

Abstract
Reading ligand structures into any simulation program is often nontrivial and time consuming, especially when the force field parameters and/or structure files of the corresponding molecules are not available. To address this problem, we have developed Ligand Reader & Modeler in CHARMM-GUI. Users can upload ligand structure information in various forms (using PDB ID, ligand ID, SMILES, MOL/MOL2/SDF file, or PDB/mmCIF file), and the uploaded structure is displayed on a sketchpad for verification and further modification. Based on the displayed structure, Ligand Reader & Modeler generates the ligand force field parameters and necessary structure files by searching for the ligand in the CHARMM force field library or using the CHARMM general force field (CGenFF). In addition, users can define chemical substitution sites and draw substituents in each site on the sketchpad to generate a set of combinatorial structure files and corresponding force field parameters for throughput or alchemical free energy simulations. Finally, the output from Ligand Reader & Modeler can be used in other CHARMM-GUI modules to build a protein-ligand simulation system for all supported simulation programs, such as CHARMM, NAMD, GROMACS, AMBER, GENESIS, LAMMPS, Desmond, OpenMM, and CHARMM/OpenMM. Ligand Reader & Modeler is available as a functional module of CHARMM-GUI at http://www.charmm-gui.org/input/ligandrm.
Keywords: molecular dynamics simulation, molecular modeling, drug design, protein-ligand interactions, CGenFF
Graphical abstract
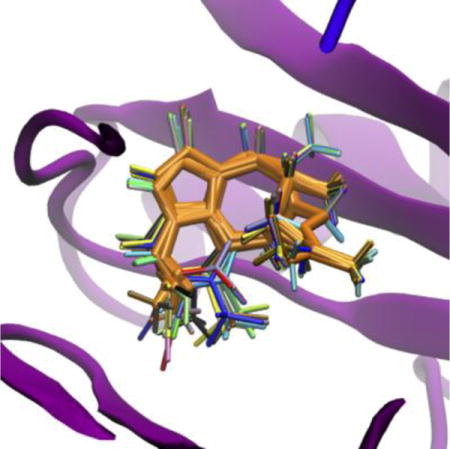
Superposition of twenty-four TIBO derivatives generated by Ligand Reader & Modeler (multiple colors) with HIV-1 RT (magenta) and the scaffold atoms (orange).
Introduction
Molecular modeling and simulation are important tools in nanoscale material and biological sciences discovery and characterization, as they provide insight into structures, dynamics, and underlying mechanisms of such systems, which are difficult to obtain from experimental data alone. The widely-adopted modeling and simulation approaches use certain force fields (FFs) to describe the energy of a molecular system. Therefore, preparation and employment of an accurate FF are a prerequisite to successful molecular modeling and simulations. Various FFs (e.g., CHARMM,1 GROMOS,2 AMBER,3 OPLS,4 and UFF5) have been developed in conjunction with advanced simulation algorithms to achieve more accurate results.6 Arguably, recent molecular simulation techniques and FF parameters are reasonably well tested and mature enough to interpret experiments and guide new experiments with testable hypotheses.7,8
The CHARMM FF9–13 has been widely used for biomolecular simulations, covering proteins, nucleic acids, lipids, carbohydrates, and small molecules. Despite the abundance of small molecules available, however, the CHARMM FF is still limited to a small subset of ligands, leaving the influx of newly designed molecules unsupported.14 To start to ameliorate this problem, software platforms like MATCH15 and the CHARMM general force field (CGenFF/ParamChem) program16,17 have been developed to cover a broader range of chemical space for molecules that are not covered by the CHARMM FF using rule based interpretation of existing FF parameter libraries. Note that the automatic parameter generation is not perfect. For example, the approaches employed in MATCH and the CGenFF program are based on the similarity between the atom types that define each required parameter and those in existing parameters. The dissimilarity is quantified in terms of the “penalty scores” associated with the partial charges and other parameters and are given in the output FF files, so that users can check the quality of the parameters for their molecules; a lower penalty score suggests a closer match and more confidence in the parameters.
Considerable efforts have also been made to facilitate the parameterization process through (web-based) graphical user interface tools such as SwissParam,18 GAAMP,19 and Force Field Toolkit (ffTK).20 MATCH and ParamChem store pre-parameterized molecules and molecular fragments on their own database and provide CHARMM FF parameters of given molecules by matching the atom and bond types with the templates in the database. In the case of SwissParam, parameters for bonds, angles, dihedral angles, improper angles, and charges are taken from Merck molecular force field,21 and the atom type and corresponding van der Waals parameters are determined by the fragment-based search with the CHARMM FF. GAAMP generates GAAMP generates parameters and charges of target molecules from quantum mechanical (QM) calculations. Finally, ffTK, a VMD plugin, was developed for experienced users who want to have more control over analogous molecules used in the process, and helps users derive missing parameters of given molecules from the CHARMM compatible parameters and QM calculations. While all these tools have helped researchers to prepare FF parameters for their customized molecules, users still need to prepare accurate MOL2-like structure file and make extra efforts to include their ligands into the biological systems for detailed studies of biomolecule-ligand interactions. In addition, the lack of an automated procedure to generate congeneric series of a compound to study the effects of substituents in specific sites in a scaffold remains a cumbersome, time-consuming and error-prone task.
