

Abstract
The phosphorylation of amylopectin by the glucan, water dikinase (GWD; EC 2.7.9.4) is an essential step within starch metabolism. This is indicated by the starch excess phenotype of GWD-deficient plants, such as the sex1-3 mutant of Arabidopsis (Arabidopsis thaliana). To identify starch-related enzymes that rely on glucan-bound phosphate, we studied the binding of proteins extracted from Arabidopsis wild-type leaves to either phosphorylated or nonphosphorylated starch granules. Granules prepared from the sex1-3 mutant were prephosphorylated in vitro using recombinant potato (Solanum tuberosum) GWD. As a control, the unmodified, phosphate free granules were used. An as-yet uncharacterized protein was identified that preferentially binds to the phosphorylated starch. The C-terminal part of this protein exhibits similarity to that of GWD. The novel protein phosphorylates starch granules, but only following prephosphorylation with GWD. The enzyme transfers the β-P of ATP to the phosphoglucan, whereas the γ-P is released as orthophosphate. Therefore, the novel protein is designated as phosphoglucan, water dikinase (PWD). Unlike GWD that phosphorylates preferentially the C6 position of the glucose units, PWD phosphorylates predominantly (or exclusively) the C3 position. Western-blot analysis of protoplast and chloroplast fractions from Arabidopsis leaves reveals a plastidic location of PWD. Binding of PWD to starch granules strongly increases during net starch breakdown. Transgenic Arabidopsis plants in which the expression of PWD was reduced by either RNAi or a T-DNA insertion exhibit a starch excess phenotype. Thus, in Arabidopsis leaves starch turnover requires a close collaboration of PWD and GWD.
Starch, as the predominant storage carbohydrate in plants, is a major constituent of human and animal diets, and it is also an important raw material for various industrial processes (Slattery et al., 2000). Amylopectin, the major constituent of starch, contains varying amounts of covalently bound phosphate, depending upon plant species and organ. Phosphate is monoesterified to the C6 position (approximately two-thirds) and to the C3 position (approximately one-third). In addition, approximately 1% of the esterified phosphate may be linked to the C2 position (Tabata and Hizukuri, 1971). The total phosphorylation level of starch is quite low. In cereal endosperm starch, the amounts of Glc-6-P and Glc-3-P residues are at or below the detection limit and even in potato (Solanum tuberosum) tuber starch that is regarded as highly phosphorylated only 0.2% to 0.5% of the Glc residues are phosphorylated (Tabata and Hizukuri, 1971; Blennow et al., 2002). In Arabidopsis (Arabidopsis thaliana) leaf starch approximately 1 in 1,000 Glc residues is phosphorylated (Yu et al., 2001). Phosphate monoesters in amylopectin strongly affect starch functionality, and high phosphate contents are desirable for many industrial applications (Slattery et al., 2000; Blennow et al., 2002).
Phosphorylation of starch like polyglucans is catalyzed by the glucan, water dikinase (GWD, formerly designated as R1; EC 2.7.9.4; Ritte et al., 2002):
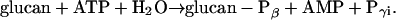 |
The catalytic mechanism includes autophosphorylation of the dikinase protein. The β-P of ATP is firstly transferred to a His residue of GWD and then to either the C6 or the C3 position of a glucosyl unit (Ritte et al., 2002; Mikkelsen et al., 2004).
In GWD-deficient plants, not only starch phosphorylation but also starch breakdown is strongly impaired. In GWD antisense potato plants (Lorberth et al., 1998) as well as in the GWD-deficient starch-excess 1 (sex1) mutants of Arabidopsis (Yu et al., 2001), leaf starch contents at the end of the day are 3 to 5 times higher than those of the respective wild-type plants. More recently, it was shown that transitory starch in Chlamydomonas and potato is mainly phosphorylated during degradation (Ritte et al., 2004). This provides direct evidence for a link between the phosphorylation of starch and its degradation, but the underlying mechanisms remained obscure. It has been postulated that the activity of certain proteins that are involved in starch degradation depends on the presence of phosphate esters within starch (Yu et al., 2001; Ritte et al., 2002). However, until now, such enzymatic activities have never been documented. These proteins are expected to display a higher affinity to phosphorylated starch than to nonphosphorylated starch. This assumption provides a possible strategy for their identification.
Here we describe the discovery of a novel protein, which preferentially binds to phosphorylated starch. Its enzymatic function was investigated in vitro using purified protein and in vivo using transgenic plants.
RESULTS
Identification of a Novel Protein That Preferentially Binds to Phosphorylated Starch Granules in Vitro
To identify proteins whose activity depends on starch-bound phosphate esters, we compared binding of proteins to phosphorylated or nonphosphorylated starch granules. Phosphate free starch granules were isolated from leaves of the GWD-deficient Arabidopsis sex1-3 mutant (Yu et al., 2001). Aliquots of the starch preparations were then in vitro phosphorylated using purified recombinant GWD from potato. Under the conditions applied, the phosphorylation levels range between 200 and 400 pmol P/mg starch. All the phosphate groups are located at the granule surface, since the recombinant GWD has no access to the granule interior. Phosphorylated and nonphosphorylated granules were then incubated with protein extracts from Arabidopsis wild-type leaves. Proteins bound to the granules were desorbed by a SDS-containing buffer and subsequently analyzed using SDS-PAGE and matrix-assisted laser-desorption ionization mass spectrometry (Fig. 1). Three proteins did bind preferentially to the phosphorylated starch. These were GWD (SEX1; At1g10760), cytosolic phosphorylase (Pho2; At3g46970), and an as-yet uncharacterized protein, preliminarily designated as OK1 (At5g26570). In the following we focus on the latter. OK1 turned out to be one of the two putative proteins with homology to GWD, whose existence was predicted from the Arabidopsis sequence (Yu et al., 2001). Database analysis using PlantGDB Blast (Dong et al., 2004) revealed the presence of putative OK1 orthologs in 14 different plant species including potato, tomato, barley, and rice.
Figure 1.

In vitro binding of proteins to phosphorylated or nonphosphorylated starch granules. Protein extracts from ecotype Columbia leaves were incubated with either nonphosphorylated (2) or previously in vitro phosphorylated (3) starch granules from sex1-3 leaves. Granules were washed, and bound proteins were released with SDS sample buffer and separated by SDS-PAGE. 1, In vitro phosphorylated starch granules incubated without extract; M, molecular mass marker. Three proteins with higher affinity to phosphorylated starch are indicated (GWD, OK1, and Pho2).
Using primers designed for the At5g26570 gene, the full-length OK1 cDNA sequence was cloned. In the sequence thereby derived, 15 additional nucleotides (1,555–1,569) were found that were not present in the already existing corresponding National Center for Biotechnology Information (NCBI) entry (NM_122538). The OK1 cDNA sequence was submitted to EMBL (accession no. AJ635427).
OK1 and GWD amino acid sequences were compared using the BLAST 2 Sequences tool (Tatusova and Madden, 1999). The C-terminal regions, ranging from amino acid 611 to 1,196 (OK1), and 860 to 1,398 (GWD), displayed 25% amino acid identity, and 41% sequence similarity. No similarity could be detected in the N-terminal regions. The overall amino acid identity, as analyzed with the AlignX program (Vector NTI, Invitrogen, Karlsruhe, Germany), is 14%, and sequence similarity is 24%.
Analysis of the OK1 sequence using TargetP (Emanuelsson et al., 2000) reveals a high probability for the existence of a signal peptide directing the protein to plastids (score 0.992). Three further domains could be detected (Fig. 2A). A starch binding domain (CBM 20) is located at the N-terminal region of OK1. The C terminus of OK1 displays homology to the nucleotide binding domains of the dikinases pyruvate, phosphate dikinase (PPDK; EC 2.7.9.1), phosphoenolpyruvate (PEP)-synthase (pyruvate, water dikinase; EC 2.7.9.2), and GWD (Marchler-Bauer et al., 2003). A region with significant homology to the phosphohistidine domains of these dikinases is also present in the OK1 sequence (Fig. 2B).
