

Abstract
Three hydroxypyridinones (HOPOs) positional isomers - 1,2-HOPO (L1H) and its water soluble analogue (L1′H), 3,2-HOPO (L2H) and 3,4-HOPO (L3H)- have been investigated for the complexation of Zr(IV). Potentiometric and UV-Vis spectrometric studies show a higher thermodynamic stability for the formation of Zr(L1′)4 in comparison with Zr(L2)4 and Zr(L3)4 as well as a higher kinetic inertness in competition studies with EDTA or Fe3+ at radiotracer concentration with 89Zr. Besides the low pKa of L1H or L1′H (pKa = 5.01) in comparison with L2H and L3H (pKa = 8.83 and 9.55, respectively), the higher stability of Zr(L1′)4 can be attributed in part to the presence of the amide group next to the chelating oxygen that induces intramolecular H-bond and amide/π interactions that were observed by X-ray crystallography and confirmed by quantum chemical calculations. The data presented here indicates that the 1,2-HOPO L1′ exhibits the best characteristics for Zr(IV) complexation. However, 2,3-HOPO and 3,4-HOPO patterns, if appropriately tuned, for instance with the addition of an amide group as in the 1,2-HOPO ligand, may also become interesting alternatives for the design of Zr(IV) chelators with improved characteristics for application in nuclear imaging with 89Zr.
Introduction
Hydroxypyridinones (HOPOs) are compounds that have long been studied for their ability to complex hard metallic cations via their oxygen atoms (Scheme 1).1 They have particularly been considered as medical tools for complexation of Fe(III) for the treatment of iron overload.2,3 Due to the electronic properties of actinides (IV) elements being similar to Fe(III), HOPO-based chelators have also been developed as sequestering agents of actinides for decontamination or decorporation applications.4 HOPOs have also recently proven useful for the stable complexation of Gd(III) associated with improved relaxivity and sensitivity of Gd-based contrast agents for magnetic resonance imaging (MRI).5 In the field of nuclear medicine, HOPOs have also been studied as ligands of Ga(III), whose radioisotopes 67Ga and 68Ga can be used for nuclear imaging.6–8
Scheme 1.

Structure of ligands discussed in this article.
The Zr(IV) cation, with its relatively small ionic radius (0.84 Å) and high positive charge number, exhibits the characteristics of a hard Lewis acid and is ideal for strong complexation by hard bases such as HOPOs. In recent years, this metal has received a growing interest in nuclear imaging of cancers with the use of the β+-emitter 89Zr. With its relatively long half-life (t1/2 = 78.4 h) and favorable positron energy (βavg = 395.5 keV), high tumor/background signal ratio can be acquired up to several days after administration to the patient when 89Zr is associated with a long blood circulating vector such as an antibody. This radionuclide broadens the panel of β+-emitters available (68Ga, t1/2 = 1.1 h; 18F, t1/2 = 1.8 h; 44Sc, t1/2 = 3.97 h; 64Cu, t1/2 = 12.7 h; 86Y, t1/2 = 14.7 h or 124I, t1/2 = 100.2 h as examples of the most studied to date),9–11 and expands the range of diseases that can be imaged by positron emission tomography (PET).
To date, the only chelating agent that has proven successful for labeling biomolecules for clinical studies with 89Zr is desferrioxamine B (DFB, see Scheme 1), a natural siderophore comprised of three hydroxamic acid subunits involved in the complexation of Zr(IV).12 A large number of antibodies has been labeled with 89Zr-DFB and used in pre-clinical and clinical studies in recent years.13 However, in a number of cases, the in vivo stability of the Zr-DFB complex has proven insufficient as seen by the accumulation of free, osteophilic 89Zr in bones 2 to 4 days after injection of the labeled antibody.14–17
After more than 15 years of imaging studies with the sub-optimal 89Zr-DFB complex, only very recently have chemistry groups initiated research programs to provide new chelating agents with improved characteristics for stable complexation of 89Zr(IV) and safer in vivo use and improved sensitivity. Recent studies have provided a better insight of the aqueous behavior of Zr(IV) in the presence of hydroxamate ligands by crystallographic, potentiometric and quantum chemical studies.18,19 Results indicate the tendency of Zr(IV) to form a more stable complex in the presence of an octadentate chelator as compared to the hexadentate DFB. Accordingly, several octadentate ligands have been proposed to fit the Zr(IV) requirements. Most of the investigations have focused on the development of chelators bearing hydroxamate groups with either the acyclic,20–22 the cyclic23 or the aromatic hydroxamate (1,2-HOPO) pattern.24 These recent advances have led to the first hydroxamate based bifunctional chelators that provide 89Zr complexes with improved in vivo stability compared to DFB25,26. Alternatively, chelators bearing catechols,27,28 phosphonates29 or 3,2-HOPOs30 have also been proposed. The development of HOPO based chelators for nuclear imaging application is not new and several examples of chelators for 67Ga or 68Ga complexation have been reported over the recent years. 7,8,31 In line with these reported applications, bifunctional HOPO based chelators for 89Zr complexation have recently been developed and studied in vivo with varying degrees of success.30,32,33 The most stable to date, p-SCN-Bn-HOPO which is based on the 1,2-HOPO subunit, released a significantly lower fraction of the 89Zr activity to the bone in comparison with the same antibody radiolabeled using DFB-SCN. This new compound is however accompanied by a significant decrease of the 89Zr activity in the tumor which indicates that further improvements are still necessary.34
Whereas the development of HOPO-based chelators appears to be a viable alternative to hydroxamates, surprisingly little is known about the aqueous structure and thermodynamic stability of such Zr-HOPO complexes. This partial vision of the aqueous chemistry of Zr(IV) is a limitation for the rational design of optimally pre-organized chelating agents based on this class of ligands. Accordingly, we have initiated an in-depth investigation on the complexation chemistry of Zr(IV) towards the 1-hydroxy-2(1H)-pyridinone (1,2-HOPO) unit as well as its structural isomers 3-hydroxy-2(1H)-pyridinone (3,2-HOPO) and 3-hydroxy-4(1H)-pyridinone (3,4-HOPO) (Scheme 1), differing in their ability to sequester Zr(IV).
