

Abstract
Introduction
Nitric oxide (NO) synthesized via neuronal nitric oxide synthase (nNOS) plays a significant role in regulation/modulation of autonomic control of circulation. Various pathological states are associated with diminished nNOS expression and blunted autonomic effects of NO in the central nervous system (CNS) including heart failure, hypertension, diabetes mellitus, chronic renal failure etc. Therefore, elucidation of the molecular mechanism/s involved in dysregulation of nNOS is essential to understand the pathogenesis of increased sympathoexcitation in these diseased states.
Areas Covered
nNOS is a highly regulated enzyme, being regulated at transcriptional and posttranslational levels via protein-protein interactions and modifications viz. phosphorylation, ubiquitination, and sumoylation. The enzyme activity of nNOS also depends on the optimal concentration of substrate, cofactors and association with regulatory proteins. This review focuses on the posttranslational regulation of nNOS in the context of normal and diseased states within the CNS.
Expert Opinion
Gaining insight into the mechanism/s involved in the regulation of nNOS would provide novel strategies for manipulating nNOS directed therapeutic modalities in the future, including catalytically active dimer stabilization and protein-protein interactions with intracellular protein effectors. Ultimately, this is expected to provide tools to improve autonomic dysregulation in various diseases such as heart failure, hypertension, and diabetes.
Keywords: cardiovascular diseases, nNOS, PVN, sympathoexcitation
1. INTRODUCTION
Anomalous regulation of the sympathetic nervous system leading to exaggerated sympathetic nerve activity is associated with many cardiovascular diseases including heart failure (CHF)[1, 2], hypertension, obesity and insulin resistance[3]. The prognosis of patients with elevated sympathoexcitation in cardiovascular diseases is dismal [1, 4, 5]. Despite major advances in the therapy directed to alleviate sympathoexcitation, the morbidity, and mortality related to it are still high for cardiovascular diseases [6, 7]. The increased sympathoexcitation is mediated by a number of important regions within the central nervous system (CNS) including the paraventricular nucleus (PVN) of the hypothalamus and the rostral ventrolateral medulla (RVLM) [8]. The PVN has been suggested to be the highest autonomic control center within the CNS for the regulation of sympathoexcitation [9, 10, 11, 12]. Various neuroanatomical, electrophysiological and functional studies have indicated an important role for the PVN in cardiovascular regulation [10, 13] via sending efferent nerves to RVLM, as well as direct projections to the intermediolateral cell column in the spinal cord[14]. A number of different neurotransmitter systems, excitatory as well as inhibitory have been shown to converge in the PVN to influence ongoing neuronal activity [10]. One important mediator identified to be sympathoinhibitory within the PVN is NO (nitric oxide)[15, 16, 17, 18, 19]. NO is a well-acknowledged, universal signaling molecule, which also acts as a neuromodulator in the central and peripheral nervous system [20, 21]. Cardiovascular actions of NO are not only limited to its direct effects on dilation of blood vessels but also affect the synaptic excitability of pre-autonomic neurons in the CNS. NO modulates the release of several neurotransmitters, such as acetylcholine, catecholamine, excitatory and inhibitory amino acids viz. glutamate and GABA (γ-aminobutyric acid) to influence overall neuronal function [22, 23]. Previous studies from our laboratory have shown that the administration of an NO donor into the PVN decreases renal sympathetic nerve activity (RSNA), whereas administration of nitric oxide synthase (NOS) inhibitor within the PVN increases RSNA [24]. NO-mediated inhibition of neurons within the PVN have been suggested to utilize a GABA-mediated mechanism [25, 26], which is also blunted in rats with CHF [27, 28, 29].
NO is synthesized by the NOS enzyme, which exists in three isoforms, neuronal nitric oxide synthase (nNOS), inducible nitric oxide synthase (iNOS), and endothelial nitric oxide synthase (eNOS). nNOS is the major source of NO in the brain and is expressed in neurons, glia as well as in the adventitia of a subset of rat cerebral blood vessels in CNS [30]. Studies in mammalian, particularly in rodents brains showed that nNOS is abundantly expressed in the various areas including those involved in the regulation of cardiovascular functions, such as, the amygdala, CA1 region, dentate gyrus of hippocampus, the hypothalamus (the supraoptic nucleus and PVN), the thalamus, the RVLM, nucleus tractus solitarius (NTS) and the cerebellum [31, 32]. NO synthesized by nNOS in the brain affects not only PVN but also other central sites, particularly the NTS, and RVLM under physiological and pathological conditions [22]. Lack of NO during various pathological conditions leads to an increase in sympathetic hyperactivity, primarily due to a blunted sympathoinhibitory effect by nNOS, such as CHF[33, 34, 35, 36], hypertension [37, 38], diabetes[39] and in hypertension associated with chronic renal failure[40]. Moreover, oxidative stress has been shown to cause an uncoupling of NOS in the brain during hypertension [41, 42] and CHF [43, 44], thus further depleting the levels of NO and aggravating the disease progression. Until recently, nNOS was considered to be a constitutively expressed enzyme, but current studies [20, 36, 45, 46, 47, 48] suggest that activity and expression of nNOS are regulated, both transcriptionally and post-transcriptionally by various physiological and pathological stimuli. Although, there are multiple reports on the dysregulation of nNOS in different physiological and pathological conditions [33, 34, 35, 36, 37, 38, 39, 40], there are limited studies examining the molecular mechanism of regulation of nNOS within the CNS. This review highlights the regulation of nNOS in the CNS with particular reference to PVN and with emphasis on post-translational modification and protein-to-protein interactions leading to the regulation of sympathoexcitation using an example of a typical sympatho-excitatory state of chronic congestive heart failure.
2. STRUCTURES AND ACTIVITY OF NO SYNTHASES
Since awarding the Nobel Prize to Furchgott, Ignarro, and Murad for the discovery of NO in 1998, the prevailing literature points towards a significant role for NO in the regulation of neurotransmission within the CNS [15, 16, 17, 18, 19, 24, 26]. NO is an important signaling molecule with a short half-life of 1-5s and is involved in the regulation of the cardiovascular, immune and nervous systems, rather than just a toxic pollutant [30, 45]. NO acts as a novel neural messenger by stimulating soluble guanylyl cyclase, thus increasing the levels of cyclic guanosine 3′, 5′-monophosphate in target cells. As mentioned, NO is synthesized by a family of enzymes known as NOS which exist as three genetically different isoforms, coded by their corresponding gene which include NOS1 for nNOS being the isoform first found in neuronal tissues, NOS2 for iNOS being the isoform induced by pro-inflammatory cytokines and inflammation and NOS3 for eNOS expressed in endothelial cell. A unique NOS isoform known as mitochondrial NOS is present in the inner mitochondrial membrane [49]. All three isoforms of NOSs are flavohaem enzymes, which share ~50–60% sequence homology and similar catalytic mechanisms [50]. NOS homodimerize through oxygenase domain and catalyze O2-dependent, five-electron oxidation of the guanidine nitrogen of L-arginine to NO and citrulline, with Nω-hydroxy-L-arginine formation as an intermediate [30]. Dimer formation is essential for the catalytic activity of the enzyme. Each monomer exhibits bi-domain structure comprising a carboxy-terminal diflavin-reductase domain and an amino-terminal oxygenase domain, which are separated by a calmodulin-binding motif. Dimerization is assumed to activate the enzyme by sequestering iron, generating high-affinity binding sites for arginine and the essential cofactor (6R)-5,6,7,8-tetrahydrobiopterin (BH4) in the oxygenase domain. The transfer of electrons during the catalytic reaction occurs from the reductase domain of one monomer to the oxygenase domain of the other monomer [46]. Further, Ca2+/calmodulin binding support electron transfer. Binding of calmodulin is dependent on calcium in eNOS and nNOS, and therefore, enzyme activity in these isoforms is calcium dependent while calmodulin is tightly bound to iNOS, making this isoform fully active even at basal intracellular calcium concentration [47]. The activity of NOS also depends on the availability of the cofactors BH4, flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), calmodulin (CaM), and iron protoporphyrin IX (heme), and nicotinamide adenine dinucleotide phosphate (NADPH) as an electron source (Figure 1A). Calmodulin binding is required to trigger the flow of electrons from the flavins to the active site. As stated previously, regions of high homology characterize the three isoforms of NOS, yet they also have some significant differences in size, as well as each isoform, has distinctive characteristics that make these forms different in their function and distribution (Figure 1B). The molecular weight of nNOS is 160kDa compared to iNOS (130kDa) and eNOS (135kDa), due to the presence of additional 300 amino-acid sequences at N-terminal containing a PDZ {postsynaptic density protein (PSD95), disc large, ZO-1} domain. The N-terminus of eNOS also has unique myristoylation and palmitoylation sites that regulate the localization of the enzyme to the plasmalemma caveolae.
Figure 1.
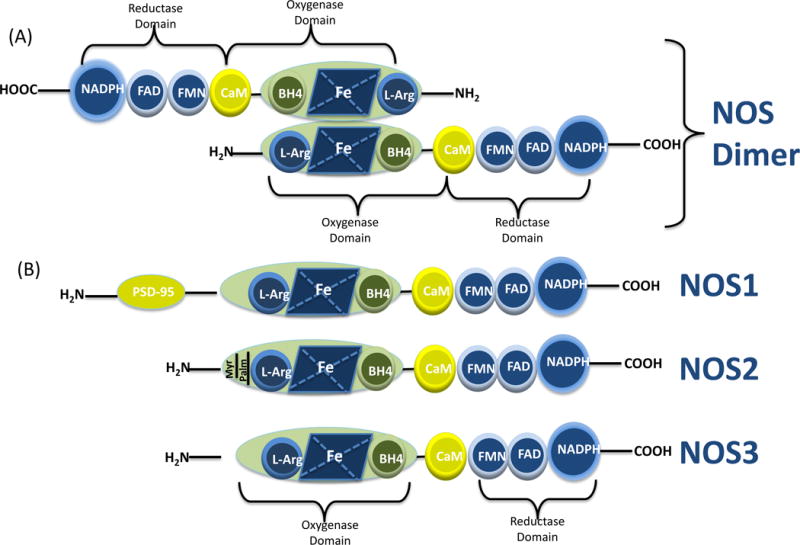
(A) Illustration of the molecular structure of NOS dimer. All NOS monomers include an oxygenase and a reductase domain and calmodulin (CaM) binding site. The oxygenase moiety comprises the L-arginine, heme, and tetrahydrobiopterin (BH4)-binding domains; whereas the reductase moiety includes the flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and nicotinamide adenine dinucleotide phosphate (NADPH)- binding domains. NOS monomers are unable to bind the cofactor BH4 or the substrate arginine to catalyze the NO production. In the presence of heme, NOS can form a functional dimer that can bind to BH4 and L-Arg that allows interdomain electron transfer. (B) Schematic illustration of the protein structure of NOS isoforms.