Since 2006, CHARMM-GUI (http://www.charmm-gui.org) has been available to make the building processes of complex biomolecular systems simple and straightforward for broader research communities to carry out innovative and novel biomolecular modeling and simulation research to acquire insight into structure, dynamics, and underlying mechanism of biomolecular systems.22–31 However, preparing biological simulation systems that contain ligands remains challenging even in CHARMM-GUI for multiple reasons such as residue name mismatch, atom name mismatch, uncertainty of protonation state, bad ligand structure SDF/MOL2 file, etc. (see reference26 and below). Therefore, an easy-to-use CHARMM-GUI functionality that reliably generates a batch of CHARMM FF parameters and simulation systems should significantly facilitate simulation studies of ligand-containing biological systems.
In this study, we present a new CHARMM-GUI module, Ligand Reader & Modeler (http://www.charmm-gui.org/input/ligandrm) that provides a user-friendly interface to prepare a set of CHARMM FF parameters, structure, and necessary input files for user-specified molecules. The module includes a chemical editor, Marvin JS,32 allowing users to interactively verify and modify the molecular structure of their interest. For preparation of multiple ligands with various substituents within a chemical scaffold, Ligand Reader & Modeler also provides the FF parameters and structure files for the combinatorial set of structures based on a user-specified core structure and substituent chemical groups. The structure file with biomolecule and (modified) ligand(s) is provided for further application in other CHARMM-GUI modules. A schematic outline of Ligand Reader & Modeler is represented in Figure 1, and its implementation is described in detail in following sections, together with illustrative protein-ligand system generation examples.
Figure 1.
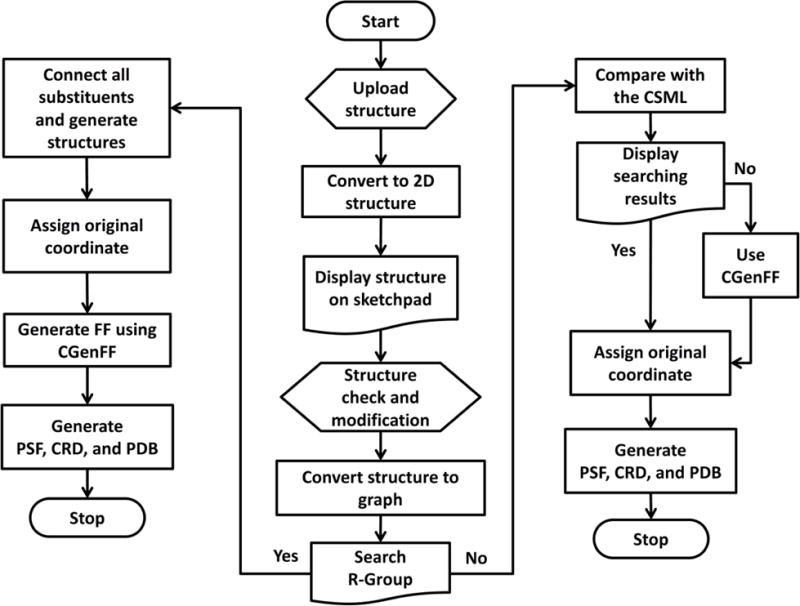
Schematic overview of Ligand Reader & Modeler.
Modus operandi of single structure generation
The standard CHARMM FF parameters for specific biomolecules and small molecules have been optimized and validated to best reproduce various experimental observables. Therefore, if applicable FF parameters exist, they should be the first choice, instead of generating new ones using CGenFF. However, finding a particular CHARMM residue that matches with a molecule of interest is not straightforward, especially when the residue name in the CHARMM topology files does not match the one in the PDB file. Even when the residue names match, it is not always guaranteed that the ligand in the PDB file is the same molecule to the one in the CHARMM FF. In addition, although the residues are the same molecule, it is still challenging to read the coordinates if atom names are different in between the PDB file and the CHARMM residue. To facilitate searching a molecule in the CHARMM FF library, we first developed a CHARMM small molecule library (CSML) in CHARMM-GUI (http://www.charmm-gui.org/docs/archive/csml), an archive containing most non-redundant small molecules available in the CHARMM FF. While CSML provides a searching toolbar to help users find a molecule of interest using its CHARMM or common name, an advanced tool was necessary for automated molecule search and/or atom name matching, which motivated us to develop Ligand Reader & Modeler.
Ligand Reader & Modeler starts with the chemical structure information uploaded in various forms: (i) PDB ID33 containing ligands, (ii) ligand ID defined in the chemical component dictionary (http://www.wwpdb.org/data/ccd),34 (iii) simplified molecular-input line-entry system (SMILES)35 notation, (iv) MOL/MOL2/SDF file,36 (v) PDB/mmCIF file, and (vi) drawing on the sketchpad powered by Marvin JS. If the uploaded PDB file contains multiple ligands, all non-identical ligands are listed in selection buttons. The selected/uploaded structure information is automatically converted to a two-dimentional (2D) structure on the sketchpad. In the case when a SMILES string is used as a user input, the ligand structure is generated by Molconverter37 based on “Daylights SMILES specification Rules”.38 Users can modify the 2D structure on the sketchpad interactively. Importantly, for accurate FF generation, users need to explicitly place all hydrogen and missing atoms at this stage. If the PDB file contains multiple identical ligands, only one structure is listed as the ligand structure for structural modification and FF generation, and the result is applied to all identical ligands based on their coordinates at the final step.