Results and discussion
Synthesis of ligands and Zr(IV) complexes
As models for the study, we chose to synthesize the most simple and unconstrained HOPOs while introducing a short chain that would mimic the electron density on the ring when attached to the backbone of a chelator. A rich synthetic chemistry of HOPOs is available from Raymond or Hider’s groups which have developed an array of HOPO-based chelators. In most cases, convenient methods were to introduce a carboxylic acid on the ligand structure for conjugation to polyamines with formation of amide bonds.2,35–38 Accordingly, the O-benzyl protected HOPOs were synthesized following previously reported procedures.35,39,40 After conversion into their N-hydroxysuccinimidyl esters, they were conjugated to methylamine and deprotected to form 1,2-HOPO-NHMe (L1H), 3,2-HOPO-NHMe (L2H) and 3,4-HOPO-NHMe (L3H). Both L2H and L3H were soluble in water whereas L1H exhibited poor solubility in this solvent. This lower solubility may be attributed to intramolecular hydrogen bonding of the amide hydrogen with the 1-hydroxy group in L1H that lowers the ability of the molecule to be solvated by water via hydrogen bonding, as reported with similar compounds.37 Consequently, a variation of L1H was also synthesized (L1′H), bearing a n-propyl instead of the methyl and reported to be water soluble.41 The improved water solubility made then possible further aqueous stability studies (Scheme 2). It is worth noting that besides the difference in the hydroxyl group position, a striking feature of (L1H) or (L1′H) that (L2H) and (L3H) do not exhibit is the presence of an amide group directly connected to the ring on the carbon next to the nitrogen. This amide group is expected to greatly influence metal complexation by modification of the electron density on the hydroxyl by inductive effect, and the pKa by stabilization of the deprotonated form due to hydrogen bonding as previously observed in other HOPOs studies.37,42 Zr(HOPO)4 complexes were formed by transchelation of zirconium(IV) acetylacetonate in the presence of a slight excess of ligand (4.5 equiv.) in refluxing methanol. All ligands formed the expected ZrL4 complexes as confirmed by NMR, mass spectrometry and elemental analyses. Additionally, Zr(L1)4 and Zr(L2)4 were isolated as crystals suitable for X-ray diffraction studies.
Scheme 2.

Synthesis of HOPOs L1H, L2H and L3H. i) EDCI, NHS, DMF, ii) R-NH2, THF, iii) AcOH/HClconc (1:1), iv) H2 (30 psi), Pd/C (10%), MeOH, v) Zr(acac)4, MeOH. L1′H was prepared in accordance with the reported procedure.41
Structure elucidation by crystallography and quantum chemical calculations
To optimally design pre-organized chelators with HOPOs, it is essential to know how the free ligands organize themselves without external constraint when forming a complex with the Zr4+ cation. Single crystal crystallography (see experimental details in ESI) shows that both L1 and L2 form a complex in which the central Zr atom is octacoordinated by four hydroxypyridinonates via their oxygen atoms (Figure 1a & 2). Zr(L2)4 differs by a lower degree of symmetry (triclinic crystal system) than Zr(L1)4 (monoclinic). Whereas each pair of oxygen atom exhibits a high symmetry in Zr(L1)4 (i.e Zr-O1 = 2.209 Å and Zr-O2 = 2.170, all Zr-O comprised between 2.165 and 2.215 Å), it is significantly lower in the case of Zr(L2)4 (Zr-O distances between 2.127 and 2.305 Å). Crystals of Zr(L3)4 could not be obtained and thus we resorted to quantum chemical calculation to obtain structural information of Zr(L3)4. This structure was built by modifying the crystal structure of Zr(L1)4 and then geometry optimized in the reaction field of water. The calculated structure is analogous to Zr(L2)4 with Zr-O distances ranging from 2.187 to 2.335 Å (Figure S4). A distinctive feature of the Zr(L1)4 complex is the H-bonds formed between the amide hydrogen atoms and the oxygen atoms coordinating Zr with i.e (N2)H-O1 = 1.978 Å and a donor/acceptor distance N2-O1 of 2.661 Å (Figure 1b). Such intramolecular H-bonds are known to provide pre-organization and subsequent extra stabilization to the complex as reported with other metals such as Gd3+.42 Additionally, whereas the four HOPO rings are equally spaced around the Zr atom in Zr(L2)4 and Zr(L3)4, the ligands in Zr(L1)4 are oriented as two pairs of stacked rings resulting in the HOPO rings being significantly pushed out of the planes formed by Zr and its chelating oxygen atoms (Figure 1c). For instance, plane (a) formed with Zr and its pair of chelating oxygen atoms (O4 and O5) intersects the ring plane (b) with an angle of 25° whereas it is only 10° in Zr(L2)4 (Figure 1c). Such a geometry can be attributed to amide/heteroarene π stacking, with the amide nitrogen to HOPO ring center distance of 3.30 Å (Figure 1d) that matches the calculated distance for similar interaction reported between N-methylacetamide and pyridinone, providing additional stabilization to the complex.43
Figure 1.