Since NO has a strong chemical reactivity, short half-life, and high diffusibility, NOS expression, and activity are tightly linked and regulated. Despite the beneficial roles of NO as the neuromodulator, as a messenger and in host defense mechanism, a dysregulated production of NO can give rise to pathological conditions. The modulations in NOS and NO metabolism leads to either decreased the production of NO, which is linked to cardiovascular diseases [35, 36, 37, 39], erectile dysfunction (ED) [51] and gastrointestinal disorders [52] or excess of NO which is related to tumor progression [53] and neurodegenerative diseases[30]. Therefore, the regulation of nNOS activity is strictly controlled to perform the variety of functions, which dictate the specificity of NO signaling and influence on the activity of neurons in regions of the CNS involved in autonomic regulation such as PVN and RVLM.
3. REGULATION OF nNOS
The activity of NOS enzymes is under the complex and integrated control of transcriptional, translational, and post-translational regulation. Hence, understanding of molecular mechanisms involved in dysregulation of nNOS leading to result ultimately in increased sympathoexcitation in various disease states is essential.
3.1 Transcriptional Regulation
nNOS gene located on chromosome 12 has a complex genomic organization, spanning over 160 kbp and consisting of an open reading frame of 29 exons extending from exon 2 to exon 30 in humans [46, 54]. nNOS gene transcribes multiple mRNA transcripts using alternate promoters usage, alternate splicing, cassette insertions/deletions, polyadenylation and different sites for 3′-UTR cleavage making transcriptional regulation of nNOS very complex [46, 55, 56, 57]. Literature suggests that cellular and tissue-specific regulation of nNOS is attributed to various transcription factors, such as the activator protein-2, the transcriptional enhancer factor-1/MCAT binding factor, cAMP response element-binding protein (CREB)/activating transcription factor/c-Fos, nuclear respiratory factor-1, Ets, nuclear factor-1and NF-kappa B [54, 58]. Binding of CREB to cAMP response element within the nNOS exon 2 promoter makes nNOS a Ca(2+)-regulated gene, which is likely to be involved in the regulation of nNOS in response to neuronal injury and activity-dependent plasticity [58]. Besides that, nNOS is subjected to alternative splicing, and more than ten different spliced variants of nNOS have been reported [59, 60, 61, 62], which potentially encompass unique structural features and catalytic properties. In the brain, the exon 2 – containing nNOSα form is the major splice variant, whereas nNOSμ is predominant splice variant in skeletal muscle and heart [60]. nNOS-2 detected in mouse brain acts as a dominant negative regulator of nNOS activity because of 105-amino acid in-frame deletion of residues 504-608 involved in arginine binding[63]. Both nNOSα and nNOSμ are anchored to subcellular structures via PDZ domain, whereas another two forms, nNOSβ, and nNOSγ are cytoplasmic. These transcriptional and post-transcriptional levels of regulation of nNOS have been reviewed elsewhere [46, 55, 56, 57].
3.2 Post-translational regulation of nNOS
The post-translational modifications including phosphorylation, ubiquitination, and interactions with other structural and regulatory proteins to form functional heterologous complex reveals an additional level of complexity in nNOS regulation.
3.2.1 Regulation of nNOS by phosphorylation
The nNOS enzymatic activity is modulated by phosphorylation at multiple sites and facilitates the cross-talk between NO and other signaling pathways via different protein kinases. Phosphorylation of nNOS can occur at multiple residues, including serine (Ser), threonine (The) and tyrosine (Tyr), which has been associated with either activation or inactivation of the enzyme directly or by modulating other regulatory domains present in nNOS. The best-characterized nNOS phosphorylation site is Ser-847, which is located in the reductase domain of the enzyme. Diverse signaling pathways regulate nNOS phosphorylation/dephosphorylation through specific protein kinases and protein phosphatases, including AMP-activated protein kinase[64] protein kinase C (PKC)[65], cyclic-nucleotide-dependent protein kinases[66] and Ca2+/calmodulin-dependent protein kinase (CAMK) [67, 68]. CaMKI and CaMKII phosphorylate nNOS at Ser-741 and Ser-847 residues, respectively, to inhibit its enzyme activity [68, 69]. nNOS has also been shown to have putative consensus sequences for phosphorylation by RSK (p90 ribosomal S6 kinases), which is reported to inhibit nNOS enzyme activity by phosphorylation at Ser 847 in human embryonic kidney cell line over-expressing nNOS and RSK1 and treated with epidermal growth factor [70]. The role of BH4 in precluding PKC- mediated phosphorylation of nNOS dimers has been demonstrated in in vitro studies via stabilization of nNOS dimers in neurons that are less susceptible to PKC-dependent phosphorylation [71]. NMDAR mediated transient activation of nNOS involved phosphorylation of nNOS at activating and inhibitory sites viz. Ser-1412 and Ser-847, respectively, in a time-dependent manner [72]. Activation of NMDAR leads to AKT (protein kinase B) dependent phosphorylation at Ser-1412 in the C-terminus of nNOS, resulting in enzyme activation with increased NO and cGMP production[73]. However, phosphorylation at Ser-1412 act in a feedback manner assisting phosphorylation at Ser-847 in the alpha helix auto-inhibitory domain of nNOS by calcium-calmodulin-dependent kinase II, resulting in enzyme deactivation[69]. Further, AngII mediated ERK1/2-RSK-nNOS signaling pathway had been demonstrated in NTS to modulate central blood pressure in spontaneous hypertensive rats[74]. The role of these phosphorylation events in nNOS regulation during other cardiovascular diseases including heart failure remains to be investigated.
3.2.2 Regulation of nNOS via ubiquitination/sumoylation
Ubiquitination and sumoylation have arisen as two major post-translational regulatory mechanisms involved in controlling the levels and function of several neuronal proteins besides phosphorylation. Ubiquitination of proteins is a signal for degradation that leads to delivery and degradation of dysfunctional proteins in the 26S proteasome [75]. The ubiquitin-proteasome system (UPS) regulates the fidelity of many proteins in the post-synaptic density [76]. The UPS is a key non-lysosomal pathway, which is known to participate in the pathogenesis of various neurodegenerative disorders like Parkinson’s, and Alzheimer’s disease, and diffuse Lewy body disease [77, 78]. The ubiquitination process is initiated by tagging of cellular proteins with multiple ubiquitin (8kDa peptide) molecules, involving series of enzymes including ubiquitin-activating enzymes, ubiquitin-conjugating enzymes and ubiquitin-isopeptide ligases[79, 80]. Ubiquitin ligases are the critical mediators to provide specificity to the reaction via substrate recognition [81]. As deduced from the literature in a cell-free system, nNOS undergoes enhanced proteolytic degradation due to suicide inactivation with metabolism-based inactivators [82], anti-hypertensive agent guanabenz [83] or by the inhibition of the HSP90-based chaperone system with geldanamycin [84]. Moreover, nNOS is found in ubiquitin conjugates in rat brain homogenates [82]Bender et al., 2000a, strongly suggesting the involvement of UPS in the regulation of nNOS in vivo. Importantly, the heme-deficient monomeric form of nNOS is preferentially ubiquitinated compared to heme-bound homodimer form. CHIP (carboxy terminus of Hsp 70 interacting protein) a chaperone-dependent E3 ligase has been proposed to act as E3 ligase for nNOS in human embryonic kidney (HEK) 293T cells [85]. As stated, nNOS requires homodimerization and association with Ca2+-calmodulin for NO production [46]. The monomeric form, however, catalyzes the production of superoxide (O2.−) that immediately forms peroxynitrite in the presence of NO. Therefore, a decrease in dimer/monomer ratio is thought to reduce NO production and its bioavailability. [82]. Therefore, conditions such as iron heme and BH4 depletion and addition of a dimerization inhibitor that favors a decrease in dimer/monomer ratio of nNOS will have an effect on the proteolytic degradation of nNOS. This suggests that ubiquitination of nNOS can potentially contribute to the regulated proteolysis of the nonfunctional enzyme [86].
Likewise, the conjugation mechanism of small ubiquitin-related modifier (SUMO ~12Kd) is very similar to those of ubiquitin, but the biological functions of sumoylation are different from ubiquitination. They do not provide a signal for proteasomal degradation, but rather inhibits polyubiquitin-mediated degradation. Levels of sumoylated proteins have been reported to be increased in animal models of neurological disorders such as brain ischemia [87]. Interestingly, studies have shown that components of the sumoylation machinery SUMO-1, Ubc9, and PIASxβ are all expressed in cerebellar granular cells of the cerebellum [88] which also express nNOS [89] suggesting that nNOS may be post-translationally modified by SUMO-1 in the brain [90]. This third posttranslational modification besides phosphorylation and ubiquitination might be potentially important in the regulation of nNOS function in the brain, and presents a significant unexplored challenge for future research.
3.2.3 Regulation of nNOS by multiple protein interactions
The catalytic activity of nNOS is influenced by the variety of regulatory and structural proteins [20, 45, 46] Using various methods to investigate protein-protein interactions including co-immunoprecipitation, yeast two-hybrid system, fluorescence resonance energy transfer and mass spectrophotometry, others and we have identified multiple interacting partners of nNOS that modulate its stability as well as catalytic activity [36, 47, 48, 91].