Note that ligand structures in PDB often have missing heavy atoms, making it difficult to identify the correct ligand structures. To address this issue, Ligand Reader & Modeler downloads the complete chemical structure (SDF file) of a given residue name from RCSB Chemical Component Dictionary (CCD) and compares the SDF structure with the PDB ligand structure using VF2 algorithm.39 The algorithm compares two graphs and determines if one graph is a subgraph of the other. If the PDB ligand structure is a subgraph of the SDF structure, the complete SDF structure from the repository is used as the reference ligand structure instead of the ligand structure in the PDB file. The coordinates of the uploaded ligand file are reassigned to the reference ligand structure at the final stage.
Ligand Reader & Modeler provides an option to “Find similar residues in the CHARMM FF” on the front page. The option is useful when users want to see whether any fragment of their molecule exists in the CHARMM FF for their own FF development. The user-specified molecule is searched in the CHARMM FF based on the structure similarity between the query molecule and CSML residues using the maximum common edge subgraph (MCES) algorithm.40,41 Figure 2 illustrates how the graph-based MCES search is performed. First, when the (edited) molecular structure on the sketchpad (e.g., 2A) is submitted for the next step, the structure information is converted into a graph representation (2C).42 In our graph representation, the nodes (atoms) are assigned according to the chemical element types and the number of associated covalent bonds, regardless of the bond order (including hydrogen atoms). Note that all residues in CSML (e.g., 2B) were pre-converted to the corresponding graph representations (2D) and stored for efficient search.
Figure 2.
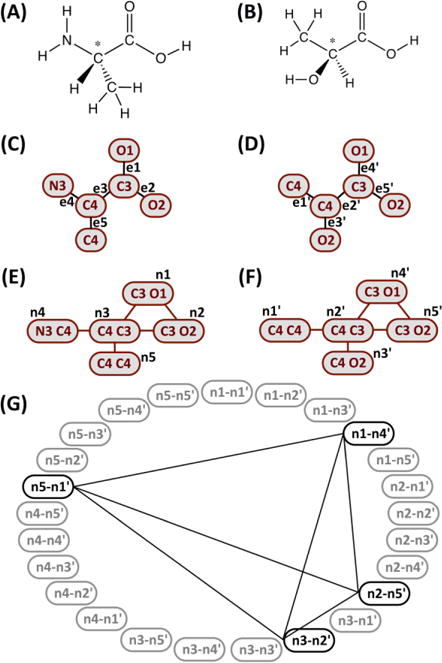
Illustrative procedure of bond degree based maximum common subgraph algorithm using the chemical structures, the graph representations, and the convoluted graphs of (A, C, E) alanine and (B, D, F) lactic acid. (G) A product graph of (E) and (F) shows the bold nodes used for the maximum clique search whose result is shown in the connecting lines; in this example, all bold nodes are involved in the maximum clique.
Next, the convoluted graphs (2E and 2F) are generated; the nodes in the convoluted graph are the pairs of neighboring nodes defined in the corresponding molecular graph, and the edges in the convoluted graph are constructed if the incident nodes share the same node type from the molecular graph. For example, the edge e1 [C3, O1] in the molecular graph (2C) is converted to the node n1 [C3 O1] in the convoluted graph (2E). In the same way, the edge e2 [C3, O2] in the molecular graph (2C) is convoluted to the node n2 [C3 O2] (2E), and an edge between nodes n1 and n2 is created because they share the incident node type [C3] in the molecular graph. Two convoluted graphs (2E and 2F) are converted into a modular product graph (2G). The edges in the product graph are created if both pairs of node components have the same adjacency property in the convoluted graph. For instance, two nodes n1-n4′ and n2-n5′ are connected because both n1/n2 and n4′/n5′ pairs are adjacent in the corresponding convoluted graphs. Also, two nodes n5-n1′ and n2-n5′ are connected because both n5/n2 and n1′/n5′ pairs are not adjacent in each convoluted graph. Finally, the maximum clique is searched using the improved approximate coloring algorithm.43 The nodes found in the maximum clique are the largest common substructure between two molecules. Based on this procedure, the query graph is compared with all CSML graphs and the Tanimoto similarity score44 (TAB) between 0 and 1 is calculated as a similarity measure: TAB = NAB/(NA + NB - NAB), where NAB is the number of the common nodes in structure A or B, and NA (or NB) is the total number of nodes in A (or B).