a) Crystal structure of Zr(L1)4. Selected bond lengths (Å) and angles (°): Zr1-O1, 2.209; Zr1-O2, 2.170; O1-Zr1-O1, 69.73; N1-O1-Zr1, 113.49; C1-O2-Zr1, 117.68. b) View of H-bond between amide proton and adjacent oxygen donor. c) Representation of the angle formed between the plane of Zr1-O4-O5 and the HOPO ring. d) View of Amide/HOPO ring π stacking. Water molecules and hydrogen atoms omitted for clarity. Crystallographic summary available in ESI, Table S1.44
Figure 2.

Crystal structure of Zr(L2)4. Selected bond lengths (Å) and angles (°): Zr1-O1, 2.272; Zr1-O2, 2.127; O1-Zr1-O1, 71.75; C1-O1-Zr1, 114.37; C2-O2-Zr1, 120.87. Water molecules and hydrogen atoms omitted for clarity. Crystallographic summary available in ESI, Table S1.44
Thermodynamic stability studies
To further study the properties of the HOPO moieties and their affinity for Zr(IV), their acid-base properties and the complexation with Zr(IV) were studied by potentiometric or UV-vis spectroscopic titrations in aqueous solution. The measured constants were compared with N-methyl-acetohydroxamic acid (Me-AHA, Scheme 1) which is the bidendate unit of DFB that we studied previously and which is known to form very stable zirconium (IV) complexes.18
Ligand protonation constants were determined by potentiometric titration carried out from low to high pH (results in Table 2). L1′ and L2 protonation constants were determined to be log Ka = 5.01 and log Ka = 8.83, respectively. L3 exhibits two successive protonation steps with a first log Ka value of 9.55 and a second one of 2.99 that were attributed to the 4-oxo group and the 3-hydroxy group, respectively, as reported in similar compounds.37 These data show that L1′ is significantly more acidic, which can be explained by the position of the hydroxyl on the pyridinonate nitrogen and by the adjacent amide group that acts on the acidity by two effects: i) the electron withdrawing effect of the amide group and ii) the intramolecular H-bond formed by the amide hydrogen that stabilizes the deprotonated form of the ligand, two features that L2 and L3 do not exhibit. All protonation constants are in good agreement with those previously reported by Raymond et al.41
Table 2.
Calculated p(Zr4+)a values for the complexes of L1′, L2, L3 and Me-AHA.
| L1′ | L2 | L3 | Me-AHA | |
|---|---|---|---|---|
| pZr4+ | 33.48 | 28.54 | 28.64 | 28.52 |
Values calculated at pH = 7.4 with [Zr4+]tot = 1 × 10−5 M and [L] = 5 × 10−5 M, based on the constants given in Table 2.
Based on the X-ray structures of the complexes indicating the favored formation of 1:4 (M:L) adducts, 0.25 equivalents of Zr(IV) relative to the ligand was used during complexation titrations. Measurements of the stability constants for L2 and L3 were performed by potentiometry. For L1′ whose Zr(IV) complexes exhibited poor aqueous solubility, we resorted to spectrophotometric titration that allows working at lower concentration than potentiometric methods and which prevented precipitation issues. L1′ complexation was monitored by a shift of the absorbance from 315 nm to 345 nm. All results are reported in Table 1.
Table 1.
Overall (log β) and stepwise (log K) thermodynamic protonation constants of L1′, L2 and L3 and their stability constants with Zr4+ in aqueous solution at 25.0 °C. Literature data for Me-AHA are provided for comparison.15
| Species | L1′ a | L2 b | L3 b | Me-AHA c | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| log β | log K | log β | log K | log β | log K | log β | log K | |
| LHd | 5.01 (7)e | 5.01 | 8.83 (4) | 8.83 | 9.55 (3) | 9.55 | 8.75 | 8.75 |
| LH2 | - | - | - | - | 12.54 (4) | 2.99 | - | - |
| ZrL3 | 31.3 (1)f | 31.3 | - | - | 41.1 (3) | 41.3 | 28.66 (5) | 3.44 |
| ZrL3H | 40.2 (1)f | 8.9 | 36.0 (6) | 36.0 (6) | 43.0 (4) | 2.4 | - | - |
| ZrL4 | 49.40 (1)f | 18.1 | 45.5 (1) | - | 49.27 (5) | 8.7 | 45.98 (1) | 17.32 |
| ZrL4(OH) | 41.97 (1)f | −7.43 | 35.9 (1) | −9.6 | - | - | - | - |
25.0 °C, I = 0.1 M in KCl;
25.0 °C, I = 0.2 M in KNO3;
I = 0.1 M KNO3, previously reported data, see Guérard et al. 18;
Charges are omitted for clarity.
Values in parentheses are standard deviations in the last significant digit;
Constants determined by UV spectrophotometric titration.
All three HOPO ligands exhibit high stability of Zr(IV) complexes with log KZrL4 > 45, but L1′ and L3 (log KZrL4 = 49.4 and 49.27, respectively) are superior to L2 and Me-AHA by four orders of magnitude (log KZrL4 = 45.5 and 45.98, respectively). Interestingly, despites its lowest log Ka, L1′ exhibits the highest log KZrL4. This observation deviates from the trends observed within other reported HOPO series in which the cumulative stability constants for metal complexation increase as proton affinity increases; although some exceptions are observed (but not as marked as in this study).37,38 This deviation from the trend with L1′ may be explained in part by the extra-stabilization by H-bond and amide/π stacking observed in the crystal structure (see previous section).
For a better comparison of Zr(IV) affinity independent of log Ka values, pZr4+ values were calculated for each ligand at physiological pH (Table 2). Results were nearly identical for L2, L3 and Me-AHA (pZr4+ ≈ 28.5) whereas L1′ exhibited a significantly higher pZr4+ value of 33.48, confirming the better ability of this ligand to complex Zr(IV).