1. Regulation of nNOS by Calmodulin
Ubiquitous calcium binding protein, calmodulin was the first NOS-associated protein identified, which act as an allosteric activator in the isoform-specific manner [92]. Ca2+/calmodulin binding to NOS is non-covalent and reversible and has been shown to be indispensable for electron transfer from the reductase domain to the heme group of oxidase domain of NOS [93]. The threshold levels of 400nM of the cytosolic concentration of calcium facilitate calmodulin and calcium binding and its subsequent interaction with nNOS. The calcium concentration below this level causes dissociation of calmodulin from the nNOS, thus acting as a metabolic switch to turn off the enzyme [20]. Decreased calmodulin levels have been demonstrated in the brains of the genetic model of spontaneously hypertensive rats and deoxycorticosterone acetate (DOCA)-salt rats compared with those in Wistar-Kyoto rats, suggesting that intracellular calcium-dependent regulatory mechanism may be impaired [94]. Similarly, decreased expression of calmodulin has been documented in failing hearts, affecting many Ca2+-dependent processes during the end-stage heart failure but the role of calmodulin in the regulation of nNOS in the brain during heart failure remains to be explored [95].
2. Regulation of nNOS by heat shock proteins
The heat shock proteins (HSP) include a family of molecular chaperones responsible for the proper folding and maturation of proteins [96]. Hsp90/Hsp70 based chaperone machinery regulates nNOS activity and turnover via modulating ligand-binding clefts[97]. The selective inhibition of Hsp90 by geldanamycin leads to inactivation of nNOS [85]. Recently, Hsp90 has been suggested to be responsible for the insertion of heme into monomeric apo-nNOS during the limited availability of heme [84]. Consecutively, the inhibition of Hsp90-based chaperones would favor the monomeric form of nNOS leading to its ubiquitin-mediated proteasomal degradation. In contrast, overexpression of Hsp70 promotes nNOS ubiquitination and degradation leading to reduce levels of NO [98]. Elevated levels of Hsp70 in plasma are correlated with the progression of CHF, which might be used as a potential diagnostic biomarker for early detection of CHF [99]. The role of HSP in the regulation of nNOS in the brain during various cardiovascular disease conditions remains to be explored.
3. Regulation of nNOS by PDZ-domain proteins
PDZ-domain containing proteins such as syntrophin, PSD-95, or PSD-93 play a central role in forming multiprotein signaling complexes and are required for the binding of nNOS to different calcium sources[100, 101]. In skeletal muscles, nNOS is attached to the sarcolemma via syntrophin and dystrophin proteins. While in the CNS, nNOS couples to NMDA-NR2 cytoplasmic tail through PSD95, which is responsible for calcium influx followed by activation of nNOS [102, 103, 104]. Subcellular fractionation of the brain tissue demonstrates that approximately half of the nNOS in the brain is soluble, and another half is associated with membranous fraction via PSD95 [100]. PSD proteins are predominantly expressed at higher synaptic densities and interact with nNOS through PDZ domains. The nNOS/PSD95 interaction involves the unique β-finger PDZ domain of nNOS and PDZ1 or PDZ2 domain of PSD95. These PDZ domain based interactions create a ternary complex, which triggers the calcium-dependent activation of nNOS[101, 105] and subsequent intracellular signaling cascade[102]. Decreased PSD95 expression and function could contribute to the loss of NO-mediated balance leading to exaggerated glutamatergic/NMDAR driven sympathoexcitation in the PVN in neurogenic cardiovascular diseases [106]. Studies have shown that nNOS-PSD-95 interaction impairs regenerative repair after stroke in rats. Dissociating nNOS-PSD-95 coupling in neurons promotes neuronal differentiation of neural stem cells and improves stroke outcome by promoting regenerative repair [107] and also can prevent cerebral ischemic damage in rodents[108]. Inhibition of nNOS–PSD-95 complex via ZL006, a drug developed, has been suggested as a potential treatment for stroke [108].
Carboxy-terminal PDZ ligand of nNOS (CAPON) competes with PSD95 for interaction with nNOS in the brain, consequently acting as a negative regulator of NO production and its downstream signaling effects [109, 110]. Hence, over-expression of CAPON results in loss of PSD95/nNOS complexes [110], and uncoupling of NMDA-nNOS complex, resulting in abrogation of NO-mediated signaling pathways. CAPON is also reported to interact selectively with monomeric G-proteins, Dexras1 (Dexamethasone-induced ras protein 1) activated by NO donors [109, 111] through the phosphotyrosine-binding domain, suggesting a potentially important role for CAPON in the regulation of neurotransmitter release and neuronal plasticity under normal and pathological conditions [112]. nNOS-CAPON-Dexras1 association contributes to the modulation of anxiety-related behavior via regulating Dexras1-ERK signaling in the hippocampus of mice subjected to chronic mild stress while targeting of nNOS-CAPON binding via small-molecule blocker Tat-CAPON-12C produced an anxiolytic effect[113]. It has been suggested that reduced CAPON expression in stellate ganglia of SHR rats before the development of hypertension compared to age-matched control rats contributes to a pre-disease neuronal phenotype, that enhances calcium handling and cardiac sympathetic neurotransmission providing a relationship between CAPON and NO-dependent pathway [114]. The role of PSD95, interactions with CAPON and Dexras1 in the regulation of nNOS in the brain during various cardiovascular diseases such as heart failure remains to be explored.
CAPON has been extensively studied in schizophrenia [115, 116] because of its role in glutamate neurotransmission, known to be involved in this disease [117]. Interestingly, nNOS expressing PVN neurons are also significantly reduced in schizophrenic and depressive patients as compared to control patients [118], similar, to decreased expression of nNOS [36] as well as significantly decreased number of NADPH-diaphorase positive neurons in the PVN of rats with heart failure [27]. nNOS expression has also been shown to be altered in other human brain areas of patients with schizophrenia and depression[119, 120]. Our recent studies revealed that enhanced expression of CAPON is associated with a decrease in nNOS in the PVN of rats with CHF (Figure 2 A, B). Further, we have demonstrated that knockdown of CAPON using siRNA in isolated NG108 cells increases the levels of nNOS (Figure 2 C). In a preliminary study, we observed that adenovirus mediated upregulation of nNOS in NG108 cells does not affect the expression of CAPON (Figure 2 D). These data taken together suggest that the decreased levels of nNOS in the PVN of CHF rats is likely due to enhanced levels of CAPON leading to nNOS degradation. These reciprocal changes in CAPON and nNOS in the PVN of rats with CHF are reversed by AT1R blocker [36]. AT1R blocker also restores the endogenous levels of functional nNOS demonstrated by typical responses in RSNA to blockade of NOS within the PVN of rats with CHF. This data establish that elevated levels of Ang II in CHF up-regulate the expression of CAPON in PVN, which sequesters and dissociate nNOS from the membrane, resulting in uncoupling of nNOS from NMDAR. This reduces the NO-mediated inhibitory influence and thus an over-activation of the sympathoexcitatory drive from the PVN in CHF. There is high co-morbidity in patients with depression, anxiety, heart failure, and other heart disease [121, 122]. However, the cause(s) for this significant correlation and coincidence of occurrence and underlying pathophysiological relation are not yet known. Depression is an established risk factor for heart disease [123] and 50% of the patients with congestive heart failure clinically manifest the symptoms of depression [121]. It may well be that the central pathways that are involved in CHF and depression/schizophrenia may utilize nNOS and CAPON as part of the neural circuitry for central processing and alterations in this interaction between CAPON and nNOS in the PVN may be common features involved in the pathogenesis of these diseases. Thus, it seems reasonable to hypothesize that the identification of candidate mediators such as CAPON and nNOS, common to heart failure, hypertension, diabetes and depression/schizophrenia may serve as an important convergence point responsible for frequent co-morbidity. The role of CAPON in the regulation of nNOS in the brain during hypertension and diabetes, both known to exhibit enhanced sympatho-excitatory states remains to be explored.
Figure 2.
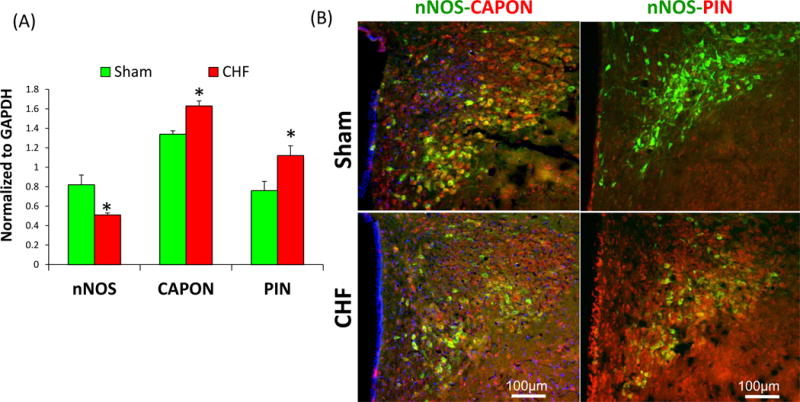
(A) Composite data were showing the expression of nNOS, CAPON, and PIN in the PVN of CHF rats. Values are mean ± SEM (n = 4–5 rats per group). *P < 0.05 vs. Sham. Modified from Sharma et al (2011)[36] and Sharma et al (2013)[48](B) Immunofluorescent photomicrographs from the sections of the PVN stained for nNOS, CAPON, and PIN. The intensity of nNOS staining (green) is decreased while the intensity of CAPON and PIN as shown in red is increased in the PVN of CHF rats. Yellow staining showed colocalization of nNOS-PIN or nNOS-CAPON in the same neuron. Blue spots showed the nucleus stained by Dap1. Modified from Sharma et al (2011)[36] and Sharma et al (2013)[48] (C) Effect of silencing of CAPON on nNOS expression in NG108 neuronal cell line. siRNA CAPON or negative control (20 pmol) was transfected in NG108 cells using Lipofectamine 2000 as per the manufacturer’s instructions. Values were mean±SE from four Independent experiments. From Sharma et al (2011)[36]. (D) Effect of upregulation of nNOS on the CAPON expression. Adenovirus vectors encoding nNOS (AdnNOS, 105~108pfu/ml, 48hrs) or adenovirus vectors encoding EGFP (AdEGFP, 105 pfu/ml, 48hrs) were transfected into NG108 cells
3.2.4 Regulation of nNOS by endogenous inhibitors
In addition to activity and localization, the other mechanism that regulates NO levels are endogenous NOS inhibitors named asymmetrical dimethyl arginine (ADMA) and its structural isomer, symmetric dimethyl arginine (SDMA). ADMA competitively displaced arginine from the substrate-binding site of NOS and, therefore, impede NOS catalytic activity, while SDMA has been shown not to alter NO production [124]. Limited information concerning the role of ADMA and SDMA in the CNS has been available to date, but in the periphery, ADMA has been associated with cardiovascular risk and clinically correlated with the burden of cardiovascular disease [125]. In renal wrap model of hypertension in rats, decreased expression of nNOS along with reduced SDMA levels, but no significant changes in ADMA levels was reported in the PVN during the onset of hypertension [91]. The role of ADMA and SDMA in the regulation of nNOS in the brain during heart failure and diabetes remain to be explored.