Figure 2.

OK1 contains a starch binding domain and domains typical for dikinases. A, Protein domains of the OK1 protein. The amino acid number indicates the location of the respective domain within the protein. CTS, Chloroplast targeting signal; SB, starch binding domain CBM 20; PH, putative phosphohistidine domain; NB, nucleotide binding domain. B, Alignment of the putative phosphohistidine domains of OK1 and different dikinases. The putative phosphohistidine regions of GWDs from Arabidopsis (At), potato (St), and Citrus reticulata (Cr) were aligned with the putative phosphohistidine regions of PPDKs from Zea mays (Zm) and E. coli, pyruvate, water dikinase (PPS) from E. coli, and the homologous region of OK1. Identical amino acids in black, conserved in gray boxes. The putative phosphohistidine is printed in boldface.
OK1 Is Localized in Plastids
The prediction of OK1 being a plastidic protein by the bioinformatic programs was further strengthened by western-blot analysis of extracts made from Arabidopsis leaf protoplasts and chloroplasts isolated from protoplasts. Equal amounts of protein were analyzed by SDS-PAGE and western blot using specific antibodies raised against ADP-Glc-pyrophosphorylase (AGPase, plastidic marker), PEP-carboxylase (cytosolic marker), and OK1. As shown in Figure 3 for both AGPase and OK1, an immunosignal was obtained in the chloroplast fraction, whereas there was hardly any signal in the protoplast fraction. In contrast, the cytosolic marker PEP-carboxylase was exclusively detected in the protoplast fraction. Nonaqueous fractionation of Arabidopsis leaf material and subsequent western-blot analysis also indicates that OK1 resides in plastids (data not shown). Furthermore, OK1 is found in the Arabidopsis chloroplast protein database (http://www.pb.ipw.biol.ethz.ch/index.php?toc=91; Kleffmann et al., 2004), thereby providing another line of evidence for plastidic localization of this protein. Thus, the interaction of OK1 with phosphorylated starch may well be of physiological relevance.
Figure 3.

OK1 is located in chloroplasts. Equal amounts of protein extracted from Arabidopsis leaf protoplasts (P) and chloroplasts (C), respectively, were separated by SDS-PAGE and examined by immunoblot analysis using antibodies against OK1, AGPase (plastidic marker), and PEPCase (cytosolic marker).
OK1 Is a Phosphoglucan, Water Dikinase
A vector was constructed allowing the expression of OK1 containing a 6×His tag at the N terminus in Escherichia coli and one step purification of the recombinant protein using a Ni-NTA agarose resin. The full-size OK1 protein is clearly predominant in the resulting protein fraction (Supplemental Fig. 1). Because of the similarity between OK1 and GWD, we tested whether or not OK1 also displays starch phosphorylating activity. As for the in vitro binding assay, nonphosphorylated or phosphorylated starch granules served as substrates. OK1 was indeed able to transfer 33P from [βγ-33P]ATP to starch. Most remarkably, however, the activity strictly depended on a preceding phosphorylation of the granules by recombinant potato GWD (Fig. 4A). We never observed OK1-catalyzed phosphorylation of the unmodified phosphate free sex1-3 starch (n = 7). Similar to GWD, OK1 transfers the β-P of ATP to starch. There was no significant phosphate incorporation of labeled phosphate into the starch substrate if [γ-33P]ATP served as phosphate donor (Fig. 4B).
Figure 4.

In vitro activity assay with recombinant OK1 protein. A, OK1 phosphorylates phosphoglucans but not phosphate free glucans. One microgram recombinant OK1 protein was incubated for 15 min with 25 μm ATP containing 0.5 μCi [βγ-33P]ATP and 5 mg of either nonphosphorylated (1) or in vitro phosphorylated (2) starch granules from sex1-3 leaves. The radioactivity incorporated into the granules was counted. -, Control without protein. Bars represent the individual measurements of two parallel samples. B, OK1 transfers the β-P of ATP to phosphoglucans. Recombinant OK1 protein (2.2 μg) was incubated for 1 h with 4.2 mg in vitro phosphorylated starch granules and either [βγ-33P]ATP or solely [γ-33P]ATP (0.5 μCi in both cases). ATP concentration, 25 μm. The radioactivity incorporated into the granules was counted. -, Control without protein.
To study the fate of the γ-P of ATP, we analyzed OK1-catalyzed incorporation of β-P into phosphorylated starch using [β-33P]ATP as phosphate donor as well as a possible release of γ-P-ortophosphate into the soluble phase using [γ-33P]ATP. For comparison, the same experiment was also conducted using recombinant potato GWD, for which we have shown before that water is the acceptor of the γ-P (Ritte et al., 2002). As shown in Table I, in both cases comparable amounts of phosphate were incorporated into starch or released as inorganic phosphate into the soluble phase, respectively. In both samples, the amount of γ-P released exceeded that of β-P detected in the starch fraction by about 20% to 30%. However, it is possible that the amount of incorporated phosphate is underestimated due to some loss of starch granules during the extensive washing procedure.
Table I.
OK1 transfers the γ-P of ATP to water
Recombinant OK1 or GWD protein (2.2 μg each) were incubated with in vitro phosphorylated sex1-3 starch granules for 80 min. Either [β-33P]ATP or [γ-33P]ATP of identical specific radioactivity served as phosphate donor. The incorporation of β-P into starch (control, buffer instead of protein) and the release of γ-Pi into the soluble phase (control, no starch) was determined.
| [β-33P]-Incorporation | [γ-33P]-Release | |
|---|---|---|
| pmol | pmol | |
| OK1 | 34.3 | 42.8 |
| GWD | 123.3 | 172.0 |
We conclude that OK1 is a phosphoglucan, water dikinase (PWD) that transfers the β-P of ATP to a phosphoglucan and the γ-P of ATP to water:
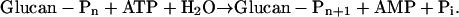 |
Therefore, we replace the preliminary term OK1 with PWD.
Under standard assay conditions (10 mg phosphorylated starch granules/mL, 25 μm ATP, 30 min), a linear correlation between PWD protein amount and phosphate incorporation into the substrate is only observed at low protein concentrations (<0.1 μg/mg starch; Supplemental Fig. 2A). For 0.05 μg PWD/mg starch, we calculated an activity of 2.55 nmol min−1 (mg protein)−1. This value is slightly underestimated because [βγ-33P]ATP was used that contains ≥80% of label in the β-position (Ritte et al., 2003). The PWD activity is similar to the specific activity of potato GWD with soluble potato amylopectin (Ritte et al., 2002) and also to the activity of GWD with unmodified sex1-3 starch granules as substrate (data not shown).
For a more quantitative analysis, starch granules containing different amounts of phosphate esters (9, 49, and 215 pmol P/mg starch) were reacted with PWD (Supplemental Fig. 2B). The granule preparation with the highest phosphate content was the most efficient phosphate acceptor. However, PWD activities with the different substrates varied about 6-fold, whereas the level of prephosphorylation varied about 24-fold. Thus, there is no linear relation between the phosphate content of a polyglucan and its capacity to serve as phosphate acceptor for PWD.
As observed with nonphosphorylated sex1-3 starch granules PWD was unable to phosphorylate solubilized sex1-3 starch (Supplemental Fig. 2C). However, even solubilized sex1-3 starch that had been prephosphorylated by GWD proved to be an extremely poor substrate for PWD (Supplemental Fig. 2C). The same holds true for soluble potato starch (Sigma S-2004) although it contains approximately 15 nmol P/mg starch (data not shown). Possibly, the structure of the starch granule or of the surface of the particle is also important for the PWD activity.