The speciation diagrams plotted over the studied pH range (Figure 3) show that ZrL4 species predominate at neutral pH with all ligands. However, only L1′ leads to predominating Zr:L ratios of 1:4 over the whole studied pH range (from 2 to 12), whereas these ratios predominate from pH 2 to 9 with L2 and only from pH 5 to 10 with L3. This suggests that derivatives based on L1′ will be useful for 89Zr biomolecule radiolabeling over a very wide pH range. In addition, it is noted that from pH 4 to 8, the presence of Zr(OH)4 is significantly higher for L2 and L3 compared to L1′ for which no Zr(IV) hydroxides were detected below pH 8. This indicates that L2 and L3 are likely to produce lower radiolabeling yields with 89Zr.
Figure 3.

Speciation diagrams of Zr species with: (a) L1′, (b) L2 and (c) L3.
Overall, a higher affinity of Zr(IV) for L1′ than for L2 and L3 was observed. These constants are very close or even higher than those of Me-AHA that mimics the chelating groups present in DFB. It is likely that an octadentate chelator based on these HOPO motifs will lead to very stable Zr(IV) complexes, potentially more stable than DFB that lacks a fourth coordinating ligand. Indeed, recent work undertaken by Deri et al. and using the 1,2-HOPO motif supports this hypothesis.24,34
Zr-89 labeling, kinetic stability
To compare the ability of these ligands to complex Zr(IV) at the radiotracer level, complexation experiments with 89Zr were performed. The Me-AHA ligand (Scheme 1) previously investigated for 89Zr complexation was also included in this study as a reference.18 [89Zr]-zirconium(IV) oxalate was incubated under various conditions in the presence of a large excess of ligand and the formation of complexed 89Zr was analyzed using ITLC-SG chromatographic strips and a 10 mM EDTA solution at pH 6 as mobile phase. These chromatographic conditions were found to be the best to distinguish free 89Zr from complexed 89Zr. However, because of the low kinetic inertness of complexes in the absence of pre-organization of the “naked” ligands, dissociation occurred with the weakest ligands during the ITLC-SG elution by transchelation with the strong competitor EDTA present in the eluent. Whereas this chromatographic method exhibits limitations to assess the actual formation of the weakest complexes, it provided essential information in terms of kinetic inertness, a crucial parameter when assaying in vivo stability of complexes (see details in ESI).45 Accordingly, the formation of 89Zr complexes was studied from pH 4 to 10 (Figure 4). Results show a drastic superiority of L1′ with 89Zr fully complexed from pH 4 to 8, with a decrease starting only from pH 9. In contrast, all other ligands tested were significantly less efficient in retaining 89Zr activity in these conditions, with a maximum of 2.5 and 3.5 % with L2 and L3, respectively. Whereas far from L1′ efficiency, the reference Me-AHA was superior to L2 and L3 with up to 10% 89Zr complexed visible at pH 8. These results are in good agreement with data of the thermodynamic stability study that also show most favorable results with L1′.
Figure 4.

Influence of pH on the % 89Zr complexed after 30 min incubation with 0.4 mM ligand at 20 °C (n ≥ 3) with L1′ (
 ), L2 (
), L2 (
 ), L3 (
), L3 (
 ) and Me-AHA (
) and Me-AHA (
 ).
).
Additional tests were then performed to better probe the kinetic inertness of the complexes of 1,2-HOPO and Me-AHA against the presence of excess EDTA or Fe3+. Although the mechanism by which 89Zr is released from its chelate in vivo has not been fully elucidated, it is reasonable to invoke transchelation by endogenous chelators and/or transmetalation by oxophilic endogenous metal cations such as Fe3+. Moreover, 89Zr complexes that have previously demonstrated higher inertness than DFB in such in vitro conditions have often led to improved in vivo stabilities, indicating that such a simple test can help probing ability of ligand to remain complexed to the metal in vivo.26,34 L2 and L3 were not considered in this test since their initial 89Zr complex stability were too weak to provide accurate and meaningful results. As expected, release of 89Zr in EDTA was much faster for Me-AHA than L1′ with a half-life of the 89Zr-Me-AHA complex < 30 s compared to 5 min for the 89Zr-L1′ complex (Figure 5a). Similarly, the presence of Fe3+ led to a faster release of 89Zr in the case of Me-AHA (Figure 5b). Overall, in addition to its better thermodynamic stability reported in the previous section, the higher inertness of Zr(L1′)4 observed at the radiotracer scale strongly supports the potential superiority of chelators based on the L1′ motif in comparison with hydroxamate based analogues.
Figure 5.

Kinetic inertness of the 89Zr complexes of L1′ and Me-AHA in the presence of a) 50 mM EDTA at pH = 7 at 20 °C (corresponds to 2000 eq. of EDTA over the ligand) and b) 1mM FeCl3 in phosphate buffer 0.1 M pH 7.2 at 20°C (40 equiv. over the ligand) (n ≥ 3).
Quantum chemical calculations
To rationalize the thermochemical stability and kinetic inertness observed experimentally with these complexes, we have resorted to quantum chemical calculations.
