sigmaB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J Bacteriol. 2002;184(19):5457–5467. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katzif S, Lee E, Law AB, Tzeng Y, Shafer WM. CspA regulates pigment production in Staphylococcus aureus through a SigB-dependent mechanism. J Bacteriol. 2005;187:8181–8184. doi: 10.1128/JB.187.23.8181-8184.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wieland B, Feil C, Gloria-Maercker E, Thumm EG, Lechner M, Bravo JM, et al. Genetic and biochemical analyses of the biosynthesis of the yellow carotenoid 4,4′-diaponeurosporene of Staphylococcus aureus. J Bacteriol. 1994;176:7719–7726. doi: 10.1128/jb.176.24.7719-7726.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuroda M, Ohta T, Hayashi H. Isolation and the gene cloning of an alkaline shock protein in methicillin resistant Staphylococcus aureus. Biochem Biophys Res Commun. 1995;207:978–984. doi: 10.1006/bbrc.1995.1281. [DOI] [PubMed] [Google Scholar]
- 15.Wu S, de Lencastre H, Tomasz A. SigmaB, a putative operon encoding alternate sigma factor of Staphylococcus aureus RNA polymerase: molecular cloning and DNA sequencing. J Bacteriol. 1996;178:6036–6042. doi: 10.1128/jb.178.20.6036-6042.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gertz S, Engelmann S, Schmid R, Ohlsen K, Hack J, Hecker M. Regulation of σB-dependent transcription of sigB and asp23 in two different Staphylococcus aureus strains. Mol Gen Genet. 1999;261:558–566. doi: 10.1007/s004380051001. [DOI] [PubMed] [Google Scholar]
- 17.Elbarasi A. Identification and characterization of msaB gene involved in biofilm formation and virulence in Staphylococcus aureus. Master’s Theses. 2014. Paper 49. http://aquila.usm.edu/masters_theses/49/. Accessed Mar 2016.
- 18.Sahukhal GS, Batte JL, Elasri MO. msaABCR operon positively regulates biofilm development by repressing proteases and autolysis in Staphylococcus. FEMS Microbiol Lett. 2015;362(4):1–10. doi: 10.1093/femsle/fnv006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batte JL, Samanta D, Elasri MO. MsaB activates capsule production at the transcription level in Staphylococcus aureus. Microbiology. 2016;162:575–589. doi: 10.1099/mic.0.000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahukhal GS, Elasri MO. Identification and characterization of an operon, msaABCR, that controls virulence and biofilm development in Staphylococcus aureus. BMC Microbiol. 2014;14:154. doi: 10.1186/1471-2180-14-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duthie ES, Lorenz LL. Staphylococcal coagulase; mode of action and antigenicity. J Gen Microbiol. 1952;6:95–107. doi: 10.1099/00221287-6-1-2-95. [DOI] [PubMed] [Google Scholar]
- 22.Kreiswirth BN, Löfdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 23.Augustin J, Götz F. Transformation of Staphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol Lett. 1990;54:203–207. doi: 10.1111/j.1574-6968.1990.tb03997.x. [DOI] [PubMed] [Google Scholar]
- 24.Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui J, Anderson KL, Dunman PM, Joyce S, Skaar EP. A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe. 2007;1(2):109–119. doi: 10.1016/j.chom.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hernandez JA, Igarashi RY, Soboh B, Curatti L, Dean DR, Ludden PW, Rubio LM. NifX and NifEN exchange NifB cofactor and the VK-cluster, a newly isolated intermediate of the iron-molybdenum cofactor biosynthetic pathway. Mol Microbiol. 2007;63:177–192. doi: 10.1111/j.1365-2958.2006.05514.x. [DOI] [PubMed] [Google Scholar]
- 26.Buss GM. Production of the cold chock protein CspA by Staphylococcus aureus. Master’s Thesis. Glendale: Midwestern University Library; 2006.
- 27.Derzelle S, Hallet B, Francis KP, Ferain T, Delcour J, Hols P. Changes in the cspL, cspP, and cspC mRNA abundance as a function of cold shock and growth phase in Lactobacillus plantarum. J Bacteriol. 2000;182:5105–5113. doi: 10.1128/JB.182.18.5105-5113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lange EA, Marques MV. Identification and transcriptional control of Caulobacter crescentus genes encoding proteins containing a cold shock domain. J Bacteriol. 2004;186:5603–5613. doi: 10.1128/JB.186.17.5603-5613.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Luong TT, Lee CY. The sbcDC locus mediates repression of type 5 capsule production as part of the SOS response in Staphylococcus aureus. J Bacteriol. 2007;189:7343–7350. doi: 10.1128/JB.01079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lioliou E, Sharma CM, Caldelari I, Helfer A, Fechter P, Vandenesch F, et al. Global regulatory function of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 2012;8(6):e1002782. doi: 10.1371/journal.pgen.1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Max KEA, Zeeb M, Bienert R, Balbach J, Heinemann U. Common mode of DNA binding to cold shock domains. Crystal structure of hexathymidine bound to the domain-swapped form of a major cold shock protein from Bacillus caldolyticus. FEBS J. 2007;274:1265–1279. doi: 10.1111/j.1742-4658.2007.05672.x. [DOI] [PubMed] [Google Scholar]
- 32.Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired methicillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 33.Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol. 2008;190:300–310. doi: 10.1128/JB.01000-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Novick RP. Staphylococcal plasmids and their replication. Ann Rev Microbiol. 1989;43:537–565. doi: 10.1146/annurev.mi.43.100189.002541. [DOI] [PubMed] [Google Scholar]
- 35.Gao P, Wang Y, Villanueva I, Ho PL, Davies J, Kao RYT. Construction of a multiplex promoter reporter platform to monitor Staphylococcus aureus virulence gene expression and the identification of usnic acid as a potent suppressor of psm gene expression. Front Microbiol. 2016;7:1–12. doi: 10.3389/fmicb.2016.01344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Francis KP, Joh D, Bellinger-Kawahara C, Hawkinson MJ, Purchio TF, Contag PR. Monitoring bioluminescent Staphylococcus aureus infections in living mice using a novel luxABCDE construct. Infect Immun. 2000;68(6):3594–3600. doi: 10.1128/IAI.68.6.3594-3600.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.