PWD Is Capable of Autophosphorylation
A phosphohistidine is an intermediate in the dikinase type reactions catalyzed by PPDK (Goss et al., 1980), PEP-synthase (Narindrasorasak and Bridger, 1977), and GWD (Ritte et al., 2002; Mikkelsen et al., 2004). The β-P of ATP is firstly transferred to the His residue and then to pyruvate (in case of PPDK and PEP-synthase) or a glucan (in case of GWD). As mentioned above an amino acid stretch displaying homology to the phosphohistidine domains of these enzymes is also present in PWD. To study whether PWD follows a similar mechanism, we incubated the purified protein with either [βγ-33P]ATP or [γ-33P]ATP in the absence of a phosphoglucan and analyzed a possible autophosphorylation by SDS-PAGE and autoradiography. Labeling of the PWD protein was observed only in the samples containing [βγ-33P]ATP but not in those containing [γ-33P]ATP (Fig. 5). Thus, PWD phosphorylates itself with the β-P. The phosphorylation was heat labile, acid labile but rather stable in alkali (Fig. 5). This is consistent with a phosphohistidine being formed, since phosphoserine, phosphotyrosine, and phosphothreonine are heat stable, acid stable, but alkali labile (Rosenberg, 1996). The results strongly indicate that the β-P of ATP is firstly transferred to a His residue of PWD and afterward to the phosphoglucan.
Figure 5.

Autocatalytic phosphorylation of recombinant PWD (OK1). Recombinant PWD protein was incubated in the absence of starch granules with either [βγ-33P]ATP or solely [γ-33P]ATP (1.4 μCi/μg protein each). Heat stability of the phosphorylated PWD was tested by incubation of the samples at either 30°C for 30 min or 95°C for 5 min. Acid or base stability of the phosphorylated PWD protein was assayed by incubation in 0.5 n NaOH or 0.5 n HCl for 30 min at RT, followed by denaturation in SDS sample buffer for 30 min at 30°C. Samples were separated by SDS-PAGE (1.8 μg PWD protein/lane) and visualized by either Coomassie Blue staining (A) or autoradiography (B).
PWD Phosphorylates Preferably the C3 Position of the Glucosyl Residues in Phosphoglucans
To analyze which positions of the Glc residues are phosphorylated by PWD, starch was prephosphorylated by GWD (using unlabeled ATP) and then phosphorylated by PWD using 33P-ATP. For comparison, an experiment in which starch was solely phosphorylated by GWD (using labeled ATP) was also performed. Following in vitro phosphorylation, Glc and Glc-Ps were released from starch by means of acid hydrolysis. To separate Glc-6-P and Glc-3-P, the samples were subjected to high performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD). Since the extent of in vitro phosphorylation is too low to allow for reliable amperometric detection of the Glc-Ps thereby formed we added authentic Glc-6-P and Glc-3-P as standards. Fractions were collected and the radioactivity was counted.
In contrast to GWD, which phosphorylates preferably the C6-position, PWD phosphorylates predominantly the C3-position (Fig. 6). Approximately 70% of the radioactivity incorporated into starch by PWD coeluted with Glc-3-P in two independent experiments. It has to be considered that Glc-3-P is a rather acid labile compound. The investigation of starch bound phosphate esters by acid hydrolysis of granular starch and subsequent analysis of the products by HPAEC-PAD was adapted from Blennow et al. (1998). They estimated that during 2 h of acid hydrolysis approximately 20% of the Glc-3-P is dephosphorylated. We found that 24% of the radioactivity in the starch hydrolysate was present as inorganic phosphate using the modified Parvin and Smith method (see “Materials and Methods”). In contrast, if starch was hydrolyzed following in vitro phosphorylation with GWD less than 5% of the label were present as inorganic phosphate. When 32P-orthophosphate was subjected to HPAEC-PAD approximately 20% of radioactivity coeluted with Glc-6-P the remaining 80% eluted in the fraction between Glc-6-P and Glc-3-P (data not shown). Thus, in the hydrolyzed PWD product the radioactivity eluting ahead of the Glc-3-P peak (Fig. 6) can, at least in part, be attributed to orthophosphate that is released from Glc-3-P during sample processing.
Figure 6.

PWD (OK1) phosphorylates preferentially the C-3 position of the Glc units. Recombinant PWD and GWD protein (first experiment: 25 μg PWD, 5 μg GWD; second experiment: 4.2 μg PWD, 0.7 μg GWD), respectively, were incubated with in vitro phosphorylated sex1-3 starch granules and [βγ-33P]ATP for 1 h. Starch granules were hydrolyzed. Aliquots of the hydrolyzates were supplemented with Glc-3-P and Glc-6-P as internal standards and subjected to HPAEC-PAD analysis (top section). Fractions were collected every minute (1 mL), except for Glc-3-P and Glc-6-P fractions, which were collected quantitatively in separate fractions. Radioactivity in the collected fractions was counted. The relative distribution of radiolabel was calculated as percentage of total radioactivity in all collected fractions (bottom section). GWD, White bars; PWD, black bars. Values are means of two independent experiments.
In Vitro Analysis of PWD Purified from Leaves Confirms the Results with the Heterologously Expressed Protein
Analysis of the recombinant PWD has revealed unique features of this enzyme. However, it has to be kept in mind that the recombinant PWD protein is not identical to PWD in plants. Since the length of the transit peptide is only predicted but not exactly known the complete coding sequence of PWD was used to generate the expression vector. Furthermore, the protein contains an N-terminal His tag and a single amino acid replacement (see “Materials and Methods”). Therefore, we analyzed the plant-derived protein in addition to the recombinant PWD. The protein was partially purified by ammoniumsulfate precipitation and affinity chromatography using immobilized maltoheptaose (Supplemental Fig. 3A). As has been shown for the recombinant protein, activity of PWD purified from leaves strictly depended on a preceding phosphorylation by GWD (Supplemental Fig. 3B) and phosphorylation predominantly (or exclusively) occurs at the C3-position of the Glc residues (Supplemental Fig. 3C).
Reduction of PWD in Transgenic Plants Causes a Starch Excess Phenotype
To investigate the in vivo function of PWD, an RNA interference (RNAi) construct was designed to repress the activity of this protein in Arabidopsis. Fifteen transgenic lines were analyzed by western blot, all of which proved to be strongly reduced in PWD protein (data not shown). Five of these lines were examined in more detail. As shown in Figure 7A the PWD protein amount in the RNAi lines 1 to 4 is below the limit of detection using western-blot analysis. In line 5 PWD expression is reduced by at least 75%. The analysis of starch contents in these plants revealed a metabolic phenotype for PWD. At the end of the day, the PWD-deficient plants contained up to 2 times more starch than ecotype Columbia wild type (Fig. 7B). Starch is nearly completely remobilized during night in wild-type but not in the transgenic plants. In the less inhibited RNAi-line 5, starch amounts are only slightly increased compared with wild-type plants. Thus, the extent of PWD inhibition correlates with the effect on starch content.
Figure 7.

Transgenic plants with reduced PWD (OK1) protein exhibit a starch excess phenotype. A, The PWD protein level is decreased in PWD RNAi plants. Leaf samples of five PWD RNAi lines and wild type were harvested at the end of the light period. Proteins were extracted and 40 μg each were separated by SDS-PAGE and examined by immunoblot analysis using an anti-PWD antibody. WT, Wild type; 1 to 5, PWD RNAi lines 1 to 5. B, Starch content in leaves of wild-type and PWD RNAi lines. Plants were grown in a 12-h-light/12-h-dark cycle. Samples were taken at the end of the light (white bars) and dark period (black bars), respectively. Values are means ± se; n = 4 plants. 1 to 5, PWD RNAi lines; WT, wild type; FW, fresh weight. C, Starch content in leaves of wild type and PWD knockout mutant. All leaves of wild-type plants (white symbols) or pwd plants (black symbols) were harvested at the times indicated. Values are means ± se; n = 4 plants.