Finally, the search results are displayed in the four categories: “Exact residue”, “Isomers”, “Different Protonation/Hydrogenation State Residues”, and “Similar Residues” with associated information such as hyperlinks to show 2D or 3D structures, molecular net charge, corresponding CHARMM FF file name, and miscellaneous comments (Fig. 3). The VF2 isomorphism algorithm39 is used to search for the exact residue, isomers, and different protonation/hydrogenation state residues to accelerate the searching process, and the MCES algorithm is used only for the similar residue searching. The all-atom graphs are used to find the exact residue or isomers, and the non-hydrogen heavy atom graphs are used to find different protonation/hydrogenation state residues. If isomorphic graphs are found in CSML, improper angles of all chiral center carbons are calculated and compared to classify them as either the exact structure or isomers. If an exact residue is not found in CSML, the CGenFF generation option appears in the “Exact residue” section, so that one can generate the ligand topology and parameter files using the CGenFF program. All CSML residues that have the same heavy atoms as the query structure but different numbers of hydrogen atoms are displayed under “Different Protonation/Hydrogenation State Residues”. The similar residues, ranked by Tanimoto scores, are listed under “Similar Residues”, and users can check the shared heavy atoms (represented in red) by clicking a hyperlink on each Tanimoto score. Note that, long acyl chains in lipid-like molecules have a lot of nodes with the same types that make the similarity search very inefficient, so if the number of carbon atoms in an acyl chain is greater than twenty, the search is skipped automatically; e.g., typical lipids such as palmitoyl-oleoyl-phosphatidylcholine (POPC) are eligible for similarity search.
Figure 3.
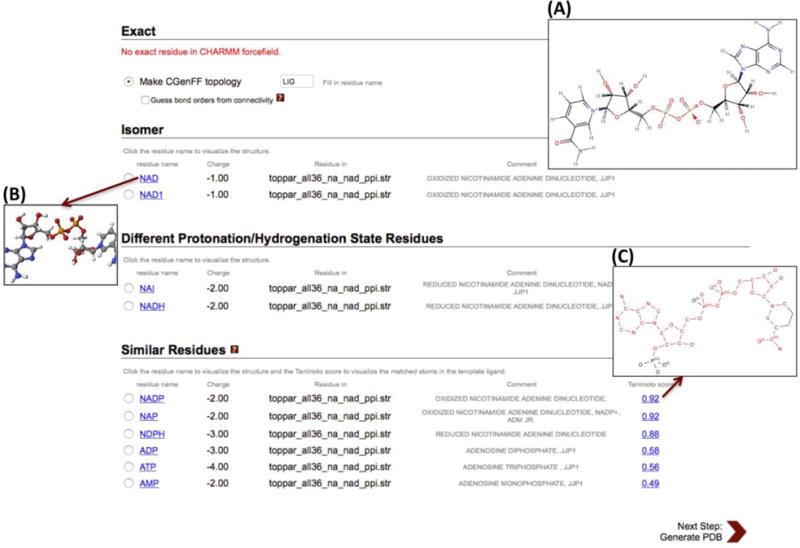
Snapshot of a CSML search result for NAD. (A) The NAD structure on the sketchpad of Ligand Reader & Modeler. One can visualize (B) the 3D structures of searched molecules and (C) the shared heavy atoms between similar residues (represented by red) on a pop-up window.
Once one of the CHARMM residues or CGenFF parameterization is selected, Ligand Reader & Modeler reads the original heavy atom coordinates from the uploaded structure file by creating a CHARMM coordinate file using the CHARMM atom types and unmodified coordinates. This coordinate file is read in a CHARMM input script “ligandrm.inp” to build a complete ligand structure (using the internal coordinate (IC) information if there are missing atoms) and to make sure that the ligand reading is successful. In addition, all other components (except the selected ligand(s)) in the input PDB file are merged with the selected ligand(s) into one CHARMM PDB format file (PDBID_modified.pdb or PDBfilename_modified.pdb). The combined CHARMM PDB file can be used for other CHARMM-GUI modules. A set of structure and topology/parameter files for CHARMM (PSF, CRD, and PDB) and GROMACS (ITP), as well as the related FF files are downloadable by clicking the “download.tgz” button.
There are three technical, unique aspects to be noted in our approach to find similar molecules in CSML. First, while direct SMILES string comparison is fast and easy, we decided not to use it because SMILES notations depend on the atom order in the structure file and the exact bond order information cannot be simply obtained from the CSML structure files (generated from the CHARMM topology files). Second, while the original MCES algorithm uses the bond order information, the degree of the node is used instead due to the lack of the exact bond order information in CSML. Third, as graph-based algorithms require more computational resources, one could employ a computationally more efficient chemical fingerprint algorithm44 in which one first compares only fingerprints, and then uses graph-based algorithms for only those entries that have high fingerprint similarity to increase search performance. However, since the number of compounds in CSML is relatively small (~1300, as of November 2016) and the compounds are generally small (<200 atoms), we decided to use the node-based graphs as described above.