Both ΔG and ΔH for the complexation reaction of Zr(OH)4 with hydroxypyridinones L were calculated using equation (1) in the reaction field of water. While the most acidic ligand L1 (or L1′) with a log Ka of 5.01 is deprotonated at neutral pH (unlike L2 and L3, log Ka of 8.83 and 9.55, respectively), the present calculations were all done in the neutral form for direct comparison.
| (1) |
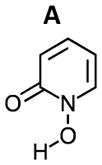
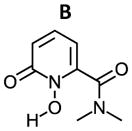
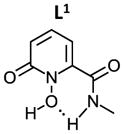
As shown in Table 3, the calculated ΔG for the formation of Zr(L1′)4, −80.8 kcal/mol, is much more favorable than for Zr(L2)4, Zr(L3)4 and Zr(Me-AHA)4. When comparing these calculated ΔGs with the experimental log Ka values, a correlation between the two appears to exist. Since each log Ka unit corresponds to ΔG of 1.36 kcal/mol, the 4 log Ka values difference between L1′ and L2 alone may confer an extra stability of 21.7 kcal/mol (4×4×1.36 kcal/mol) to Zr(L1′)4 over Zr(L2)4 (ΔG = −56.8 kcal/mol). This in turn indicates that the log Ka of HOPOs is a key determinant in modulating the binding affinity of HOPOs to Zr(IV). Such log Ka tuning can be achieved by inducing a H-bond. For example, the calculated log Ka of L1 (that is able to form intramolecular H-bond via the amide hydrogen) is 1.3 unit lower than B (in which the possibility of H-bond formation is removed by replacement of the amide hydrogen by a methyl, see structures in table 4); such H-bond was observed in the crystal structure of Zr(L1)4 (see Fig. 1B). This log Ka difference between L1 and B translates to 7.1 kcal/mol in terms of ΔΔG, and thus largely accounts for the calculated stability of 10.3 kcal/mol for Zr(L1)4 over Zr(B)4. Whereas the calculated ΔG = −80.8 kcal/mol for Zr(L1′)4 is more negative than the reported cyclic Zr-C7 (ΔG = −71.0 kcal/mol) and acyclic Zr-L7 (ΔG = −70.6 kcal/mol) tetrahydroxamate complexes (Scheme 3),20 it is not as kinetically inert against EDTA. Our previous study has shown that both Zr-C7 and Zr-L7 remain largely intact (> 80 %) against EDTA even after 7 days while the current work shows that only 22 % of Zr(L1′)4 remain intact against EDTA after 15 min. The strong Zr-C7 stability thus indicates that the energy barrier for the dissociation of Zr-C7 must be substantially higher than that of Zr(L1′)4, which highlights the strong impact of the pre-organization on kinetic inertness of such complexes. This in turn suggests that L1, L2, and L3 might form complexes of Zr(IV) which can become as inert or even more inert than the highly stable Zr-C7 if suitably pre-organized. Of the three, the ligand of choice for further modification to achieve desirable optimal kinetic inertness appears to be of the L1 type.
Table 3.
Calculated ΔG and ΔH for the Zr(L)4 complexes formation at 298.15K in the reaction field of water.
| Ligand | L1′ | L2 | L3 | Me-AHA |
|---|---|---|---|---|
| ΔG (kcal/mol) | −80.8 | −56.8 | −58.5 | −56.1 |
| ΔH (kcal/mol) | −99.8 | −76.8 | −67.7 | −68.9 |
| log Ka(exp) | 5.01 | 8.83 | 9.46 | 8.75 |
Table 4.
Calculated impact of the modulation of the 1,2-HOPO ring on the log Ka of ligands and ZrL4 complex stability ΔG and ΔH in the reaction field of water at 298.15 K (calculated structure of complexes available in Figures S5, S6 and S7).
|
|
|
|
|---|---|---|---|
|
|
|||
| log Ka(calc) | 5.8 | 5.8 | 4.5 |
| ΔGZrL4(kcal/mol) | −72.9 | −78.1 | −88.4 |
| ΔHZrL4(kcal/mol) | −82.3 | −91.7 | −99.0 |
Scheme 3.

Chemical structure of Zr-C7 and Zr-L7.20
Comparison of HOPOs with hydroxamates
Most data reported herein can be compared to results reported previously for hydroxamate ligands.18,20 Both ligand classes exhibit a strong predominancy of Zr(IV) octacoordination by four ligands at physiological pH, suggesting that chelators made of 4 of these ligands are likely to provide optimal stability for in vivo use. The thermodynamic stability study did not highlight noticeable differences between the HOPOs L2 and L3 and the hydroxamate ligand (pZr4+ in the 28.5–28.6 range), whereas L1′ was significantly superior (pZr4+ = 33.48). Differences appear more marked when comparing the inertness of complexes. At radiotracer concentrations, L1′ exhibited by far the highest inertness in presence of competitive ligand or metal, followed by Me-AHA that was in turn significantly superior to L2 and L3. Such results provide good indications of the potential of these ligands for radiopharmaceutical applications with 89Zr. Despite the lower inertness of Me-AHA complexes than L1′, hydroxamates offer the advantage of smaller and simpler structures. This may be a criterion of choice for designing chelators. They benefit of being already used in clinical trials for several years with 89Zr with the FDA approved DFB ligand, which might facilitate the clinical transfer of future new chelators based on that same pattern. Recent works undertaken in several research group highlight the interest of working with the hydroxamate motif.25,26 On the other hand, the hydroxypyridinonate structure offers the advantage of a higher modularity since the hydroxyl group or the substituents position can be varied, offering more options to tune the characteristics of Zr(IV) chelators. This is reflected in the variety of HOPO isomers used in chelators that have been reported recently30,32,34 and we hope results presented herein will guide future developments in that field.