We also obtained an insertion mutant (SALK_110814) and selected a homozygous line for the At5g26570 gene. No PWD protein could be detected in the mutant (data not shown). As observed with the RNAi plants, the PWD knockout mutant (pwd) contains considerably more starch than wild-type plants throughout the day/night cycle (Fig. 7C). Starch turnover occurred in the PWD-deficient plants, but the rate of starch degradation in the pwd plants was lower than that of the wild-type plants (Fig. 7C). This effect was also observed using an independent batch of plants grown under a 14-h-light period (data not shown).
The high starch phenotype resembles that of the GWD-deficient sex1 mutants. However, when grown under the same conditions the sex1 mutants accumulate more starch than the pwd plants. The lack of PWD had a minor effect on plant development, whereas the GWD knockout mutant sex1-3 is strongly retarded in growth when cultivated under a 12-h-light/12-h-dark regime (Supplemental Fig. 4).
Starch-bound phosphates in wild-type and PWD RNAi plants (lines 1–5) were analyzed by HPAEC-PAD and quantification of the peak areas following acid hydrolysis of the starch granules. The Glc-6-P:Glc-3-P ratio increased from 2.1 in wild type to 2.5 in the transgenic plants (mean of lines 1–5), ranging from 2.2 in line 5 to 2.7 in line 1. This increase, however, was caused by slightly elevated Glc-6-P levels, whereas the Glc-3-P content was essentially unchanged (data not shown). The Glc-6-P:Glc-3-P ratio in starch of pwd plants was also increased compared with wild type (data not shown). It has to be considered that the Glc phosphate levels in starch hydrolyzates mainly reflect starch phosphorylation during biosynthesis and not the transient phosphorylation of the granule surface during breakdown (Ritte et al., 2004).
PWD Binds to Leaf Starch Granules during Their Breakdown
To analyze whether binding of PWD to transitory starch is affected by the physiological state of the cell, granule-bound and soluble protein was extracted from leaves of wild-type plants that had been harvested either in the light or dark period. As revealed by western-blot analysis, binding of PWD to the surface of transitory starch granules strongly increases during starch mobilization in darkness (Fig. 8). In contrast, the PWD level in the buffer soluble fraction was equal in the light and dark samples. Binding of GWD to transitory starch in Arabidopsis also significantly increases in darkness (Fig. 8), in agreement with earlier results using leaves of potato and pea, respectively (Ritte et al., 2000a).
Figure 8.

PWD (OK1) binds to the surface of leaf starch granules during their degradation. Leaves of Arabidopsis wild-type plants were harvested 2 h before (L) and 2 h after the end of the light period (D). Soluble proteins and starch were extracted. Proteins bound to the surface of the starch granules were released by incubation in SDS-sample buffer at RT. Proteins released from 6.5 mg starch (bound) or 40 μg of soluble protein (soluble) were subjected to SDS-PAGE and immunoblot analysis using antibodies against PWD and GWD. The experiment was repeated with an independently grown batch of plants and yielded the same result.
DISCUSSION
The factors and mechanisms leading to the degradation of the crystalline starch granule are largely unknown, but there is increasing evidence that phosphorylation of starch by GWD is involved (Zeeman et al., 2004). In a first approach to unravel the link between the GWD-catalyzed phosphorylation of starch and the degradation of the polyglucans, we searched for proteins displaying enhanced affinity to phosphorylated starch compared with nonphosphorylated starch. We identified a novel protein (PWD) that preferentially binds to phosphorylated starch and is capable of glucan phosphorylation. The reaction catalyzed by PWD differs from that of GWD and, thus, represents a novel enzymatic activity. In contrast to GWD, phosphorylating activity of PWD was only observed using substrates that already contained glucan-bound phosphate. In vitro phosphorylated sex1-3 granules were a suitable substrate for PWD, whereas we did not detect PWD-catalyzed phosphate incorporation into the unmodified phosphate free sex1-3 starch. Likewise, starch granules from wheat (Sigma S-5127) were not phosphorylated by PWD, but following prephosphorylation with GWD they can serve as substrate for PWD (data not shown).
Why is PWD active only on phosphorylated starch? It is reasonable to assume that PWD either phosphorylates glucan chains that were previously phosphorylated by GWD, or it phosphorylates unphosphorylated chains within a phosphorylated matrix. Analysis of the phosphorylated glucan chains following in vitro phosphorylation with 33P-ATP shows that the latter predominates. Approximately 80% of the incorporated label was recovered in singly phosphorylated glucan chains; the remaining radioactivity was found in doubly phosphorylated chains. Probably phosphate incorporation by GWD locally alters the starch structure and thereby generates phosphorylation sites that can be used by PWD.
Whereas activity of PWD strictly depends on the presence of glucan bound phosphate binding of the protein to carbohydrates does not. PWD can bind to the unmodified sex1-3 granules, albeit with low efficiency. The protein also binds to immobilized maltoheptaose (S. Orzechowski, unpublished data), and we made use of this to enrich the protein from leaf extracts. These maltoheptaose beads were also not at all phosphorylated by the recombinant PWD (data not shown).
An important difference between PWD and GWD is the site of phosphate incorporation. GWD phosphorylates both the C3 and the C6 position, with a clear preference of the latter. In contrast, PWD phosphorylates preferably the C3 position (Fig. 6). A low extent of C6-phosphorylation cannot be ruled out; the same holds true for C2-phosphorylation. It has been suggested by Tabata and Hizukuri (1971) that about 1% of the phosphate in potato tuber starch could be linked to the C2 position. Glc-2-P is far more acid labile than Glc-3-P (Tabata and Hizukuri, 1971; Kokesh et al., 1978), and complete dephosphorylation during acid hydrolysis as applied here can be expected. Therefore, we cannot exclude that Glc-2-P residues may also be formed by PWD (or GWD as well). NMR analysis could provide further information whether or not C3 is the single phosphorylation site of PWD. However, due to the low level of PWD-catalyzed phosphate incorporation in vitro and the high background level of phosphate esters in the starch substrate, NMR-analysis of the PWD products is not practicable at present.
Starch is phosphorylated during its biosynthesis (Nielsen et al., 1994; Wischmann et al., 1999) but in addition also during its degradation (Ritte et al., 2004). The amounts of Glc-6-P and Glc-3-P determined in hydrolyzed starch mainly reflect starch phosphorylation during starch synthesis (Ritte et al., 2004). The absolute level of starch-bound Glc-3-P is not significantly altered in the PWD-deficient plants. This may indicate that the enzyme is not involved in biosynthesis associated phosphorylation. However, GWD can attach phosphate at both C3 and C6 positions of Glc residues in amylopectin, and may thus partly compensate for lacking PWD activity. The amount of GWD protein is unaltered in the PWD-deficient plants as revealed by western-blot analysis (data not shown), but it is possible that more glucan targets become available for GWD if PWD is lacking.
The increased phosphorylation of the granule surface during breakdown of starch in chloroplasts (Ritte et al., 2004) should favor activity of PWD during starch mobilization. In fact, binding of PWD to transitory starch granules is strongly increased during degradation of the starch particle (Fig. 8). Phosphorylation during starch mobilization is restricted to the outermost layer of the granule, and the phosphate esters introduced during breakdown underlie a rapid turnover (Ritte et al., 2004). Consequently, the reduction of C3-phosphorylation during the period of starch mobilization is not expected to noticeably affect the total Glc-3-P level of starch.