CSML search in PDB Reader & Manipulator
PDB Reader & Manipulator provides several options for ligand modeling. One option is to find an identical molecule in the CHARMM FF and match the residue name to the found one. In this case, as mentioned above, the atom names of the PDB residue should also match those in the CHARMM FF, which can be a cumbersome task. To make the search and match process seamless, a “CSML Search” option has been introduced in PDB Reader & Manipulator. With this option, the reference structure file corresponding to a given PDB residue name is received from RCSB CCD and used for the CSML search as in Ligand Reader & Modeler. The search results are displayed on a pop-up window. If a match is found, the PDB ligand residue and atom names are changed to the corresponding CHARMM residue using a user-selected CHARMM residue. The advantages of the new feature in PDB Reader & Manipulator are as follows: 1) one can easily check if a PDB ligand exist in the CHARMM FF; 2) Missing atoms in PDB ligands can be easily identified and their coordinates can be generated based on the CHARMM IC information; 3) The result is applied to all multiple identical ligands that have the same residue name in the PDB file; and 4) users can still use all other PDB Reader & Manipulator options.
Combinatorial structure generation workflow
Preparing combinatorial structures by introducing diverse chemical groups into a ligand scaffold is a first step for throughput protein-ligand simulations or alchemical free energy simulations such as λ-dynamics45 and multi-site λ dynamics,46 which are promising applications to lead optimization in drug discovery. However, such a preparation is often time consuming and challenging. Ligand Reader & Modeler provides a combinatorial structure generation functionality to support multiple ligand preparation. On the sketchpad, users can define chemical substitution sites (using “Attachment points”) and substituents (using “R-group”) to design combinatorial structures of R-groups (Fig. 4). The core scaffold is defined by comparing all derivative pairs using our MCES searching scheme, and its heavy atom coordinates (from the uploaded structure) are preserved. The FF parameters for each derivative are generated using CGenFF (i.e., no similarity search for this mode). The combinatorial structures are generated by CHARMM, and each structure and related FF parameters are placed in different directories (ld1, ld2, etc). If the uploaded PDB file contains proteins and unselected ligands, they are merged with each combinatorial structure into the one CHARMM PDB file (PDBID_modified.pdb or PDBfilename_modified.pdb) that is copied to the corresponding directory. Finally, a set of CHARMM (PSF, CRD, and PDB), GROMACS (ITP), and FF files for all derivatives is freely downloadable by clicking the “download.tgz” button.
Figure 4.
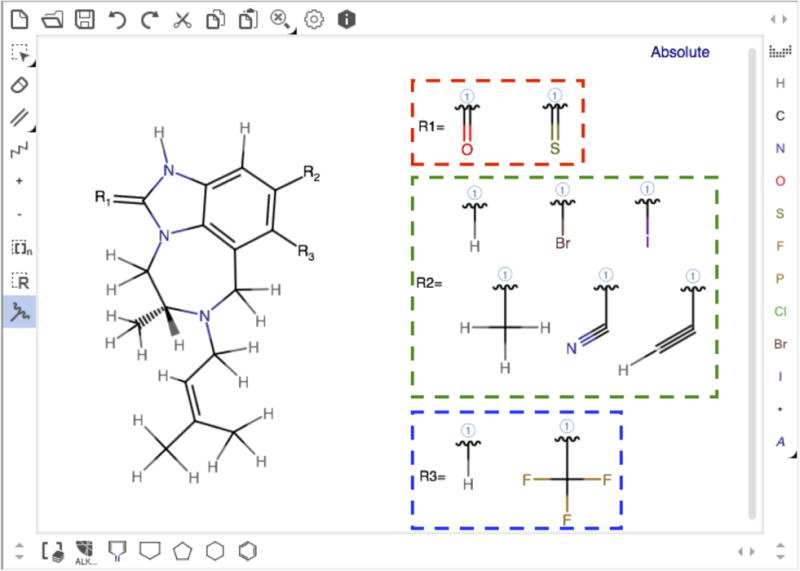
Snapshot of a sketchpad drawn for the combinatorial structure generation with TIBO in PDB:1TVR.
Applications
Several protein-ligand systems were built to illustrate the functionality of Ligand Reader & Modeler in combination with other CHARMM-GUI modules. An example of single ligand structure generation is nicotinamide adenine dinucleotide (NAD) whose oxidized (NAD+) and reduced (NADH) forms are used as a cofactor in glyceraldehyde 3-phosphate dehydrogenase (GAPDH). GAPDH is conserved in all species, playing an important role in glycolysis and gluconeogenesis. The crystal structure of holo-GAPDH (PDB: 5JY6)47 contains a GAPDH tetramer, and each subunit has a ligand named NAD in its binding site (Fig. 5A). Ligand Reader & Modeler found a match for NAD+ and NADH under “Different Protonation/Hydrogenation State Residues”, which were used to separately generate NAD+ or NADH structures. Because the PDB NAD structure has different atom names from the CHARMM residues (NAD/NADH), Ligand Reader & Modeler changes the atom names in the PDB NAD to those in the CHARMM FF. All heavy atom coordinates of PDB NAD structure (Fig. 6A) were transferred to NAD+ (Fig. 6B) and NADH (Fig. 6B), and all missing hydrogen atoms were generated using the IC information in the CHARMM FF. Ligand Reader & Modeler provided the CHARMM PDB file “5JY6_modified.pdb” (Fig. 5B) that contains the original tetrameric protein, four modified ligands (NAD+ or NADH), four magnesium ions, and crystal water. This file can be used for other CHARMM-GUI modules such as Quick MD Simulator or Membrane Builder to build a biological simulation system. Fig. 5C is a solvated simulation system generated using Quick MD Simulator with “5JY6_modified.pdb”. Note that, “PDBID_modified.pdb” is written in the CHARMM PDB format, so that users need to check the “PDB Format” option to “CHARMM” while uploading the structure to the modules.