Conclusions
Recent literature shows a strong interest in improving the complexation of 89Zr for PET imaging applications. From only 1 available and suboptimal ligand (desferioxamine B) until 2013, more than 10 chelators have now been developed by several research groups to provide enhanced in vivo stability of the radiolabeling. Whereas several HOPO-based chelators have been proposed, with very promising results for some of them, the literature strongly lacks data on the basics of the complexation of Zr(IV) by this class of bidentate ligands. Yet, such data is essential to finely tune the characteristics of chelators to match the coordination sphere of the Zr(IV) cation. In this study, which compares three hydroxypyridinone positional isomers, we observed that the selected 1,2-HOPO pattern combines the best features to provide high thermodynamic stability and high kinetic inertness for Zr(IV) complexation. The origin of this superiority arises in part from its higher acidity, but also from H-bonding and amide-π interactions that were not observed in the 2,3-HOPO and 3,4-HOPO used in this study. The 1,2-HOPO isomer, in combination with a H-donor amide group next to the hydroxyl, appears as a powerful pattern to build octadentate chelators for Zr(IV) complexation. These observations are in line with the promising results reported recently by Deri et al with a similar 1,2-HOPO based chelator that showed high in vivo inertness.34 However, it must be kept in mind that the 3,2-HOPO and 3,4-HOPO rings we reported herein have the advantage of providing ZrL4 complexes of higher aqueous solubility. Thus, by tuning them to provide lower pKa and/or generate H-bond and amide-π interactions as in 1,2-HOPO, they can also become highly adequate for the design of Zr(IV) chelators. We believe that the crystallographic, thermochemical and quantum chemical studies presented herein will provide a firm basis for such task and for further advances in the growing field of PET imaging with 89Zr.
Experimental
General
Procedures for ligands preparation, crystal structure analysis, thermodynamic stability constants determination and complexation at the radiotracer level with 89Zr are available in the Supplementary Information file.
Preparation of natZrL4 complexes
Zr(L1)4
To zirconium(IV) acetylacetonate (acac) (102 mg, 205 μmol) in dry DMF (10 mL) was added L1H (155 mg, 927 μmol) dissolved in dry MeOH (10 mL) and the solution was refluxed for 18 h. After cooling down to room temperature, the white precipitate was filtered and dried in vacuo (85 mg, 55%). 1H NMR (DMSO-d6, 300 MHz, ppm): δ 2.94 (d, 3H), 7.04 (d, 1H, J = 8.7 Hz), 7.34 (d, 1H, J = 7.5 Hz), 7.67 (m, 1H), 9.57 (s, 1H broad). 13C NMR (DMSO-d6, 75 MHz, ppm): δ 26.2, 113.3, 115.5, 135.8, 137.1, 159.1, 160.3. ESI-MS: m/z = 781.1 [M+Na]+, 675.1 [M-L1+DMSO-d6]+ (+ Zr isotopic distribution, see spectrum in ESI). Elemental analyzes: Calculated for C28H28N8O12Zr + 4 H2O: C, 40.43; H, 4.36; N, 13.47%. Found: C, 40.23; H, 4.42; N, 13.09. Crystals suitable form X-ray diffraction were obtained by recrystallization in hot acetonitrile (Details in ESI).
Zr(L1′)4
Was prepared from L1′H and Zr(acac)4 using the same procedure as Zr(L1)4, except that the product did not precipitate upon formation. After evaporation of methanol, the residue was dissolved in chloroform and washed twice with water. The chloroform was evaporated, affording a yellowish solid (97 %). 1H NMR (CDCl3, 300 MHz, ppm): δ 1.02 (t, 3H, J = 7.2 Hz), 1.64 (m, 2H), 3.37 (m, 2H), 6.90 (d, 1H, J = 8.4 Hz), 7.51 (m, 1H), 7.61 (m, 1H) 9.95 (t, broad, 1H). 13C NMR (CDCl3, 75 MHz, ppm): δ 11.8, 22.7, 42.0, 114.7, 116.2, 135.2, 137.1, 158.8, 161.1. ESI-MS: m/z = 870.9 [M+H]+, 892.9 [M+Na]+ (+ Zr isotopic distribution, see spectrum in ESI). Elemental analyzes: Calculated for C36H44N8O12Zr + 6 H2O: C, 44.12; H, 5.76; N, 11.43%. Found: C, 43.96; H, 5.88; N, 11.80.
Zr(L2)4
Was prepared from L2H and Zr(acac)4 using the same procedure as Zr(L1)4, except that the product did not precipitate upon formation. After evaporation of methanol, the residue was partitioned between water and chloroform. The water was evaporated, affording a white solid (88 %). 1H NMR (DMSO, 300 MHz, ppm): δ 2.15 (t, 2H, J = 7.2 Hz), 2.55 (d, 3H), 3.90 (t, 2H, J = 7.2 Hz), 6.35 (d, 1H), 6.88 (m, 1H), 7.58 (d, 1H). 13C NMR (DMSO-d6, 75 MHz, ppm): δ 25.4, 33.7, 45.8, 110.8, 113.3, 122.2, 156.9, 163.0, 169.5. ESI-MS: m/z = 675.1 [M-L2]+, 757.1 [M-L2+2MeOH+H2O]+, 897.1 [M+H2O+H]+ (+ Zr isotopic distribution, see spectrum in ESI). Elemental analyses: Calculated for C36H44N8O12Zr + 6 H2O: C, 42.12; H, 5.76; N, 11.43%. Found: C, 44.22; H, 5.42; N, 11.51. Crystals suitable for X-ray diffraction were obtained by slow evaporation from a 50:50 mixture of ethyl acetate and methanol (Details in ESI).