A small molecular weight protein, dynein light chain, also known as PIN was identified by Jaffrey and Snyder in 1996 using the N-terminal domain of nNOS in a yeast two-hybrid system. PIN was reported to bind to a 17-residue peptide fragment of nNOS (Met-228 to His-244) and destabilizes the dimeric structure of nNOS [126] leading to catalytically inactive monomeric form. Because of homology of PIN with the light chain of myosin and dynein, it is conceivable that PIN may be involved in nNOS’s association with the neuronal cytoskeleton during axonal transport [127, 128]. PIN has also been shown to be co-expressed and colocalizes with nNOS in the cavernosal and dorsal nerves of the penis as well as in the hypothalamic regions of the rat, particularly in the regions of PVN that controls penile erection [129]. Overexpression of PIN leads to a reduced erectile response to electrical field stimulation while shRNA-mediated knockdown of PIN helps reverses age-related ED [130]. Recently, we have shown AngII-mediated increased expression of PIN in the PVN of CHF rats (Figure 2A, B). PIN colocalizes with nNOS in neurons, and also interacts physically with nNOS as demonstrated by our co-immunoprecipitation studies and destabilizes the nNOS dimers to catalytically impeded monomers susceptible to degradation[48]. Functional experiments also established the significance of PIN in nNOS regulation as microinjection of plasmid construct of PIN cDNA in the in the rat corpora cavernosa, partially reduced the erectile response to electrical field stimulation of the cavernosal nerve supporting role for PIN in inhibiting the nitrergic transmission for erection by reducing NO synthesis in vivo [129]. Furthermore, RNA silencing of PIN in the kidney in the young SHRs attenuated the development of hypertension and restored the normal levels of nNOS [131]. PIN also plays a role in nNOS uncoupling in the RVLM via decreasing the ratio of nNOS dimer/monomer and knockdown of PIN restores nNOS dimer/monomer ratio, restored NO content and alleviated oxidative stress in the RVLM resulting in decreased sympathoexcitation and hypertension associated with metabolic syndrome [132]. Other studies suggest that the function of PIN may be a dynein light chain involved in nNOS axonal [133]. In these experiments with recombinant PIN peptide and nNOS protein showed no alteration in nNOS dimer to monomeric ratio, which leads the authors to conclude that PIN does not have any role in the inhibition of nNOS via interfering with dimerization. This conclusion was based on the fact that the PIN interacting interphase sequence in nNOS lies outside its catalytic core, and thus is not a part of the dimerization region of nNOS [134]. The possibility that PIN may have a role in dimer assembly via interfering with nNOS dimerization even outside the dimerization region or via some yet unknown mechanism/s remains to be explored. Examining the effect of PIN silencing on NO-mediated sympathoinhibition in the PVN of CHF rats, for instance, will help to explore the molecular mechanism further and potentially provide therapeutic targets for intervention in sympatho-excitatory states. The role of PIN, regulated by Ang II, in the regulation of nNOS in the brain during various cardiovascular disease conditions, such as hypertension remains to be explored.
Work from our lab has shown that increased PIN expression (Figure 2A) with concomitant decrease in dimer to monomer ratio in the PVN during CHF (Figure 3A, C), points towards the accumulation of monomeric inactive nNOS enzyme, which leads to ubiquitin-mediated proteolytic degradation, and hence a decrease in levels of nNOS in CHF[48](Figure 3B,C). We also demonstrated an increased accumulation of Ub-nNOS conjugates in the PVN of CHF rats compared to control rats and also provided evidence for Ub-colocalization with nNOS in vivo (Figure 3D). Taken together our studies have revealed that nNOS in the PVN is regulated post-translationally by multiple protein-protein interactions as summarized in Figure 4. Monomerization of nNOS via PIN causes the formation of catalytically impaired enzyme leading to reduced levels of NO. Decreased NO level is expected to reduce GABA release and its inhibitory actions [16] resulting in reduced inhibitory influences on neurons in the PVN, and thereby, causing enhanced sympathoexcitation during CHF. NO would also be expected to counteract the enhanced glutamatergic actions observed in rats with CHF [16]. Recently, we demonstrated that up-regulation of nNOS transgenically, significantly decreased the enhanced renal sympathetic activity, arterial blood pressure, and heart rate responses to N-methyl-D-aspartic acid in the rats with CHF [23] which suggests that maybe PIN indirectly regulates NMDA receptors via manipulating functional nNOS levels. However, the details of these complex interactions remain to be explored. Nevertheless, taken together, these observations provide a significant insight into the possible mechanism(s) within the PVN that may contribute to the increased sympathetic drive commonly observed during CHF and offer a novel target for treatment of the enhanced sympathoexcitation in CHF and other cardiovascular diseases.
Figure 3.
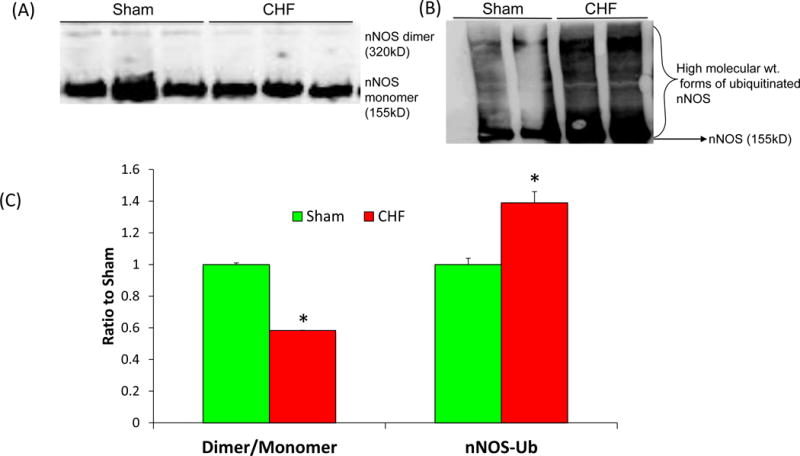
(A) Assessment of monomers and dimers of nNOS represented as a ratio of dimer to monomer and (B) high molecular weight ubiquitinated forms of immuno-detectable nNOS in the PVN of Sham and CHF rats. Dimeric and monomeric nNOS were separated by low temperature-PAGE in the cold room and visualized by immunoblotting using an anti-nNOS antibody. (C) Composite data of Dimer/Monomer ratio and nNOS-Ub in the PVN of Sham and CHF rats. Values are mean ± SEM (n = 4–5 rats per group). *P < 0.05 vs. Sham. (D) High magnification images of the coronal sections of PVN (63X) showing the colocalization of Ub(red) and nNOS (green). From Sharma et al (2013)[48]
Figure 4.
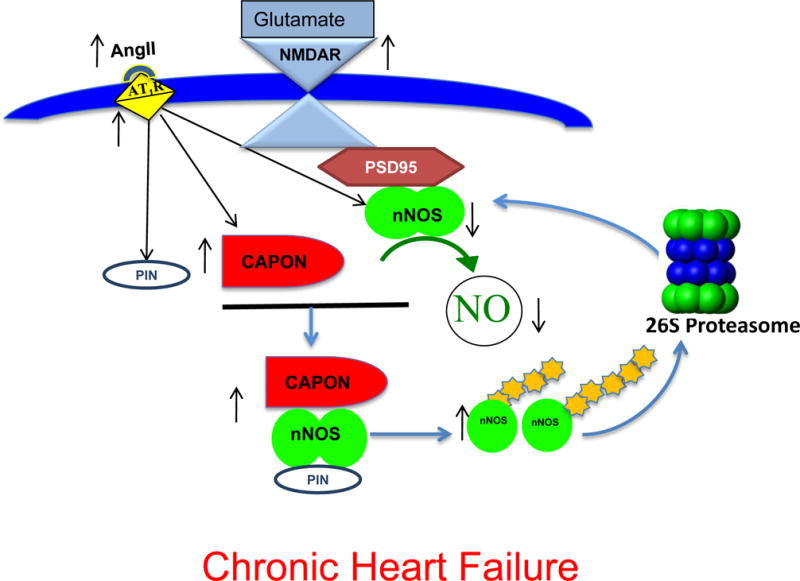
Schematic depicting presumptive pathway of nNOS regulation in the PVN of CHF rats. CAPON and PIN are over-expressed due to augmented levels of Ang II and AT1 receptors in the PVN. Increased CAPON competes with PSD95 for binding to nNOS and sequesters nNOS therefore decreasing NMDAR/PSD95/nNOS complexes at the synaptic membrane. Binding of PIN to nNOS in cytosol destabilizes nNOS dimers, which renders nNOS catalytically inactive, either by interfering with the assembly or dimer stability. Inactive nNOS monomers are susceptible to ubiquitination and subsequent proteasomal degradation. This results in decreased levels of nNOS as well as NO production in the PVN during CHF causing an increase in sympathetic nerve activity (SNA). Modified from Sharma et al (2013)[48]
4. Expert Opinion
Prevailing evidence indicates that the CNS, particularly the PVN is critically responsible for activation of neurohumoral drive in various cardiovascular diseases including CHF [9, 10, 11, 12]. Numerous studies have shown a significant role for nNOS in the regulation of sympathoexcitation [33, 34, 35, 36, 37, 38, 39, 40, 135]. Decrease in nNOS expression and NO bioavailability are evident in several autonomic regions suggesting that decreased nNOS activity may give rise to an overdrive of pre-autonomic neurons within the CNS which have detrimental pathological consequences in these cardiovascular diseases [33, 34, 35, 36, 37, 38, 39]. Despite the intensive research and attempts to develop NO-donors and NOS inhibitors, no significant success in the use of NO-related drugs in cardiovascular medicine has been achieved to date. The studies discussed in this review provide valuable insight into the possible post-translational modifications of nNOS including phosphorylation, ubiquitination, sumoylation, interaction with endogenous proteins viz. CAPON, PIN, HSP etc. in the regulation of nNOS. The importance of these molecules as critical regulator/s of nNOS function suggests that these molecules or their interactions may be the rational therapeutic targets in pathologies associated with dysregulation of nNOS. As literature[30, 35, 36, 37, 52, 53] suggest that NO is a Janus-faced molecule having two contrasting aspects, and incongruous release of this mediator has been linked to a number of pathologies, therefore, a fine regulation of nNOS within specific key brain nuclei such as the PVN is critical for maintaining cardiovascular homeostasis. The knowledge of isoform-specific structural features of the catalytic site, characterization of the interactions with modulating intracellular proteins and application of modern inhibitor design approaches in combination with mutagenesis should result in novel nNOS selective targeting with discrete pharmacological effects. The biochemical pathways known to regulate nNOS in the autonomic nervous system have not been investigated adequately under normal or pathological conditions to date. Our work provides significant insight into the possible molecular mechanism(s), specifically in the PVN, involved in regulation nNOS in CHF via ubiquitin-mediated mechanisms to regulate sympathetic outflow [36, 48]. Although restoration of nNOS centrally via targeted viral vectors or exercise training has shown beneficial effects in animal models [23, 136], further studies are required to determine whether nNOS or factors that regulate nNOS such as CAPON, PIN and CHIP are responsible for the observed benefits. Deciphering the underlying regulatory mechanisms involved in the upregulation of PIN and CAPON in the brain and identification of E3 ligase for nNOS ubiquitination during various cardiovascular diseases will yield further insight into the regulation of nNOS. Targeting nNOS and its upstream, as well as downstream regulatory molecules in the CNS, may be a worthwhile therapeutic strategy in CHF, hypertension, and other diseases associated with increased sympathoexcitation. The design of inhibitors or nanoparticles to target nNOS-CAPON, nNOS-PIN interphase, inhibition of E3 ligase involved in ubiquitination of nNOS or supplementation of food with BH4 to restore the active dimers can help to produce adequate NO in patients. On the whole, understanding molecular and cellular mechanisms involved in regulation of nNOS and NO signaling may provide novel therapeutic targets to reduce sympathetic outflow for better management of these autonomic abnormalities in patients with cardiovascular diseases.