Figure 5.
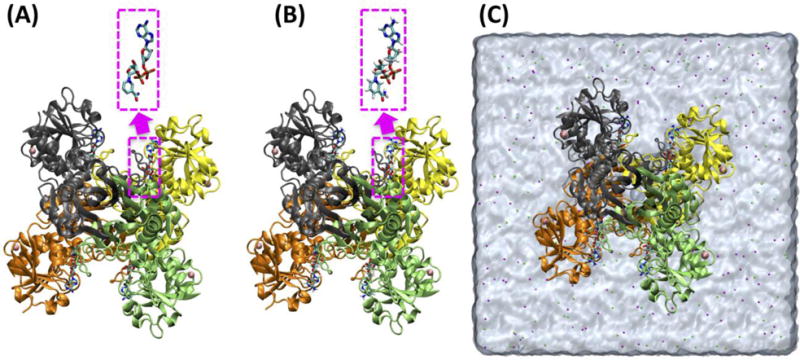
Snapshots of (A) the structure of 5JY6.pdb, (B) 5JY6_modified.pdb that contains NAD+, and (C) a solvated system of 5JY6_modified.pdb using Quick MD Simulator. The NAD heavy atom coordinates were preserved (pink dashed squares).
Figure 6.
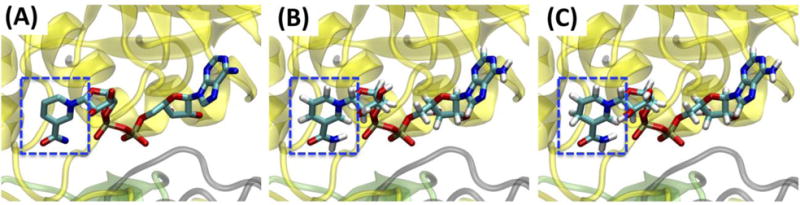
Snapshots of (A) NAD in 5JY6.pdb, (B) NAD+, and (C) NADH in glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The ligand heavy atom coordinates were preserved with the redox site hydrogen atoms (blue dashed squares).
For the above PDB:5JY6 example in which there is no need to modify the ligand, one can use the “CSML search” option in PDB Reader & Manipulator to build a solvated simulation system using Quick MD Simulator. For example, during the PDB reading and manipulation step, one can click the “CSML Search” button for the ligand NAD (Fig. 7A), and then the CHARMM residues with the identical heavy-atom structure are searched in the CSML, and the results are displayed on a pop-up window (Fig. 7B). Given a user-selected CHARMM residue, the PDB ligand residue and atom names are changed to those of the corresponding CHARMM residue for ligand reading. Another example is a structure of cellulose synthase/translocation intermediate (PDB: 4HG6), where two lauryldimethylamine-N-oxide (LDAO) molecules are bound to the crystal structure, but the tail of one LDAO ligand is missing (Fig. 8A). The “CSML Search” option identified the CHARMM LDAO residue, and the atom names and IC information in the CHARMM residue were used to read two LDAO molecules and to generate all missing atoms (Fig. 8B).
Figure 7.
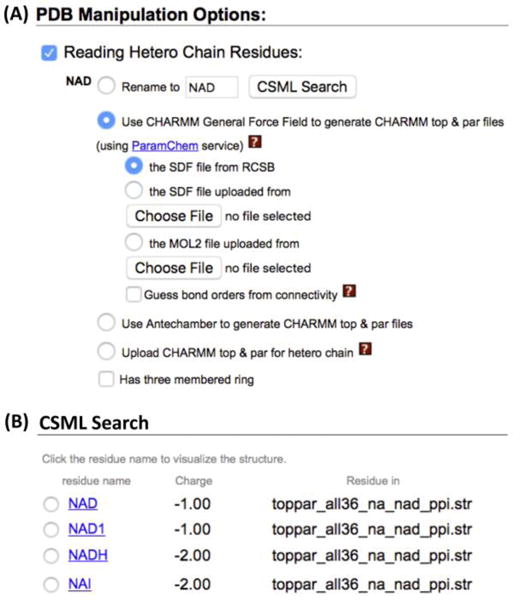
(A) “CSML Search” in PDB Manipulation Options. (B) The search results for NAD in holo-GAPDA (PDB: 5JY6)
Figure 8.