Zr(L3)4
Was prepared from L3H and Zr(acac)4 using the same procedure as Zr(L1)4, except that the product did not precipitate upon formation. The reaction solution was reduced to about 2 mL under reduced pressure and the product precipitated as a yellowish solid upon addition of Et2O. The solid was filtered and dried in vacuo (69%). 1H NMR (DMSO, 300 MHz, ppm): δ 2.40 (s, 3H), 2.54 (d, 3H), 2.63 (t, 2H, J = 6.6 Hz), 4.38 (t, 2H), 6.49 (m, broad, 1H), 7.77 (m, broad, 1H), 8.04 (m, broad, 1H). 13C NMR (DMSO-d6, 75 MHz, ppm): δ 11.1, 25.4, 36.1, 50.0, 106.0, 126.6, 133.0, 133.3, 158.6, 169.1. ESI-MS: m/z = 717.2 [M-L3]+ (+ Zr isotopic distribution, see spectrum in ESI). Calculated for C40H52N8O12Zr + 6 H2O: C, 46.36; H, 6.23; N, 10.81%. Found: C, 46.02; H, 5.87; N, 10.57.
Quantum chemical calculations
Crystal structure coordinates of Zr(L1)4 were used as a template to build Zr(L1′)4, Zr(L3)4, Zr(A)4 and Zr(B)4. These structures and the crystal structure coordinates of Zr(L1)4 and Zr(L2)4 were then geometry optimized by utilizing the M06L functional46 with the pseudopotential LanL2DZ47 and its valence basis set for the Zr atom and the 6–31+G* basis set for the rest of the atoms, as implemented in Gaussian 09 software48. All calculations were carried out in the reaction field of water with the polarizable continuum model.49
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and Center for Information Technology and in part by grants from the French National Agency for Research, called “Investissements d’Avenir” IRON Labex n° ANR-11-LABX-0018-01 and ArronaxPlus Equipex n°ANR-11-EQPX-0004 and the Cancéropôle Grand Ouest. The quantum chemical study utilized PC/linux clusters at the center for Molecular Modeling of the National Institutes of Health (http://cit.nih.gov). The authors are grateful to Dr. L. P. Szajek at the PET Dept, clinical center, for providing 89Zr.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Amélia Santos M. Coord Chem Rev. 2002;228:187–203. [Google Scholar]
- 2.Abergel RJ, Raymond KN. Inorg Chem. 2006;45:3622–3631. doi: 10.1021/ic052111a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turcot I, Stintzi A, Xu J, Raymond KN. J Biol Inorg Chem. 2000;5:634–641. doi: 10.1007/s007750000149. [DOI] [PubMed] [Google Scholar]
- 4.Gorden AEV, Xu J, Raymond KN, Durbin P. Chem Rev. 2003;103:4207–4282. doi: 10.1021/cr990114x. [DOI] [PubMed] [Google Scholar]
- 5.Datta A, Raymond KN. Acc Chem Res. 2009;42:938–947. doi: 10.1021/ar800250h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clevette DJ, Lyster DM, Nelson WO, Rihela T, Webb GA, Orvig C. Inorg Chem. 1990;29:667–672. [Google Scholar]
- 7.Chaves S, Mendonça AC, Marques SM, Prata MI, Santos AC, Martins AF, Geraldes CFGC, Santos MA. J Inorg Biochem. 2011;105:31–38. doi: 10.1016/j.jinorgbio.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Ma MT, Cullinane C, Imberti C, Baguña Torres J, Terry SYA, Roselt P, Hicks RJ, Blower PJ. Bioconjugate Chem. 2016;27:309–318. doi: 10.1021/acs.bioconjchem.5b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, Baidoo KE, Brechbiel MW. Adv Drug Delivery Rev. 2013;65:1098–1111. doi: 10.1016/j.addr.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gambhir SS. Nat Rev Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 11.Price TW, Greenman J, Stasiuk GJ. Dalton Trans. 2016;45:15702–15724. doi: 10.1039/c5dt04706d. [DOI] [PubMed] [Google Scholar]
- 12.Vosjan MJWD, Perk LR, Visser GWM, Budde M, Jurek P, Kiefer GE, van Dongen GAMS. Nat Protoc. 2010;5:739–743. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- 13.van de Watering FCJ, Rijpkema M, Perk L, Brinkmann U, Oyen WJG, Boerman OC. BioMed Research International. 2014;2014:e203601. doi: 10.1155/2014/203601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuo F, Histed S, Xu B, Bhadrasetty V, Szajek LP, Williams MR, Wong K, Wu H, Lane K, Coble V, Vasalatiy O, Griffiths GL, Paik CH, Elbuluk O, Szot C, Chaudhary A, StCroix B, Choyke P, Jagoda EM. Mol Pharmaceutics. 2014;11:3996–4006. doi: 10.1021/mp500056d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheal SM, Punzalan B, Doran MG, Evans MJ, Osborne JR, Lewis JS, Zanzonico P, Larson SM. Eur J Nucl Med Mol Imaging. 2014;41:985–994. doi: 10.1007/s00259-013-2679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans MJ, Holland JP, Rice SL, Doran MG, Cheal SM, Campos C, Carlin SD, Mellinghoff IK, Sawyers CL, Lewis JS. J Nucl Med. 2013;54:90–95. doi: 10.2967/jnumed.112.109777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abou DS, Ku T, Smith-Jones PM. Nucl Med Biol. 2011;38:675–681. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guérard F, Lee YS, Tripier R, Szajek LP, Deschamps JR, Brechbiel MW. Chem Commun. 2013;49:1002–1004. doi: 10.1039/c2cc37549d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holland JP, Vasdev N. Dalton Trans. 2014;43:9872–9884. doi: 10.1039/c4dt00733f. [DOI] [PubMed] [Google Scholar]