Highlights.
Anomalous regulation of the sympathetic nervous system leading to exaggerated sympathetic nerve activity is implicated in a broad spectrum of cardiovascular diseases.
nNOS is the primary source for NO synthesized in the brain and is abundantly expressed in the various areas including those involved in the regulation of cardiovascular functions, such as the paraventricular nucleus of the hypothalamus.
Various pathological cardiovascular states are associated with diminished expression of nNOS within key central sites involved in regulation of the central nervous system (CNS) including heart failure, hypertension, diabetes mellitus and chronic renal failure.
This review focuses on the regulation of nNOS at the posttranslational level within the CNS in the context of healthy and diseased states.
Gaining further insight into the mechanism/s involved in the regulation of nNOS would provide potential novel strategies for manipulating nNOS directed therapeutic modalities in the future.
Acknowledgments
Funding: This paper has been supported by funding from American Heart Association, Scientist Development Grant (14SDG19980007) and grants from the U.S. Department of Health and Human Services - National Institutes of Health, Heart, Lung, & Blood Institute, (R56 HL124104) and (PO1 HL62222).
Footnotes
Declaration of Interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Ferguson DW, Berg WJ, Sanders JS. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: evidence from direct microneurographic recordings. JAm Coll Cardiol. 1990;16:1125–34. doi: 10.1016/0735-1097(90)90544-y. [DOI] [PubMed] [Google Scholar]
- 2••.Patel KP. Role of paraventricular nucleus in mediating sympathetic outflow in heart failure. Heart Fail Rev. 2000;5:73–86. doi: 10.1023/A:1009850224802. This study identified a critical role for the PVN in heart failure. [DOI] [PubMed] [Google Scholar]
- 3.Kishi TaH, Y. Cardiac Complications in Hypertension and Diabetes: Role of Sympathetic Nervous Activity. Current Hypertens Rev. 2013;9:4. [Google Scholar]
- 4.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. New Engl J Med. 1984;311:819–23. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 5.Packer M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation. 1988;77:721–30. doi: 10.1161/01.cir.77.4.721. [DOI] [PubMed] [Google Scholar]
- 6.Porter JP, Brody MJ. Neural projections of paraventricular nucleus that subserve vasomoter functions. Am J Physiol Regul Integr Comp Physiol. 1985;248:R271–R81. doi: 10.1152/ajpregu.1985.248.3.R271. [DOI] [PubMed] [Google Scholar]
- 7.Vasan RS. Biomarkers of cardiovascular disease: molecular basis and practical considerations. Circulation. 2006;113:2335–62. doi: 10.1161/CIRCULATIONAHA.104.482570. [DOI] [PubMed] [Google Scholar]
- 8.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–46. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 9.Swanson LW, Sawchenko PE. Hypothalamic integration: Organization of the paraventricular and supraoptic nuclei. Ann Rev Neurosci. 1983;6:269–324. doi: 10.1146/annurev.ne.06.030183.001413. [DOI] [PubMed] [Google Scholar]
- 10.Swanson LW, Sawchenko PE. Paraventricular nucleus: a site for the integration of neuroendocrine and autonomic mechanisms. Neuroendocrinology. 1980;31:410–7. doi: 10.1159/000123111. [DOI] [PubMed] [Google Scholar]
- 11.Kannan H, Niijima A, Yamashita H. Effects of stimulation of the hypothalamic paraventicular nucleus on blood pressure and renal sympathetic nerve activity. Brain Res Bull. 1988;20:779–83. doi: 10.1016/0361-9230(88)90091-3. [DOI] [PubMed] [Google Scholar]
- 12.Kenney MJ, Weiss ML, Haywood JR. The paraventricular nucleus: an important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol Scand. 2003;177:7–15. doi: 10.1046/j.1365-201X.2003.01042.x. [DOI] [PubMed] [Google Scholar]
- 13.Commons KG, Pfaff DW. Ultrastructural evidence for enkephalin mediated disinhibition in the ventromedial nucleus of the hypothalamus. J Chem Neuroanat. 2001;21:53–62. doi: 10.1016/s0891-0618(00)00093-4. [DOI] [PubMed] [Google Scholar]
- 14.Pyner S, Coote JH. Identification of branching paraventricular neurons of the hypothalamus that project to the rostroventrolateral medulla and spinal cord. Neuroscience. 2000;100:549–56. doi: 10.1016/s0306-4522(00)00283-9. [DOI] [PubMed] [Google Scholar]
- 15.Schuman EM, Madison DV. Nitric oxide and synaptic function. Ann Rev Neurosci. 1994;17:153–83. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- 16••.Patel KP, Li YF, Hirooka Y. Role of nitric oxide in central sympathetic outflow. Proc Soc Exp Biol Med. 2001;226:814–24. doi: 10.1177/153537020122600902. A comprehensive review describing that the central NO mechanism is altered in disease states such as hypertension and heart failure. [DOI] [PubMed] [Google Scholar]
- 17.Zucker IH, Wang W, Pliquett RU, Liu JL, Patel KP. The regulation of sympathetic outflow in heart failure. The roles of angiotensin II, nitric oxide, and exercise training. Ann NY Acad Sci. 2001;940:431–43. [PubMed] [Google Scholar]
- 18.Liu JL, Zucker IH. Regulation of sympathetic nerve activity in heart failure: a role for nitric oxide and angiotensin II. Circ Res. 1999;84:417–23. doi: 10.1161/01.res.84.4.417. [DOI] [PubMed] [Google Scholar]
- 19*.Zhang K, Mayhan WG, Patel KP. Nitric oxide within the paraventricular nucleus mediates changes in renal sympathetic nerve activity. Am J Physiol. 1997;273:R864–R72. doi: 10.1152/ajpregu.1997.273.3.R864. This study identified roles of nitric oxide in PVN in controlling sympathetic nerve discharge. [DOI] [PubMed] [Google Scholar]
- 20.Luo CX, Zhu DY. Research progress on neurobiology of neuronal nitric oxide synthase. Neurosci Bull. 2011;27:23–35. doi: 10.1007/s12264-011-1038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–75. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Golledge J. Neuronal nitric oxide synthase and sympathetic nerve activity in neurovascular and metabolic systems. Curr Neurovasc Res. 2013;10:81–9. doi: 10.2174/156720213804805963. [DOI] [PubMed] [Google Scholar]
- 23•.Zheng H, Liu X, Li Y, Sharma NM, Patel KP. Gene transfer of neuronal nitric oxide synthase to the paraventricular nucleus reduces the enhanced glutamatergic tone in rats with chronic heart failure. Hypertension. 2011;58:966–73. doi: 10.1161/HYPERTENSIONAHA.111.176222. An important study demonstrating a role for restoring nNOS within the PVN to altering enhanced sympathoexcitation during heart failure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horn T, Smith PM, McLaughlin BE, Bauce L, Marks GS, Pittman QJ, Ferguson AV. Nitric oxide actions in paraventricular nucleus: Cardiovascular and neurochemical implications. Am J Physiol Regul Integr Comp Physiol. 1994;266:R306–R13. doi: 10.1152/ajpregu.1994.266.1.R306. [DOI] [PubMed] [Google Scholar]
- 25.Zhang K, Patel KP. Effect of nitric oxide within the paraventricular nucleus on renal sympathetic nerve discharge:role of GABA. Am J Physiol. 1998;275:R728–R34. doi: 10.1152/ajpregu.1998.275.3.R728. [DOI] [PubMed] [Google Scholar]
- 26.Biancardi VC, Son SJ, Sonner PM, Zheng H, Patel KP, Stern JE. Contribution of central nervous system endothelial nitric oxide synthase to neurohumoral activation in heart failure rats. Hypertension. 2011;58:454–63. doi: 10.1161/HYPERTENSIONAHA.111.175810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27••.Li Y-F, Patel KP. Paraventricular nucleus of the hypothalamus and elevated sympathetic activity in heart failure: altered inhibtory mechanisms. Acta Physiol Scand. 2003;177:17–26. doi: 10.1046/j.1365-201X.2003.01043.x. This paper highlighted the critical inhibitory role for nNOS in the PVN of rats with CHF. [DOI] [PubMed] [Google Scholar]
- 28.Zhang K, Li YF, Patel KP. Reduced endogenous GABA-mediated inhibition in the PVN on renal nerve discharge in rats with heart failure. Am J Physiol. 2002;282:R1006–R15. doi: 10.1152/ajpregu.00241.2001. [DOI] [PubMed] [Google Scholar]
- 29.Li YF, Jackson KL, Stern JE, Rabeler B, Patel KP. Interaction between glutamate and GABA systems in the integration of sympathetic outflow by the paraventricular nucleus of the hypothalamus. Am J Physiol Heart Circ Physiol. 2006;291:H2847–2856. doi: 10.1152/ajpheart.00625.2005. [DOI] [PubMed] [Google Scholar]