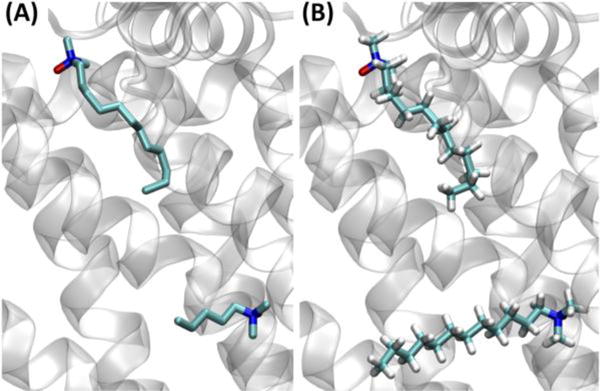
Structures of (A) PDB: 4HG6 with two lauryldimethylamine-N-oxide (LDAO) ligands, and (B) the PDB file after the PDB reading and manipulation step using “CSML Search”.
To illustrate the combinatorial structure generation in Ligand Reader & Modeler, the complex structure of human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT)-TIBO (PDB: 1TVR) was selected as an example. HIV-1 RT is responsible for viral replication and thus considered an important drug target for the treatment of AIDS.48 TIBO derivatives are drug candidates to inhibit HIV-1 RT. In 2011, Knight et al. estimated the relative binding affinities among three hybrid TIBO molecules (representing 14 unique inhibitors) to HIV-1 RT by calculating binding free energy differences using the multi-site λ dynamics approach.46 Based on the functional group and substitution site information from Knight’s paper, structures of twenty-four TIBO derivatives (all possible combinations of the given functional groups at each substitution site in Fig. 4) were generated at once using the PDB TIBO structure. After the TIBO derivative structures were generated, each derivative structure was combined with HIV-1 RT into a single CHARMM PDB structure (1TVR_modified.pdb) (Fig. 9). Note that all parameters and partial charges for the congeneric series of molecules were assigned using CGenFF. It would be very time consuming and laborious to upload each structure file to CHARMM-GUI for system building if users want all combinatorial structures. To facilitate this process, Ligand Reader & Modeler provides a python script to automate the browser actions for Quick MD Simulator (quickmd_combinatorial.py), making it much easier to generate all simulation systems that contain different combinatorial structures. Users can modify the script to use it in other modules.
Figure 9.

Superposition of twenty-four TIBO derivatives generated by Ligand Reader & Modeler (multiple colors) with HIV-1 RT (magenta) and the scaffold atoms (orange).
Conclusions
We have introduced and illustrated Ligand Reader & Modeler, a new functional module in CHARMM-GUI to help users to generate ligand FF parameter, structure, and other necessary files for various ligand-containing biomolecular simulations. The ligand FF parameters can be obtained either by searching for small molecules in the verified CHARMM FF or by using the CGenFF program. Ligand Reader & Modeler can also be used to get a set of combinatorial structures and their parameters by introducing different chemical functional groups to a ligand scaffold, which will be useful for throughput protein-ligand and alchemical free energy simulations such as the multi-site λ dynamics simulation.
Acknowledgments
We are grateful to Eufrozina Hoffmann, Marvin GUI Product Owner in ChemAxon Ltd., for allowing us to use Marvin JS in CHARMM-GUI. The work was supported in part by grants from XSEDE MCB-070009, NIH GM087519 (to WI), GM103695 (to CLB and WI), GM037554 (to CLB).
References
- 1.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 2.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. J Comput Chem. 2004;25(13):1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 3.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117(19):5179–5197. [Google Scholar]
- 4.Jorgensen WL, Maxwell DS, TiradoRives J. J Am Chem Soc. 1996;118(45):11225–11236. [Google Scholar]
- 5.Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. J Am Chem Soc. 1992;114(25):10024–10035. [Google Scholar]
- 6.González M. École thématique de la Société Française de la Neutronique. 2011;12:169–200. [Google Scholar]
- 7.Durrant JD, McCammon JA. BMC Biol. 2011;9:71. doi: 10.1186/1741-7007-9-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hospital A, Goni JR, Orozco M, Gelpi JL. Adv Appl Bioinform Chem. 2015;8:37–47. doi: 10.2147/AABC.S70333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, Mackerell AD., Jr J Chem Theory Comput. 2012;8(9):3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hart K, Foloppe N, Baker CM, Denning EJ, Nilsson L, Mackerell AD., Jr J Chem Theory Comput. 2012;8(1):348–362. doi: 10.1021/ct200723y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denning EJ, Priyakumar UD, Nilsson L, Mackerell AD., Jr J Comput Chem. 2011;32(9):1929–1943. doi: 10.1002/jcc.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. J Phys Chem B. 2010;114(23):7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guvench O, Mallajosyula SS, Raman EP, Hatcher E, Vanommeslaeghe K, Foster TJ, Jamison FW, 2nd, Mackerell AD., Jr J Chem Theory Comput. 2011;7(10):3162–3180. doi: 10.1021/ct200328p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AD., Jr J Comput Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yesselman JD, Price DJ, Knight JL, Brooks CL., III J Comput Chem. 2012;33(2):189–202. doi: 10.1002/jcc.21963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vanommeslaeghe K, MacKerell AD., Jr J Chem Inf Model. 