The starch excess phenotype observed in the PWD-deficient plants demonstrates that this enzyme plays an important metabolic role, and lack of PWD-catalyzed starch phosphorylation cannot be (fully) compensated for by other enzymes. Since the activity of PWD depends on a preceding starch phosphorylation by GWD, the lack of GWD in mutant plants should also abolish starch phosphorylation by PWD. Consistently, no Glc-3-P residues could be detected in starch of the GWD-free Arabidopsis mutant sex1-3 (Yu et al., 2001). The starch excess phenotype in the GWD-deficient plants is probably attributable to a combined reduction of GWD and PWD activity, which might explain the more severe phenotype compared with PWD mutants.
Further studies are required to explore the link between phosphorylation and degradation of starch. It has recently been reported that the in vitro degradation of granules isolated from turions of the duckweed by starch associated proteins (including a putative GWD ortholog) could be increased by addition of ATP, thereby enabling starch phosphorylation (Reimann et al., 2004). We have obtained similar results using potato leaf starch. Furthermore, we know from in vitro studies that as-yet unidentified proteins exist in Arabidopsis that degrade starch more efficiently if it is phosphorylated (G. Ritte, unpublished data). Future work will focus on the characterization of these proteins to evaluate their role in starch degradation in vivo.
We propose that the newly identified PWD acts downstream of GWD and is involved in starch breakdown in leaves. Based on molecular modeling it has been suggested that phosphate linked to the C6 position aligns with the surface of double helical motifs in amylopectin, whereas phosphate esterified to the C3-position protrudes from the double helical structure. Thus, double helix packing should be more affected by C3-phosphorylation (Blennow et al., 2002; Engelsen et al., 2003). In addition to the GWD-catalyzed phosphate incorporation, phosphorylation of the C3 position by PWD could play an important role in rendering the starch granule accessible for degrading enzymes by disturbing the helix packing and increasing the hydrophilicity.
MATERIALS AND METHODS
Plant Material and Growth Conditions
Arabidopsis (Arabidopsis thaliana) plants were cultivated in a growth cabinet under controlled conditions (12 h light/12 h dark, 20°C/16°C, 60%/70% relative humidity [day/night], and approximately 150 μmol quanta m−2 s−1). Seeds of the mutant SALK_110814 were obtained from the Nottingham Arabidopsis Stock Center (http://arabidopsis.info, Nottingham, UK). Seeds of the Arabidopsis sex1-1 and sex1-3 mutants (Yu et al., 2001) were a kind gift of Dr. Samuel Zeeman (University of Berne, Switzerland).
Chemicals and Enzymes
[γ-33P]ATP (10 mCi/mL; 3,000 Ci/mmol), [β-33P]ATP (10 mCi/mL; 800 Ci/mmol), and [32P]phosphoric acid (54 mCi/mL, carrier free) were all purchased from Hartmann Analytic (Braunschweig, Germany). Glc-3-P was synthesized as described elsewhere (Ritte et al., 2002).
Preparation of Leaf Starch Granules
Leaves (10–30 g) were frozen in liquid nitrogen and homogenized in a mortar. For the analysis of granule-bound proteins in vivo, starch was extracted as described (Ritte et al., 2000a). For structural analysis of starch and for isolation of starch granules serving as raw material for binding or activity assays, the above method was modified as follows. The extraction buffer consisted of 20 mm HEPES-KOH, pH 8.0, 0.2 mm EDTA, 0.5% (w/v) Triton X-100. Following passage through a Percoll cushion (Ritte et al., 2000a), the starch pellet was washed once in extraction buffer. Proteins bound to the starch granule surface were then removed by incubation with 0.5% (w/v) SDS on a rotating wheel (approximately 10 μL SDS-solution/mg starch, 3 × 15 min, room temperature [RT]). Subsequently SDS was removed by washing the granules three times (15 min each) in 50 mm HEPES-KOH, pH 7.2, and once in water. Starch granules were either used immediately or dried under vacuum and stored at −20°C.
Radioactive Starch Phosphorylation Assay
Five milligrams starch were resuspended in 50 mm HEPES-KOH, pH 7.5, 1 mm EDTA, 6 mm MgCl2, 0.025% Triton X-100, and radiolabeled ATP as indicated in a total volume of 0.5 mL if not otherwise stated. The radiolabel was either [β-33P]ATP, [γ-33P]ATP, or [βγ-33P]ATP. The latter was obtained by enzymatic randomization of [γ-33P]ATP according to Ritte et al. (2004) and is predominantly labeled at the β-position (Ritte et al., 2003). Reactions were started by addition of protein. The samples were agitated on a rotating wheel at RT for the times indicated, and the reaction was terminated by adding SDS (final concentration, 2%). The starch was washed two times in water and at least four times in 2 mm ATP as described (Ritte et al., 2004). Subsequently, the granules were resuspended in 0.1 mL water, mixed with 3 mL scintillation fluid, and the radioactivity was counted.
In Vitro Phosphorylation of Leaf Starch Granules by Recombinant GWD
Unless otherwise stated, dried Arabidopsis sex1-3 leaf starch granules were resuspended in 50 mm HEPES-KOH, pH 7.5, 1 mm EDTA, 6 mm MgCl2, 0.5 mm ATP, and purified recombinant potato (Solanum tuberosum) GWD (Ritte et al., 2002) was added to give final concentrations of 10 mg starch/mL buffer and 0.25 μg GWD/mg starch. Following incubation overnight on a rotating wheel, the reaction was stopped by adding SDS (final concentration, 2%). Removal of recombinant protein and washing of starch was done as described above (starch granule preparation). The amount of phosphate incorporated was estimated in parallel samples in which, however, [βγ-33P]ATP (0.5–1 μCi) was included under otherwise identical conditions. In the [βγ-33P]ATP preparation, ≥80% of the label is normally present in the β-position (Ritte et al., 2003). Estimations were done as if 100% of the label were present in the β-position. Thus, the indicated phosphate incorporation is slightly underestimated.
Protein Extraction
Arabidopsis leaves were harvested and immediately frozen in liquid nitrogen. Leaves were ground in a mortar, and 3 to 4 volumes (v/w) binding buffer (50 mm HEPES-KOH, pH 7.2, 1 mm EDTA, 2 mm dithioerythritol, 2 mm benzamidine, 2 mm ɛ-aminocaproic acid, 0.5 mm phenylmethylsulfonylfluoride, 0.02% Triton X-100) were added (4°C). All following steps were carried out at 4°C. Plant material was additionally homogenized in an Ultraturrax (2 × 10 s, maximum speed), passed through a 100-μm nylon mesh, and centrifuged for 20 min (20,000g). Proteins were precipitated by adding ammonium sulfate (75% saturation). Following centrifugation, the precipitate was resolved in binding buffer and desalted using Sephadex G25 (Amersham Bioscience, Freiburg, Germany).
In Vitro Binding Assay
Both in vitro-phosphorylated and nonphosphorylated sex1-3 leaf starch granules (50 mg each) were hydrated in binding buffer and were then mixed with freshly prepared Arabidopsis protein extract (total volume 0.8 mL, 4–10 mg protein mL−1). Following incubation for 15 min at 4°C, unbound proteins were removed by centrifugation through a 4-mL Percoll-cushion (see above). The pelleted starch was washed in binding buffer (2 × 5 min, 4°C). Bound proteins were solubilized by incubating the starch granules with SDS sample buffer (62.5 mm Tris-HCl, pH 6.8, 2% [w/v] SDS, 10% [w/v] glycerol, 0.01% [w/v] bromphenol blue) for 15 min at RT with shaking. After centrifugation (5 min, 20,000g), the supernatant was transferred to a new tube and incubated at 95°C for 5 min. Equal amounts of both samples were separated by SDS-PAGE (9% acrylamide in the separation gel). Gels were stained with colloidal Coomassie Blue (Roth, Karlsruhe, Germany), and protein bands were cut out and subjected to tryptic digestion and matrix-assisted laser-desorption ionization mass spectrometry analysis as described (Ritte et al., 2000b).