- 30.Guix FX, Uribesalgo I, Coma M, Munoz FJ. The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol. 2005;76:126–52. doi: 10.1016/j.pneurobio.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 31.Dawson TM, Snyder SH. Gases as biological messengers: nitric oxide and carbon monoxide in the brain. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1994;14:5147–59. doi: 10.1523/JNEUROSCI.14-09-05147.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM, Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron. 1991;7:615–24. doi: 10.1016/0896-6273(91)90374-9. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Patel KP, Cornish KG, Channon KM, Zucker IH. nNOS gene transfer to RVLM improves baroreflex function in rats with chronic heart failure. Am J Physiol Heart Circ Physiol. 2003;285:H1660–7. doi: 10.1152/ajpheart.00239.2003. [DOI] [PubMed] [Google Scholar]
- 34.Li YL, Li YF, Liu D, Cornish KG, Patel KP, Zucker IH, Channon KM, Schultz HD. Gene transfer of neuronal nitric oxide synthase to carotid body reverses enhanced chemoreceptor function in heart failure rabbits. Circ Res. 2005;97:260–7. doi: 10.1161/01.RES.0000175722.21555.55. [DOI] [PubMed] [Google Scholar]
- 35.Hirooka Y, Shigematsu H, Kishi T, Kimura Y, Ueta Y, Takeshita A. Reduced nitric oxide synthase in the brainstem contributes to enhanced sympathetic drive in rats with heart failure. J Cardio Pharmacol. 2003;42(Suppl 1):S111–5. doi: 10.1097/00005344-200312001-00023. [DOI] [PubMed] [Google Scholar]
- 36•.Sharma NM, Zheng H, Mehta PP, Li YF, Patel KP. Decreased nNOS in the PVN leads to increased sympathoexcitation in chronic heart failure: role for CAPON and Ang II. Cardiovasc Res. 2011;92:342–57. doi: 10.1093/cvr217. This was the first article revealing the role of CAPON in the regulation of nNOS in the PVN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tandai-Hiruma M, Horiuchi J, Sakamoto H, Kemuriyama T, Hirakawa H, Nishida Y. Brain neuronal nitric oxide synthase neuron-mediated sympathoinhibition is enhanced in hypertensive Dahl rats. J Hypertens. 2005;23:825–34. doi: 10.1097/01.hjh.0000163152.27954.7a. [DOI] [PubMed] [Google Scholar]
- 38.Tan DY, Meng S, Manning J. Role of neuronal nitric oxide synthase in dahl salt-sensitive hypertension. Hypertension. 1999;33:456–61. doi: 10.1161/01.hyp.33.1.456. [DOI] [PubMed] [Google Scholar]
- 39.Zheng H, Mayhan WG, Bidasee KR, Patel KP. Blunted Nitric Oxide-Mediated Inhibition of Sympathetic Nerve Activity Within the Paraventricular Nucleus in Diabetic Rats. Am J Physiol Regul Integr Comp Physiol. 2006;290:R992–R1002. doi: 10.1152/ajpregu.00363.2005. [DOI] [PubMed] [Google Scholar]
- 40.Koomans HA, Blankestijn PJ, Joles JA. Sympathetic hyperactivity in chronic renal failure: a wake-up call. J Am Soc Nephrol. 2004;15:524–37. doi: 10.1097/01.asn.0000113320.57127.b9. [DOI] [PubMed] [Google Scholar]
- 41.Francis J, Davisson RL. Emerging concepts in hypertension. Antioxid Redox Signal. 2014;20:69–73. doi: 10.1089/ars.2013.5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hirooka Y, Kishi T, Sakai K, Takeshita A, Sunagawa K. Imbalance of central nitric oxide and reactive oxygen species in the regulation of sympathetic activity and neural mechanisms of hypertension. Am J Physiol Regul Integr Comp Physiol. 2011;300:R818–26. doi: 10.1152/ajpregu.00426.2010. [DOI] [PubMed] [Google Scholar]
- 43•.Patel KP, Schultz HD. Angiotensin peptides and nitric oxide in cardiovascular disease. Antioxid Redox Signal. 2013;19:1121–32. doi: 10.1089/ars.2012.4614. This paper provides details on the role of Ang II and NO in regulation of sympathoexcitation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zucker IH, Pliquett RU. Novel mechanisms of sympatho-excitation in chronic heart failure. Heart Fail Monit. 2002;3:2–7. [PubMed] [Google Scholar]
- 45.Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Europ Heart J. 2012;33:829–37. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. A comprehensive review demonstrating the stucture of NOS enzymes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kone BC. Protein-protein interactions controlling nitric oxide synthases. Acta Physiol Scand. 2000;168:27–31. doi: 10.1046/j.1365-201x.2000.00629.x. [DOI] [PubMed] [Google Scholar]
- 48••.Sharma NM, Llewellyn TL, Zheng H, Patel KP. Angiotensin II-mediated posttranslational modification of nNOS in the PVN of rats with CHF: role for PIN. Am J Physiol Heart Circ Physiol. 2013;305:H843–55. doi: 10.1152/ajpheart.00170.2013. This is a novel study conferring the posttranslational regulation of nNOS in the PVN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elfering SL, Sarkela TM, Giulivi C. Biochemistry of mitochondrial nitric-oxide synthase. J Biol Chem. 2002;277:38079–86. doi: 10.1074/jbc.M205256200. [DOI] [PubMed] [Google Scholar]
- 50.Lamas S, Marsden PA, Li GK, Tempst P, Michel T. Endothelial nitric oxide synthase: molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc Natl Acad Sci U S A. 1992;89:6348–52. doi: 10.1073/pnas.89.14.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng H, Bidasee KR, Mayhan WG, Patel KP. Lack of central nitric oxide triggers erectile dysfunction in diabetes. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1158–64. doi: 10.1152/ajpregu.00429.2006. [DOI] [PubMed] [Google Scholar]
- 52.Stanek A, Gadowska-Cicha A, Gawron K, Wielkoszynski T, Adamek B, Cieslar G, Wiczkowski A, Sieron A. Role of nitric oxide in physiology and pathology of the gastrointestinal tract. Mini Rev Med Chem. 2008;8:1549–60. doi: 10.2174/138955708786786462. [DOI] [PubMed] [Google Scholar]
- 53.Burke AJ, Sullivan FJ, Giles FJ, Glynn SA. The yin and yang of nitric oxide in cancer progression. Carcinogenesis. 2013;34:503–12. doi: 10.1093/carcin/bgt034. [DOI] [PubMed] [Google Scholar]
- 54.Hall AV, Antoniou H, Wang Y, Cheung AH, Arbus AM, Olson SL, Lu WC, Kau CL, Marsden PA. Structural organization of the human neuronal nitric oxide synthase gene (NOS1) J Biol Chem. 1994;269:33082–90. [PubMed] [Google Scholar]
- 55.Wang Y, Newton DC, Marsden PA. Neuronal NOS: gene structure, mRNA diversity, and functional relevance. Crit Rev Neurobiol. 1999;13:21–43. doi: 10.1615/critrevneurobiol.v13.i1.20. [DOI] [PubMed] [Google Scholar]
- 56.Brenman JE, Xia H, Chao DS, Black SM, Bredt DS. Regulation of neuronal nitric oxide synthase through alternative transcripts. Dev Neurosci. 1997;19:224–31. doi: 10.1159/000111211. [DOI] [PubMed] [Google Scholar]
- 57.Boissel JP, Schwarz PM, Forstermann U. Neuronal-type NO synthase: transcript diversity and expressional regulation. Nitric oxide: Biol Chem. 1998;2:337–49. doi: 10.1006/niox.1998.0189. [DOI] [PubMed] [Google Scholar]
- 58.Sasaki M, Gonzalez-Zulueta M, Huang H, Herring WJ, Ahn S, Ginty DD, Dawson VL, Dawson TM. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc Natl Acad Sci USA. 2000;97:8617–22. doi: 10.1073/pnas.97.15.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH. Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc Natl Acad Sci USA. 1997;94:3396–401. doi: 10.1073/pnas.94.7.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–86. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 61.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–67. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 62.Xie J, Roddy P, Rife TK, Murad F, Young AP. Two closely linked but separable promoters for human neuronal nitric oxide synthase gene transcription. Proc Natl Acad Sci USA. 1995;92:1242–6. doi: 10.1073/pnas.92.4.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ogura T, Yokoyama T, Fujisawa H, Kurashima Y, Esumi H. Structural diversity of neuronal nitric oxide synthase mRNA in the nervous system. Biochem Biophys Res Commu. 1993;193:1014–22. doi: 10.1006/bbrc.1993.1726. [DOI] [PubMed] [Google Scholar]
- 64.Chen ZP, McConell GK, Michell BJ, Snow RJ, Canny BJ, Kemp BE. AMPK signaling in contracting human skeletal muscle: acetyl-CoA carboxylase and NO synthase phosphorylation. American journal of physiology Endocrinol Metab. 2000;279:E1202–6. doi: 10.1152/ajpendo.2000.279.5.E1202. [DOI] [PubMed] [Google Scholar]
- 65.Bredt DS, Ferris CD, Snyder SH. Nitric oxide synthase regulatory sites. Phosphorylation by cyclic AMP-dependent protein kinase, protein kinase C, and calcium/calmodulin protein kinase; identification of flavin and calmodulin binding sites. J Biol Chem. 1992;267:10976–81. [PubMed] [Google Scholar]
- 66.Brune B, Lapetina EG. Phosphorylation of nitric oxide synthase by protein kinase A. Biochem Biophys Res Commu. 1991;181:921–6. doi: 10.1016/0006-291x(91)91279-l. [DOI] [PubMed] [Google Scholar]
- 67.Nakane M, Mitchell J, Forstermann U, Murad F. Phosphorylation by calcium calmodulin-dependent protein kinase II and protein kinase C modulates the activity of nitric oxide synthase. Biochem Biophys Res Commu. 1991;180:1396–402. doi: 10.1016/s0006-291x(05)81351-8. [DOI] [PubMed] [Google Scholar]