2012;52(12):3144–3154. doi: 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vanommeslaeghe K, Raman EP, MacKerell AD., Jr J Chem Inf Model. 2012;52(12):3155–3168. doi: 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zoete V, Cuendet MA, Grosdidier A, Michielin O. J Comput Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 19.Huang L, Roux B. J Chem Theory Comput. 2013;9(8) doi: 10.1021/ct4003477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayne CG, Saam J, Schulten K, Tajkhorshid E, Gumbart JC. J Comput Chem. 2013;34(32):2757–2770. doi: 10.1002/jcc.23422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Halgren TA. J Comput Chem. 1996;17(5–6):490–519. [Google Scholar]
- 22.Jo S, Kim T, Iyer VG, Im W. J Comput Chem. 2008;29(11):1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 23.Jo S, Lim JB, Klauda JB, Im W. Biophys J. 2009;97(1):50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jo S, Jiang W, Lee HS, Roux B, Im WJ. Chem Inf Model. 2013;53(1):267–277. doi: 10.1021/ci300505n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng X, Jo S, Lee HS, Klauda JB, Im W. J Chem Inf Model. 2013;53(8):2171–2180. doi: 10.1021/ci4002684. [DOI] [PubMed] [Google Scholar]
- 26.Qi Y, Cheng X, Han W, Jo S, Schulten K, Im W. J Chem Inf Model. 2014;54(3):1003–1009. doi: 10.1021/ci500007n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jo S, Cheng X, Islam SM, Huang L, Rui H, Zhu A, Lee HS, Qi Y, Han W, Vanommeslaeghe K, MacKerell AD, Jr, Roux B, Im W. Adv Protein Chem Struct Biol. 2014;96:235–265. doi: 10.1016/bs.apcsb.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu EL, Cheng X, Jo S, Rui H, Song KC, Davila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, Klauda JB, Im W. J Comput Chem. 2014;35(27):1997–2004. doi: 10.1002/jcc.23702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi Y, Ingolfsson HI, Cheng X, Lee J, Marrink SJ, Im W. J Chem Theory Comput. 2015;11(9):4486–4494. doi: 10.1021/acs.jctc.5b00513. [DOI] [PubMed] [Google Scholar]
- 30.Qi Y, Cheng X, Lee J, Vermaas JV, Pogorelov TV, Tajkhorshid E, Park S, Klauda JB, Im W. Biophys J. 2015;109(10):2012–2022. doi: 10.1016/j.bpj.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, Jo S, Pande VS, Case DA, Brooks CL, III, MacKerell AD, Jr, Klauda JB, Im W. J Chem Theory Comput. 2016;12(1):405–413. doi: 10.1021/acs.jctc.5b00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marvin JS was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin JS 16.6.6, 2016, ChemAxon (http://www.chemaxon.com/).
- 33.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westbrook JD, Shao C, Feng Z, Zhuravleva M, Velankar S, Young J. Bioinformatics. 2015;31(8):1274–1278. doi: 10.1093/bioinformatics/btu789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weininger D. J Chem Inform Comput Sci. 1988;28(1):31–36. [Google Scholar]
- 36.Dalby A, Nourse JG, Hounshell WD, Gushurst AKI, Grier DL, Leland BA, Laufer J. J Chem Inform Comput Sci. 1992;32(3):244–255. [Google Scholar]
- 37.Molconverter was used for converting molecule files, Molconverter 15.11.23, 2016, ChemAxon (http://www.chemaxon.com/).
- 38.SMILES - A Simplified Chemical Language. http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html (accessed Apr 10, 2017)
- 39.Cordella LP, Foggia P, Sansone C, Vento M. IEEE Trans Pattern Anal Mach Intell. 2004;26(10):1367–1372. doi: 10.1109/TPAMI.2004.75. [DOI] [PubMed] [Google Scholar]
- 40.Durand PJ, Pasari R, Baker JW, Tsai CC. Internet J Chem. 1999;2(17) art. no.-17. [Google Scholar]
- 41.Raymond JW, Willett P. J Comput Aided Mol Des. 2002;16(7):521–533. doi: 10.1023/a:1021271615909. [DOI] [PubMed] [Google Scholar]
- 42.Diestel R. Graph Theory. Springer-Verlag; Berlin Heidelberg: 2010. [Google Scholar]
- 43.Konc J, Janezic D. Match-Commun Math Co. 2007;58(3):569–590. [Google Scholar]
- 44.Willett P, Barnard JM, Downs GM. J Chem Inform Comput Sci. 1998;38(6):983–996. [Google Scholar]
- 45.Kong XJ, Brooks CL., III J Chem Phys. 1996;105(6):2414–2423. [Google Scholar]
- 46.Knight JL, Brooks CL., III J Chem Theory Comput. 2011;7(9):2728–2739. doi: 10.1021/ct200444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schormann N, Ayres CA, Fry A, Green TJ, Banerjee S, Ulett GC, Chattopadhyay D. PLoS One. 2016;11(11):e0165917. doi: 10.1371/journal.pone.0165917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Das K, Ding JP, Hsiou Y, Clark AD, Moereels H, Koymans L, Andries K, Pauwels R, Janssen PAJ, Boyer PL, Clark P, Smith RH, Smith MBK, Michejda CJ, Hughes SH, Arnold E. J Mol Biol. 1996;264(5):1085–1100. doi: 10.1006/jmbi.1996.0698. [DOI] [PubMed] [Google Scholar]