Antibodies
Antibodies were raised in rabbits. A polyclonal antibody against the purified recombinant OK1 (PWD) was produced by Eurogentec (Seraing, Belgium). For AGPase detection, an antibody raised against recombinant potato AGPase (Tiessen et al., 2002) was used. The PEPCase antibody was raised against the purified sorghum (Sorghum vulgare) enzyme (Vidal et al., 1983). The GWD antibody was raised against the purified protein from potato (Ritte et al., 2000a).
Analysis of Protoplast and Chloroplast Extracts
Chloroplasts were isolated from Arabidopsis protoplasts using a protocol adapted from Kunst (1998). The healthy green leaves of two mature Arabidopsis plants (approximately 4 g fresh weight) were harvested at the end of the dark period. Leaves were cut into 1-mm slices and the slices were washed for 5 min with 20 mL protoplast isolation buffer (PIB; 0.5 m sorbitol, 1 mm CaCl2, 10 mm MES-KOH, pH 6.0). Cell wall digestion was done in 10 mL PIB containing degrading enzymes (100 mg cellulase Onozuka, 25 mg macerozyme Onozuka) and 100 mg polyviniylpolypyrrolidone. The slices were vacuum infiltrated and incubated without shaking at RT for 3 h. The solution was then poured over a common kitchen sieve and the digested leaf material was carefully washed drop-wise with 30 mL PIB to release the protoplasts. The protoplast suspension was passed through a nylon net with 100-μm mesh width and was then centrifuged for 5 min at 40g using a swing-out rotor at 4°C. The supernatant was removed with a pipette and the protoplasts were carefully resuspended in 5 mL protoplast lysis buffer (PLB; 0.4 m sorbitol, 10 mm NaHCO3, 10 mm EDTA, 20 mm Tricine-KOH, pH 8.0).
For chloroplast isolation, 25 mL PLB were added to the protoplast suspension, which was hand shaken vigorously for 1 min and then passed through a nylon net with 30-μm mesh width to rupture the protoplasts. The suspension was then centrifuged at 400g for 2 min at 4°C and the supernatant was removed. The chloroplast pellet was carefully resuspended in 4 mL PLB. As judged by microscopic inspection, the chloroplasts were highly intact.
Three volumes of protoplast or chloroplast suspension were mixed with 1 volume of 4-fold concentrated SDS-sample buffer. Equal amounts of proteins extracted from protoplasts or chloroplasts, respectively, and Mr marker proteins were separated by SDS-PAGE (10% polyacrylamide). Western blots were performed essentially as described by Tiessen et al. (2002).
Cloning of the OK1 (PWD) cDNA
RNA was isolated from leaves of Arabidopsis wild type (ecotype Columbia) according to Logemann et al. (1987) and cDNA was prepared with the SuperScript one-step RT-PCR system (Invitrogen) using OK1rev1 primer (5′-GACTCAACCACATAACACACAAAGATC-3′). The complete OK1 coding sequence including 22 bp upstream of ATG and 69 bp downstream of TAG was amplified in a PCR reaction with the cDNA as template, the primers OK1fwd2 (5′-ATCTCTTATCACACCACCTCCAATG-3′) and OK1rev2 (5′-TGGTAACGAGGCAAATGCAGA-3′), using the Expand High Fidelity PCR System (Roche, Mannheim, Germany). The PCR product was subcloned into a pGEM-T cloning vector (Promega, Mannheim, Germany). Sequencing of OK1pGEM-T-1 revealed an Escherichia coli insertion sequence (IS1) at 540 bp. Another PCR was done and a second clone (OK1pGEM-T-2) was sequenced and revealed four base substitutions compared with the published genomic sequence of At5g26570 resulting in one amino acid substitution (L854→R). A 1,081-bp EcoRI/BamHI fragment from OK1pGEM-T-2 was replaced by the corresponding EcoRI/BamHI fragment from OK1pGEM-T-1. Sequencing of the resulting OK1pGEM-T revealed two remaining base substitutions (G208→A; C1116→G) but no amino acid replacement.
Cloning of the OK1 (PWD) Expression Vector
The expression vector containing the OK1 coding sequence was constructed by means of the GATEWAY technology (Invitrogen) according to the manufacturer's protocols. The attB recombination sites were added to the OK1 cDNA in a PCR with OK1pGEM-T as template and the primers OK1EntryB1 (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCGAGAGCATTGGCAGCCATTG-3′) and OK1EntryB2 (5′-GGGGACCACTTTGTACAAGAAAGCTGGGTCTACAGAGGTTGTGGCCTTGAC-3′), using the Expand High Fidelity PCR System (Roche). The Entry Clone OK1pSPECTRE was obtained via the BP reaction with OK1attB PCR product and the Entry Clone vector pSPECTRE, which was derived from pDONR201 (Invitrogen) by replacing the PvuI/NruI fragment of the kanamycin resistance gene by the spectinomycin resistance gene. The OK1pDEST17 expression vector was created in the LR reaction with OK1pSPECTRE and pDEST17 (Invitrogen). Sequencing of OK1pDEST17 revealed a base transposition leading to an amino acid substitution (D143→N) in the recombinant protein.
Purification of Recombinant OK1 (PWD)
E. coli BL21 Star (DE3; Invitrogen) cells were transformed with the OK1pDEST17 plasmid and incubated in 1L TB medium containing 100 μg/mL ampicillin overnight (30°C, 250 rpm). Expression of the OK1 protein was induced by adding isopropylthio-β-galactoside to a final concentration of 1 mm at an OD600 of approximately 0.8. Cells were harvested at an OD600 of approximately 1.6 by centrifugation and stored at −80°C until use. For purification of recombinant OK1 protein cells were resuspended (0.25 g/mL) in lysis buffer (50 mm HEPES, pH 8.0, 300 mm NaCl, 10 mm imidazole, 1 mg/mL lysozyme; 1 tablet/40 mL Complete EDTA free protease inhibitors, Roche), incubated for 30 min on ice, and additionally lysed by sonification. Cell debris was removed by centrifugation and the supernatant was passed through a 0.45-μm filter. Purification of the recombinant protein was achieved using a column with 1 mL Ni-NTA agarose resin (Qiagen, Hilden, Germany). After washing with 8 mL lysis buffer, recombinant protein bound to Ni-NTA agarose was eluted with elution buffers as follows: 2 × 1 mL E1, 1 mL E2, 3 × 1 mL E3 (50 mm HEPES, pH 8.0; 300 mm NaCl; 50, 75, 250 mm imidazole for E1, E2, and E3, respectively). Fractions with adequate amount and purity of recombinant OK1 protein were pooled and concentrated by ultrafiltration (Diaflo PM30, Amicon, Millipore, Bedford, MA). The buffer in the OK1 preparation was changed using a HiTrap-Desalting column (Amersham) equilibrated with 50 mm HEPES-KOH, pH 7.5, 1 mm EDTA, 1 mm DTE. Aliquots were stored at −80°C until use.