- 20.Guérard F, Lee YS, Brechbiel MW. Chem Eur J. 2014;20:5584–5591. doi: 10.1002/chem.201304115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patra M, Bauman A, Mari C, Fischer CA, Blacque O, Häussinger D, Gasser G, Mindt TL. Chem Commun. 2014;50:11523–11525. doi: 10.1039/c4cc05558f. [DOI] [PubMed] [Google Scholar]
- 22.Zhai C, Summer D, Rangger C, Franssen GM, Laverman P, Haas H, Petrik M, Haubner R, Decristoforo C. Mol Pharmaceutics. 2015;12:2142–2150. doi: 10.1021/acs.molpharmaceut.5b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jewula P, Chambron JC, Penouilh MJ, Rousselin Y, Meyer M. RSC Adv. 2014;4:22743–22754. [Google Scholar]
- 24.Deri MA, Ponnala S, Zeglis BM, Pohl G, Dannenberg JJ, Lewis JS, Francesconi LC. J Med Chem. 2014;57:4849–4860. doi: 10.1021/jm500389b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vugts DJ, Klaver C, Sewing C, Poot AJ, Adamzek K, Huegli S, Mari C, Visser GWM, Valverde IE, Gasser G, Mindt TL, van Dongen GAMS. Eur J Nucl Med Mol Imaging. 2016;1–10 doi: 10.1007/s00259-016-3499-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rudd SE, Roselt P, Cullinane C, Hicks RJ, Donnelly PS. Chem Commun. 2016;52:11889–11892. doi: 10.1039/c6cc05961a. [DOI] [PubMed] [Google Scholar]
- 27.Pandya DN, Pailloux S, Tatum D, Magda D, Wadas TJ. Chem Commun. 2015;51:2301–2303. doi: 10.1039/c4cc09256b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Captain I, Deblonde GJP, Rupert PB, An DD, Illy MC, Rostan E, Ralston CY, Strong RK, Abergel RJ. Inorg Chem. 2016;55:11930–11936. doi: 10.1021/acs.inorgchem.6b02041. [DOI] [PubMed] [Google Scholar]
- 29.Price EW, Zeglis BM, Lewis JS, Adam MJ, Orvig C. Dalton Trans. 2013;43:119–131. doi: 10.1039/c3dt51940f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tinianow J, Pandya DN, Pailloux SL, Ogasawara A, Vanderbilt AN, Gill HS, Williams SP, Wadas TJ, Magda D, Marik J. Theranostics. 2016;6:511–521. doi: 10.7150/thno.14261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Santos MA, Gil M, Gano L, Chaves S. J Biol Inorg Chem. 2005;10:564. doi: 10.1007/s00775-005-0003-7. [DOI] [PubMed] [Google Scholar]
- 32.Ma MT, Meszaros LK, Paterson BM, Berry DJ, Cooper MS, Ma Y, Hider RC, Blower PJ. Dalton Trans. 2015;44:4884–4900. doi: 10.1039/c4dt02978j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boros E, Holland JP, Kenton N, Rotile N, Caravan P. ChemPlusChem. 2016;81:274–281. doi: 10.1002/cplu.201600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deri MA, Ponnala S, Kozlowski P, Burton-Pye BP, Cicek HT, Hu C, Lewis JS, Francesconi LC. Bioconjugate Chem. 2015;26:2579–2591. doi: 10.1021/acs.bioconjchem.5b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu J, Durbin PW, Kullgren B, Ebbe SN, Uhlir LC, Raymond KN. J Med Chem. 2002;45:3963–3971. doi: 10.1021/jm010564t. [DOI] [PubMed] [Google Scholar]
- 36.Pierre VC, Botta M, Aime S, Raymond KN. J Am Chem Soc. 2006;128:9272–9273. doi: 10.1021/ja061323j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piyamongkol S, Ma YM, Kong XL, Liu ZD, Aytemir MD, van der Helm D, Hider RC. Chem Eur J. 2010;16:6374–6381. doi: 10.1002/chem.200902455. [DOI] [PubMed] [Google Scholar]
- 38.Liu ZD, Piyamongkol S, Liu DY, Khodr HH, Lu SL, Hider RC. Bioorg Med Chem. 2001;9:563–573. doi: 10.1016/s0968-0896(00)00273-x. [DOI] [PubMed] [Google Scholar]
- 39.Lambert TN, Chittamuru S, Jacobs HK, Gopalan AS. Tetrahedron Letters. 2002;43:7379–7383. doi: 10.1016/S0040-4039(02)01730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobbin PS, Hider RC, Hall AD, Taylor PD, Sarpong P, Porter JB, Xiao G, van der Helm D. J Med Chem. 1993;36:2448–2458. doi: 10.1021/jm00069a002. [DOI] [PubMed] [Google Scholar]
- 41.Xu J, Whisenhunt DW, Veeck AC, Uhlir LC, Raymond KN. Inorg Chem. 2003;42:2665–2674. doi: 10.1021/ic0259888. [DOI] [PubMed] [Google Scholar]
- 42.Xu J, Franklin SJ, Whisenhunt DW, Raymond KN. J Am Chem Soc. 1995;117:7245–7246. [Google Scholar]
- 43.Harder M, Kuhn B, Diederich F. ChemMedChem. 2013;8:397–404. doi: 10.1002/cmdc.201200512. [DOI] [PubMed] [Google Scholar]
- 44.CCDC 1508469 and 1508470 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk).
- 45.Woodin KS, Heroux KJ, Boswell CA, Wong EH, Weisman GR, Niu W, Tomellini SA, Anderson CJ, Zakharov LN, Rheingold AL. Eur J Inorg Chem. 2005;2005:4829–4833. [Google Scholar]
- 46.Zhao Y, Truhlar DG. J Chem Phys. 2006;125:194101-194101–18. doi: 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- 47.Wadt WR, Hay PJ. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 48.Frisch MJ, et al. Gaussian 09, revision A.02. Gaussian Inc; Wallingford CT: 2009. [Google Scholar]
- 49.Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.