- 68.Komeima K, Hayashi Y, Naito Y, Watanabe Y. Inhibition of neuronal nitric-oxide synthase by calcium/calmodulin-dependent protein kinase IIalpha through Ser847 phosphorylation in NG108-15 neuronal cells. J Biol Chem. 2000;275:28139–43. doi: 10.1074/jbc.M003198200. [DOI] [PubMed] [Google Scholar]
- 69.Hayashi Y, Nishio M, Naito Y, Yokokura H, Nimura Y, Hidaka H, Watanabe Y. Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J Biol Chem. 1999;274:20597–602. doi: 10.1074/jbc.274.29.20597. [DOI] [PubMed] [Google Scholar]
- 70.Song T, Sugimoto K, Ihara H, Mizutani A, Hatano N, Kume K, Kambe T, Yamaguchi F, Tokuda M, Watanabe Y. p90 RSK-1 associates with and inhibits neuronal nitric oxide synthase. Biochem J. 2007;401:391–8. doi: 10.1042/BJ20060580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Okada D. Tetrahydrobiopterin-dependent stabilization of neuronal nitric oxide synthase dimer reduces susceptibility to phosphorylation by protein kinase C in vitro. FEBS Lett. 1998;434:261–4. doi: 10.1016/s0014-5793(98)00993-4. [DOI] [PubMed] [Google Scholar]
- 72.Mount PF, Power DA. Nitric oxide in the kidney: functions and regulation of synthesis. Acta Physiol (Oxf) 2006;187:433–46. doi: 10.1111/j.1748-1716.2006.01582.x. [DOI] [PubMed] [Google Scholar]
- 73.Rameau GA, Tukey DS, Garcin-Hosfield ED, Titcombe RF, Misra C, Khatri L, Getzoff ED, Ziff EB. Biphasic coupling of neuronal nitric oxide synthase phosphorylation to the NMDA receptor regulates AMPA receptor trafficking and neuronal cell death. J Neurosci. 2007;27:3445–55. doi: 10.1523/JNEUROSCI.4799-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cheng WH, Lu PJ, Ho WY, Tung CS, Cheng PW, Hsiao M, Tseng CJ. Angiotensin II inhibits neuronal nitric oxide synthase activation through the ERK1/2-RSK signaling pathway to modulate central control of blood pressure. Circ Res. 2010;106:788–95. doi: 10.1161/CIRCRESAHA.109.208439. [DOI] [PubMed] [Google Scholar]
- 75.Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999;68:1015–68. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 76.Rinetti GV, Schweizer FE. Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. J Neurosci. 2010;30:3157–66. doi: 10.1523/JNEUROSCI.3712-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klimaschewski L. Ubiquitin-dependent proteolysis in neurons. New Physiol Sci. 2003;18:29–33. doi: 10.1152/nips.01408.2002. [DOI] [PubMed] [Google Scholar]
- 78.Lopez Salon M, Morelli L, Castano EM, Soto EF, Pasquini JM. Defective ubiquitination of cerebral proteins in Alzheimer’s disease. J Neurosci Res. 2000;62:302–10. doi: 10.1002/1097-4547(20001015)62:2<302::AID-JNR15>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 79.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 80.Hochstrasser M. Ubiquitin- dependent protein degradation. Annu Rev Genet. 1996;30:405–39. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 81.Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–33. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 82•.Dunbar AY, Kamada Y, Jenkins GJ, Lowe ER, Billecke SS, Osawa Y. Ubiquitination and degradation of neuronal nitric-oxide synthase in vitro: dimer stabilization protects the enzyme from proteolysis. Mol Pharmacol. 2004;66:964–9. doi: 10.1124/mol.104.000125. In vitro studies suggesting that the dimeric structure is a major determinant of nNOS stability and proteolysis. [DOI] [PubMed] [Google Scholar]
- 83.Noguchi S, Jianmongkol S, Bender AT, Kamada Y, Demady DR, Osawa Y. Guanabenz-mediated inactivation and enhanced proteolytic degradation of neuronal nitric oxide synthase. J Biol Chem. 2000;275:2376–80. doi: 10.1074/jbc.275.4.2376. [DOI] [PubMed] [Google Scholar]
- 84.Osawa Y, Lowe ER, Everett AC, Dunbar AY, Billecke SS. Proteolytic Degradation of Nitric Oxide Synthase: Effect of Inhibitors and Role of hsp90-Based Chaperones. J Pharmacol Exp Ther. 2003;304:493–7. doi: 10.1124/jpet.102.035055. [DOI] [PubMed] [Google Scholar]
- 85.Bender AT, Silverstein AM, Demady DR, Kanelakis KC, Noguchi S, Pratt WB, Osawa Y. Neuronal nitric oxide synthase is regulated by the hsp90-based chaperone system in vivo. J Biol Chem. 1999;274:1472–8. doi: 10.1074/jbc.274.3.1472. [DOI] [PubMed] [Google Scholar]
- 86.Bender AT, Demady DR, Osawa Y. Ubiquitination of neuronal nitric oxide synthase in vitro and in vivo. J Biol Chem. 2000;275:17407–11. doi: 10.1074/jbc.M000155200. [DOI] [PubMed] [Google Scholar]
- 87.Cimarosti H, Lindberg C, Bomholt SF, Ronn LC, Henley JM. Increased protein SUMOylation following focal cerebral ischemia. Neuropharmacology. 2008;54:280–9. doi: 10.1016/j.neuropharm.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 88.Shalizi A, Bilimoria PM, Stegmuller J, Gaudilliere B, Yang Y, Shuai K, Bonni A. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. J Neurosci. 2007;27:10037–46. doi: 10.1523/JNEUROSCI.0361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89•.Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–70. doi: 10.1038/347768a0. The evidence suggesting the association of NO with discrete neuronal populations. [DOI] [PubMed] [Google Scholar]
- 90.Watanabe M, Itoh K. Characterization of a novel posttranslational modification in neuronal nitric oxide synthase by small ubiquitin-related modifier-1. Biochim Biophys Acta. 2011;1814:900–7. doi: 10.1016/j.bbapap.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 91.Northcott CA, Billecke S, Craig T, Hinojosa-Laborde C, Patel KP, Chen AF, D’Alecy LG, Haywood JR. Nitric oxide synthase, ADMA, SDMA, and nitric oxide activity in the paraventricular nucleus throughout the etiology of renal wrap hypertension. Am J Physiol Heart Circ Physiol. 2012;302:H2276–84. doi: 10.1152/ajpheart.00562.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA. 1990;87:682–5. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Su Z, Blazing MA, Fan D, George SE. The calmodulin-nitric oxide synthase interaction. Critical role of the calmodulin latch domain in enzyme activation. J Biol Chem. 1995;270:29117–22. doi: 10.1074/jbc.270.49.29117. [DOI] [PubMed] [Google Scholar]
- 94.Higaki J, Ogihara T, Kumahara Y, Bravo EL. Calmodulin levels in hypertensive rats. Clin Sci (Lond) 1985;68:407–10. doi: 10.1042/cs0680407. [DOI] [PubMed] [Google Scholar]
- 95.Jeck CD, Zimmermann R, Schaper J, Schaper W. Decreased expression of calmodulin mRNA in human end-stage heart failure. J Mol Cell Cardiol. 1994;26:99–107. doi: 10.1006/jmcc.1994.1011. [DOI] [PubMed] [Google Scholar]
- 96.Li Z, Srivastava P. Heat-shock proteins. Curr Protoc Immunol. 2004 doi: 10.1002/0471142735.ima01ts58. Appendix 1T. [DOI] [PubMed] [Google Scholar]
- 97.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–33. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 98.Peng HM, Morishima Y, Pratt WB, Osawa Y. Modulation of heme/substrate binding cleft of neuronal nitric-oxide synthase (nNOS) regulates binding of Hsp90 and Hsp70 proteins and nNOS ubiquitination. J Biol Chem. 2012;287:1556–65. doi: 10.1074/jbc.M111.323295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li Z, Song Y, Xing R, Yu H, Zhang Y, Li Z, Gao W. Heat shock protein 70 acts as a potential biomarker for early diagnosis of heart failure. PloS One. 2013;8:e67964. doi: 10.1371/journal.pone.0067964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bredt DS. Targeting nitric oxide to its targets. Proc Soc Exp Biol Med. 1996;211:41–8. doi: 10.3181/00379727-211-43950f. [DOI] [PubMed] [Google Scholar]
- 101.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu ZQ. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha-1 syntrophin mediated by PDZ domains. Cell. 1996;84:757–67. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 102•.Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–73. doi: 10.1074/jbc.274.39.27467. This was the first paper to provide evidence that PSD95 is involved in the NMDAR medaited activation of nNOS. [DOI] [PubMed] [Google Scholar]
- 103.Garthwaite J, Garthwaite G, Palmer RMJ, Moncada S. NMDA receptor activation induces nitric oxide synthesis from arginine in rat brain slices. Eur J Pharmacol. 1989;172:413–6. doi: 10.1016/0922-4106(89)90023-0. [DOI] [PubMed] [Google Scholar]
- 104.Kornau HC, Seeburg PH, Kennedy MB. Interactions of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–73. doi: 10.1016/s0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- 105.Brenman JE, Christopherson KS, Craven SE, McGee AW, Bredt DS. Cloning and characterization of postsynaptic density 93, a nitric oxide synthase interacting protein. J Neurosci. 1996b;16:7407–15. doi: 10.1523/JNEUROSCI.16-23-07407.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Megan Bardgett MAA, Alfredo Calderon, Glenn Toney. SD95 Coupling of NMDA Receptor Activation and Nitric Oxide Activity in the Hypothalamic PVN. The FASEB Journal. 2015;29:987.7. [Google Scholar]