RNAi Plants
The RNAi construct for silencing of the OK1 (PWD) gene was established by cloning a pair of short PCR-amplified OK1 cDNA fragments in opposite orientation into pHannibal (Wesley et al., 2001). The two 302-bp fragments, representing the region 2,153 to 2,454 of the complete OK1 cDNA, were obtained from PCR reactions with two primer pairs: (1) OK1-R1a-fw (5′-TCCGATGGATCCAGCAACTTCTGGTGGTCCTAT-3′) and OK1-R1a-re (5′-TTGCGCATCGATGGTCGCACTGGATTTGGAAG-3′); and (2) OK1-R1b-fw (5′-TCCGATCTCGAGACTAGTCCAGCAACTTCTGGTGGTCCT-3′) and OK1-R1b-re (5′-TTGCGCGGTACCGGTCGCACTGGATTTGGAA-3′). Appropriate restriction enzyme sites linked to the primers permitted subsequent two-step cloning of the PCR products in opposite directions into pHannibal. After digestion of the resulting vector with NotI restriction enzyme, the RNAi constructs were ligated into the NotI restriction site of the binary vector pART27 (Gleave, 1992) giving OK1-R1-pART27.
Arabidopsis plants were transformed by the dipping method of Clough and Bent (1998). Fifteen kanamycin resistant T1 RNAi plants were analyzed by western blot for reduction of OK1 protein levels. All RNAi plants showed drastically reduced levels of OK1 protein compared with the wild type. Seeds were harvested and further analysis on starch content was done with five lines of the T2 progeny.
Autocatalytic Phosphorylation
In vitro phosphorylation of PWD (OK1) was analyzed essentially as described for GWD (Ritte et al., 2002).
Determination of 33P-Orthophosphate
A photometric assay for the detection of orthophosphate (Parvin and Smith, 1969) was modified to allow for the detection of radiolabeled orthophosphate. This assay bases on the formation of a phosphomolybdovanadate complex, followed by its subsequent extraction into butanol. In the original protocol, the maximum absorbancy of the phosphomolybdovanadate complex in butanol is measured at 310 nm against a blank, and the amount of inorganic phosphate is determined using standard curves (Parvin and Smith, 1969). Since the amounts of orthophosphate released in our enzymatic assays were too low to allow for accurate detection using the photometric assay, the activities of OK1 (PWD) or GWD were analyzed using radiolabeling assays with 33P-ATP as substrate (see above) and the radioactivity in the butanol phase was determined. The activity of OK1 and GWD was tested in assays containing 10 mg starch in 0.5 mL 50 mm HEPES-KOH (pH 7.5), 1 mm EDTA, 6 mm MgCl2, 0.025% Triton X-100, 10 μm ATP containing 1.65 × 106 cpm of either [γ-33P]ATP or [β-33P]ATP. Reactions were started by adding 20 μL (2.2 μg) of OK1 or GWD, respectively. After 80 min of incubation at RT on a rotating wheel, the samples were centrifuged (2 min, 13,000g) and 400 μL of the supernatant were removed and boiled for 5 min. Forty microliters of the boiled supernatant were diluted with 360 μL water. Subsequently, 800 μL butanol and 400 μL ammonium metavanadate-molybdate reagent (reagent I; Parvin and Smith, 1969) were added, the samples were mixed on a vortex for 10 s, and briefly centrifuged to achieve quick phase separation. Aliquots of the upper butanol phase (2 × 200 μL) were immediately mixed with 8 mL scintillation fluid each and the radioactivity was counted. Under the conditions applied, the recovery of inorganic phosphate in the butanol layer was ≥95% as determined in controls with known amounts of 32P orthophosphate as internal standards. These controls are essential, as the buffer composition can substantially affect the recovery.
HPAEC-PAD Analysis
HPAEC-PAD analysis was performed essentially as described (Ritte et al., 2000b). However, a Dionex DX 600 equipped with a CarboPac PA 100 column was used. Five milligrams of in vitro phosphorylated starch were hydrolyzed with 90 μL of 0.7 n HCl for 2 h at 95°C and subsequently neutralized with 0.7 n NaOH. Prior to HPAEC-PAD analysis, samples were centrifuged through 10-kD membranes (Ultrafree MC, Millipore) that had been washed once with water.
Analysis of Starch-Bound Phosphate in Wild-Type and Transgenic Plants
All leaves from wild-type and transgenic plants (8–10 plants each) were harvested at the end of the 12-h-light period. Starch granules were isolated. Seven milligrams starch each were hydrolyzed in 150 μL 0.7 n HCl for 2 h at 95°C. Three aliquots of each granule preparation were hydrolyzed. Following neutralization and filtration through 10-kD membranes (see above), Glc was determined and samples equivalent to 5 μmol Glc each were analyzed by HPAEC-PAD. The three different hydrolyzates per starch sample yielded highly reproducible results. The elution of Glc-3-P and Glc-6-P was monitored using authentic standards.
Purification of OK1 (PWD) from Leaves of the Arabidopsis sex1-3 Mutant
Leaves of the Arabidopsis sex1-3 mutant were harvested at the end of the light period (20 g fresh weight), proteins were extracted, precipitated, and desalted as described. However, ammonium sulfate precipitation was from 0% to 50% saturation. Further purification was achieved by affinity chromatography using 0.5 mL maltoheptaose immobilized on agarose beads (M-9676, Sigma-Aldrich, Steinheim, Germany) in a column with gravity flow. All steps were carried out at 4°C. The column was washed with 10 mL binding buffer, 2.5 mL protein extract (19.5 mg protein) was applied, and the flowthrough was applied once more. After washing with 10 mL binding buffer, bound proteins were eluted with Dextri maltose (ICN, Eschwege, Germany) dissolved in binding buffer (1 mL 10 mg/mL followed by 1 mL 50 mg/mL). Eluted proteins were further concentrated using spin column filters with an exclusion limit of 10 kD (Amicon YM-10,Microcon, Millipore), washed with 1 volume binding buffer, and again concentrated 4-fold with spin columns to give a final volume of 0.5 mL with a protein concentration of 50 μg/mL.
Analysis of Starch Content in Leaves
Measuring of the leaf starch content was done basically as described by Abel et al. (1996). All leaves of 4- to 5-week-old plants were harvested, frozen in liquid nitrogen, and homogenized in a mortar. Samples of 40 to 60 mg homogenized material were extracted 2 times, each with 1 mL 80% (v/v) ethanol for 20 min at 80°C. Insoluble material was washed in 1 mL water, vacuum dried, resuspended in 0.5 mL 0.2 n KOH, and incubated at 95°C or 1 h. After neutralization with 1 n acetic acid and centrifugation, 25 μL of the supernatant were mixed with 50 μL amyloglucosidase solution (starch determination kit, R-Biopharm, Darmstadt, Germany). However, we supplemented the amyloglucosidase solution with 1 unit of α-amylase from Bacillus amyloliquefaciens (Roche). Enzymatic hydrolysis of starch and subsequent enzymatic determination of Glc was performed according to the provider's protocol.
Upon request, all novel materials described in this publication will be made available in a timely manner for noncommercial research purposes, subject to the requisite permission from any third-party owners of all or parts of the material. Obtaining any permissions will be the responsibility of the requestor.
Sequence data from this article have been deposited with the EMBL/GenBank data libraries under accession number AJ635427.
Supplementary Material
Acknowledgments
We thank Anja Fröhlich and Torsten Schulze (MPI, Golm, Germany) for helping with plant transformation, Silke Gopp for the maintenance of plants, Anke Scharf for technical assistance, and Nora Eckermann (Plant Physiology, University of Potsdam, Germany) for advice in the HPAEC-PAD analysis. We are grateful to Jean Vidal (University of Paris) and Maria Ines Zanor (MPI) for the gift of the anti-PEPcarboxylase antibody, and Ben Trevaskis (Commonwealth Scientific and Industrial Research Organization, Canberra, Australia) for the gift of pSPECTRE vector plasmid. We thank the Salk Institute and the Nottingham Arabidopsis Stock Center for provision of the T-DNA insertion line.
This work was supported by the Deutsche Forschungsgemeinschaft (grant nos. SFB 429 TP–B2 to M.S. and TP–B7 to G.R. and P.G.).