- 107.Luo CX, Lin YH, Qian XD, Tang Y, Zhou HH, Jin X, Ni HY, Zhang FY, Qin C, Li F, Zhang Y, Wu HY, Chang L, Zhu DY. Interaction of nNOS with PSD-95 negatively controls regenerative repair after stroke. J Neurosci. 2014;34:13535–48. doi: 10.1523/JNEUROSCI.1305-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jones N. Stroke: Disruption of the nNOS-PSD-95 complex is neuroprotective in models of cerebral ischemia. Nat Rev Neurol. 2011;7:61. doi: 10.1038/nrneurol.2010.203. [DOI] [PubMed] [Google Scholar]
- 109.Jaffrey SR, Benfenati F, Snowman AM, Czernik AJ, Snyder SH. Neuronal nitric-oxide synthase localization mediated by a ternary complex with synapsin and CAPON. Proc Natl Acad Sci USA. 2002;99:3199–204. doi: 10.1073/pnas.261705799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110••.Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–24. doi: 10.1016/s0896-6273(00)80439-0. A novel paper leading to discovery of CAPON. [DOI] [PubMed] [Google Scholar]
- 111.Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH. Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–93. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 112.Che YH, Tamatani M, Tohyama M. Changes in mRNA for post-synaptic density-95 (PSD-95) and carboxy-terminal PDZ ligand of neuronal nitric oxide synthase following facial nerve transection. Mol Brain Res. 2000;76:325–35. doi: 10.1016/s0169-328x(00)00013-9. [DOI] [PubMed] [Google Scholar]
- 113.Zhu LJ, Li TY, Luo CX, Jiang N, Chang L, Lin YH, Zhou HH, Chen C, Zhang Y, Lu W, Gao LY, Ma Y, Zhou QG, Hu Q, Hu XL, Zhang J, Wu HY, Zhu DY. CAPON-nNOS coupling can serve as a target for developing new anxiolytics. Nat Med. 2014;20:1050–4. doi: 10.1038/nm.3644. [DOI] [PubMed] [Google Scholar]
- 114.Lu CJ, Hao G, Nikiforova N, Larsen HE, Liu K, Crabtree MJ, Li D, Herring N, Paterson DJ. CAPON modulates neuronal calcium handling and cardiac sympathetic neurotransmission during dysautonomia in hypertension. Hypertension. 2015;65:1288–97. doi: 10.1161/HYPERTENSIONAHA.115.05290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kremeyer B, Garcia J, Kymalainen H, Wratten N, Restrepo G, Palacio C, Miranda AL, Lopez C, Restrepo M, Bedoya G, Brzustowicz LM, Ospina-Duque J, Arbelaez MP, Ruiz-Linares A. Evidence for a role of the NOS1AP (CAPON) gene in schizophrenia and its clinical dimensions: an association study in a South American population isolate. Hum Hered. 2009;67:163–73. doi: 10.1159/000181154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu B, Wratten N, Charych EI, Buyske S, Firestein BL, Brzustowicz LM. Increased expression in dorsolateral prefrontal cortex of CAPON in schizophrenia and bipolar disorder. PLoS Med. 2005;2:e263. doi: 10.1371/journal.pmed.0020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coyle JT, Tsai G, Goff D. Converging Evidence of NMDA Receptor Hypofunction in the Pathophysiology of Schizophrenia. Ann NY Acad Sci. 2003;1003:318–27. doi: 10.1196/annals.1300.020. [DOI] [PubMed] [Google Scholar]
- 118.Bernstein HG, Stanarius A, Baumann B, Henning H, Krell D, Danos P, Falkai P, Bogerts B. Nitric oxide synthase-containing neurons in the human hypothalamus: reduced number of immunoreactive cells in the paraventricular nucleus of depressive patients and schizophrenics. Neuroscience. 1998;83:867–75. doi: 10.1016/s0306-4522(97)00461-2. [DOI] [PubMed] [Google Scholar]
- 119.Karson CN, Griffin WS, Mrak RE, Husain M, Dawson TM, Snyder SH, Moore NC, Sturner WQ. Nitric oxide synthase (NOS) in schizophrenia: increases in cerebellar vermis. Mol Chem Neuropathol. 1996;27:275–84. doi: 10.1007/BF02815109. [DOI] [PubMed] [Google Scholar]
- 120.Gentleman SM, Leclercq P, von Bussmann KA, Garey LJ, Royston MC. Differential distribution of nitric oxide synthase neurons in the cerebral cortex of schizophrenics. Schizophrenia Res. 1995;15:28. [Google Scholar]
- 121.Johnson AK, Grippo AJ. Sadness and broken hearts: neurohumoral mechanisms and co-morbidity of ischemic heart disease and psychological depression. J Physiol Pharmacol. 2006;57:5–29. [PubMed] [Google Scholar]
- 122.Chapa DW, Akintade B, Son H, Woltz P, Hunt D, Friedmann E, Hartung MK, Thomas SA. Pathophysiological relationships between heart failure and depression and anxiety. Crit Care Nurse. 2014;34:14–24. doi: 10.4037/ccn2014938. [DOI] [PubMed] [Google Scholar]
- 123.Depression Can Break Your Heart. National Institute of Mental Health; Bethesda: Published. [Google Scholar]
- 124.Vallance P, Leone A, Calver A, Collier J, Moncada S. Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. Journal of cardiovascular pharmacology. 1992;20(Suppl 12):S60–2. doi: 10.1097/00005344-199204002-00018. [DOI] [PubMed] [Google Scholar]
- 125.Zoccali C, Mallamaci F, Maas R, Benedetto FA, Tripepi G, Malatino LS, Cataliotti A, Bellanuova I, Boger R, Investigators C Left ventricular hypertrophy, cardiac remodeling and asymmetric dimethylarginine (ADMA) in hemodialysis patients. Kidney Int. 2002;62:339–45. doi: 10.1046/j.1523-1755.2002.00437.x. [DOI] [PubMed] [Google Scholar]
- 126••.Jaffrey SR, Snyder SH. PIN: an associated protein inhibitor of neuronal nitric oxide synthase. Science. 1996;274:774–7. doi: 10.1126/science.274.5288.774. A novel study leading to discovery of PIN. [DOI] [PubMed] [Google Scholar]
- 127.Gillardon F, Krep H, Brinker G, Lenz C, Bottiger B, Hossmann KA. Induction of protein inhibitor of neuronal nitric oxide synthase/cytoplasmic dynein light chain following cerebral ischemia. Neuroscience. 1998;84:81–8. doi: 10.1016/s0306-4522(97)00479-x. [DOI] [PubMed] [Google Scholar]
- 128.Chang YW, Jakobi R, McGinty A, Foschi M, Dunn MJ, Sorokin A. Cyclooxygenase 2 promotes cell survival by stimulation of dynein light chain expression and inhibition of neuronal nitric oxide synthase activity. Mol Cell Biol. 2000;20:8571–9. doi: 10.1128/mcb.20.22.8571-8579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Magee TR, Ferrini MG, Davila HH, Zeller CB, Vernet D, Sun J, Lalani R, Burnett AL, Rajfer J, Gonzalez-Cadavid NF. Protein inhibitor of nitric oxide synthase (NOS) and the N-methyl-D-aspartate receptor are expressed in the rat and mouse penile nerves and colocalize with penile neuronal NOS. Biol Reprod. 2003;68:478–88. doi: 10.1095/biolreprod.102.007310. [DOI] [PubMed] [Google Scholar]
- 130.Magee TR, Kovanecz I, Davila HH, Ferrini MG, Cantini L, Vernet D, Zuniga FI, Rajfer J, Gonzalez-Cadavid NF. Antisense and short hairpin RNA (shRNA) constructs targeting PIN (Protein Inhibitor of NOS) ameliorate aging-related erectile dysfunction in the rat. J Sex Med. 2007;4:633–43. doi: 10.1111/j.1743-6109.2007.00459.x. [DOI] [PubMed] [Google Scholar]
- 131.Wang SC, Lin KM, Chien SJ, Huang LT, Hsu CN, Tain YL. RNA silencing targeting PIN (protein inhibitor of neuronal nitric oxide synthase) attenuates the development of hypertension in young spontaneously hypertensive rats. J Am Soc Hypertens. 2014;8:5–13. doi: 10.1016/j.jash.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 132.Wu KL, Chao YM, Tsay SJ, Chen CH, Chan SH, Dovinova I, Chan JY. Role of nitric oxide synthase uncoupling at rostral ventrolateral medulla in redox-sensitive hypertension associated with metabolic syndrome. Hypertension. 2014;64:815–24. doi: 10.1161/HYPERTENSIONAHA.114.03777. [DOI] [PubMed] [Google Scholar]
- 133.Rodriguez-Crespo I, Straub W, Gavilanes F, Ortiz de Montellano PR. Binding of dynein light chain (PIN) to neuronal nitric oxide synthase in the absence of inhibition. Arch Biochem Biophys. 1998;359:297–304. doi: 10.1006/abbi.1998.0928. [DOI] [PubMed] [Google Scholar]
- 134.Fan JS, Zhang Q, Li M, Tochio H, Yamazaki T, Shimizu M, Zhang M. Protein inhibitor of neuronal nitric-oxide synthase, PIN, binds to a 17-amino acid residue fragment of the enzyme. J Biol Chem. 1998;273:33472–81. doi: 10.1074/jbc.273.50.33472. [DOI] [PubMed] [Google Scholar]
- 135.Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, Victor RG. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–23. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zheng H, Li Y-F, Liu XF, Patel KP. Exercise training improve endogenous nitric oxide machnisms within the paraventricular nucleus involved in regulating renal sympathetic nerve activity in rats with diabetes. Am J Physiol Heart Circ Physiol. 2003;288:H2332–H41. doi: 10.1152/ajpheart.00473.2004. [DOI] [PubMed] [Google Scholar]