The online version of this article contains Web-only data.
Article, publication date, and citation information can be found at www.plantphysiol.org/cgi/doi/10.1104/pp.104.055954.
References
- Abel GJ, Springer F, Willmitzer L, Kossmann J (1996) Cloning and functional analysis of a cDNA encoding a novel 139 kDa starch synthase from potato (Solanum tuberosum L.). Plant J 10: 981–991 [DOI] [PubMed] [Google Scholar]
- Blennow A, Bay-Smidt AM, Olsen CE, Møller BL (1998) Analysis of starch-bound glucose 3-phosphate and glucose 6-phosphate using controlled acid treatment combined with high-performance anion-exchange chromatography. J Chromatogr A 829: 385–391 [Google Scholar]
- Blennow A, Nielsen TH, Baunsgaard L, Mikkelsen R, Engelsen SB (2002) Starch phosphorylation: a new front line in starch research. Trends Plant Sci 7: 445–450 [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J 16: 735–743 [DOI] [PubMed] [Google Scholar]
- Dong Q, Schlueter SD, Brendel V (2004) PlantGDB, plant genome database and analysis tools. Nucleic Acids Res 32: D354–D359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300: 1005–1016 [DOI] [PubMed] [Google Scholar]
- Engelsen SB, Madsen AO, Blennow A, Motawia MS, Møller BL, Larsen S (2003) The phosphorylation site in double helical amylopectin as investigated by a combined approach using chemical synthesis, crystallography and molecular modeling. FEBS Lett 541: 137–144 [DOI] [PubMed] [Google Scholar]
- Gleave AP (1992) A versatile binary vector system with a T-DNA organisational structure conducive to efficient integration of cloned DNA into the plant genome. Plant Mol Biol 20: 1203–1207 [DOI] [PubMed] [Google Scholar]
- Goss NH, Evans CT, Wood HG (1980) Pyruvate phosphate dikinase: sequence of the histidyl peptide, the pyrophosphoryl and phosphoryl carrier. Biochemistry 19: 5805–5809 [DOI] [PubMed] [Google Scholar]
- Kleffmann T, Russenberger D, von Zychlinski A, Christopher W, Sjölander K, Gruissem W, Baginsky S (2004) The Arabidopsis thaliana chloroplast proteome reveals pathway abundance and novel protein functions. Curr Biol 14: 354–362 [DOI] [PubMed] [Google Scholar]
- Kokesh FC, Cameron DA, Kakuda Y, Kuras PV (1978) Hydrolysis of alpha-D-glucopyranose 1,2-cyclic phosphate: the effect of pH and temperature on product distribution, and position of opening of phosphate diester ring in formation of D-glucose 2-phosphate. Carbohydr Res 62: 289–300 [Google Scholar]
- Kunst L (1998) Preparation of physiologically active chloroplasts from Arabidopsis. Methods Mol Biol 82: 43–48 [DOI] [PubMed] [Google Scholar]
- Logemann J, Schell J, Willmitzer L (1987) Improved method for the isolation of RNA from plant tissues. Anal Biochem 163: 16–20 [DOI] [PubMed] [Google Scholar]
- Lorberth R, Ritte G, Willmitzer L, Kossmann J (1998) Inhibition of a starch-granule-bound protein leads to modified starch and repression of cold sweetening. Nat Biotechnol 16: 473–477 [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, et al (2003) CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res 31: 383–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen R, Baunsgaard L, Blennow A (2004) Functional characterization of alpha-glucan,water dikinase, the starch phosphorylating enzyme. Biochem J 377: 525–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narindrasorasak S, Bridger WA (1977) Phosphoenolypyruvate synthetase of Escherichia coli: molecular weight, subunit composition, and identification of phosphohistidine in phosphoenzyme intermediate. J Biol Chem 252: 3121–3127 [PubMed] [Google Scholar]
- Nielsen TH, Wischmann B, Enevoldsen K, Møller BL (1994) Starch phosphorylation in potato tubers proceeds concurrently with de novo biosynthesis of starch. Plant Physiol 105: 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvin R, Smith RA (1969) Determination of inorganic phosphate in the presence of labile organic phosphates. Anal Biochem 27: 65–72 [DOI] [PubMed] [Google Scholar]
- Reimann R, Hippler M, Machelett B, Appenroth KJ (2004) Light induces phosphorylation of glucan water dikinase, which precedes starch degradation in turions of the duckweed Spirodela polyrhiza. Plant Physiol 135: 121–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritte G, Eckermann N, Haebel S, Lorberth R, Steup M (2000. b) Compartmentation of the starch-related R1 protein in higher plants. Starch-Stärke 52: 179–185 [Google Scholar]
- Ritte G, Lloyd JR, Eckermann N, Rottmann A, Kossmann J, Steup M (2002) The starch-related R1 protein is an alpha-glucan, water dikinase. Proc Natl Acad Sci USA 99: 7166–7171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritte G, Lorberth R, Steup M (2000. a) Reversible binding of the starch-related R1 protein to the surface of transitory starch granules. Plant J 21: 387–391 [DOI] [PubMed] [Google Scholar]
- Ritte G, Scharf A, Eckermann N, Haebel S, Steup M (2004) Phosphorylation of transitory starch is increased during degradation. Plant Physiol 135: 2068–2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritte G, Steup M, Kossmann J, Lloyd JR (2003) Determination of the starch-phosphorylating enzyme activity in plant extracts. Planta 216: 798–801 [DOI] [PubMed] [Google Scholar]
- Rosenberg IM (1996) Modified proteins and peptides. B. Phosphorylation. In IM Rosenberg, ed, Protein Analysis and Purification: Benchtop Techniques. Birkhäuser, Boston, pp 224–244
- Slattery CJ, Kavakli IH, Okita TW (2000) Engineering starch for increased quantity and quality. Trends Plant Sci 5: 291–298 [DOI] [PubMed] [Google Scholar]
- Tabata S, Hizukuri S (1971) Studies on starch phosphate. 2. Isolation of glucose 3-phosphate and maltose phosphate by acid hydrolysis of potato starch. Starch-Stärke 23: 267–272 [Google Scholar]
- Tatusova TA, Madden TL (1999) BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett 174: 247–250 [DOI] [PubMed] [Google Scholar]
- Tiessen A, Hendriks JH, Stitt M, Branscheid A, Gibon Y, Farré EM, Geigenberger P (2002) Starch synthesis in potato tubers is regulated by post-translational redox modification of ADP-glucose pyrophosphorylase: a novel regulatory mechanism linking starch synthesis to the sucrose supply. Plant Cell 14: 2191–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal J, Godbillon G, Gadal P (1983) Influence of light on phosphoenol pyruvate-carboxylase in sorghum leaves. 2. Immunochemical study. Physiol Plant 57: 124–128 [Google Scholar]
- Wesley SV, Helliwell CA, Smith NA, Wang MB, Rouse DT, Liu Q, Gooding PS, Singh SP, Abbott D, Stoutjesdijk PA, et al (2001) Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J 27: 581–590 [DOI] [PubMed] [Google Scholar]
- Wischmann B, Nielsen TH, Møller BL (1999) In vitro biosynthesis of phosphorylated starch in intact potato amyloplasts. Plant Physiol 119: 455–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TS, Kofler H, Häusler RE, Hille D, Flügge UI, Zeeman SC, Smith AM, Kossmann J, Lloyd J, Ritte G, et al (2001) The Arabidopsis sex1 mutant is defective in the R1 protein, a general regulator of starch degradation in plants, and not in the chloroplast hexose transporter. Plant Cell 13: 1907–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeman SC, Smith SM, Smith AM (2004) The breakdown of starch in leaves. New Phytol 163: 247–261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.