

Summary
Eukaryotic genes are marked by conserved post-translational modifications on the RNA pol II CTD and the chromatin template. How the 5′–3′ profiles of these marks are established is poorly understood. Using pol II mutants in human cells we found that slow transcription repositioned specific co-transcriptionally deposited chromatin modifications; H3K36me3 shifted within genes toward 5′ ends and H3K4me2 extended further upstream of start sites. Slow transcription also evoked a hyperphosphorylation of CTD Ser2 residues at 5′ ends of genes that is conserved in yeast. We propose a “dwell-time in the target zone” model to explain the effects of transcriptional dynamics on establishment of co-transcriptionally deposited protein modifications. Promoter-proximal Ser2 phosphorylation is associated with longer pol II dwell time at start sites and reduced transcriptional polarity due to strongly enhanced divergent antisense transcription at promoters. These results demonstrate that pol II dynamics help govern the decision between sense and divergent antisense transcription.
Keywords: Histone methylation, K3K36me2, H3K4me2, pol II CTD S2 phosphorylation, bidirectional transcription, transcription elongation rate, kinetic coupling
Graphical Abstract
Introduction
Eukaryotic transcription units are characterized by conserved 5′ to 3′ profiles of specific pol II CTD phospho-isoforms and histone modifications that are deposited co-transcriptionally. Phosphorylation of CTD heptad repeats (YSPTSPS) on S5 is normally high at 5′ ends of genes where it facilitates mRNA capping, whereas S2 phosphorylation is high at 3′ ends where it is important for mRNA 3′ end formation (Bentley, 2014; Buratowski, 2009; Heidemann et al., 2013). However, some human genes have non-canonical peaks of Ser2-P near their 5′ ends (Schwartz et al., 2012). Furthermore knockdown of the CTD ligand FUS re-positioned Ser2-P toward 5′ ends (Schwartz et al., 2012), but the mechanism for this phenomenon is unclear. While, the potential function of CTD Ser2 phosphorylation at 5′ ends of genes is poorly understood, this modification has been found on polymerases engaged in divergent antisense transcription, which occurs at numerous human promoters (Core et al., 2008; Preker et al.; Preker et al., 2008), suggesting 5′ Ser2-P may play a functional role in divergent transcription. Several gene features correlate with the extent of divergent antisense transcription including nucleosome positioning and histone H3K4 dimethylation (Duttke et al., 2015; Marquardt et al., 2014; Scruggs et al., 2015), histone H4 de-acetylation coupled to H3K36 methylation (Churchman and Weissman, 2011) promoter structure (Chen et al., 2016), proximity to CpG islands (Core et al., 2008), and the frequency of polyA sites and U1snRNP binding sites in 5′ flanking sequences (Almada et al., 2013; Ntini et al., 2013). No components of the transcription machinery have yet been identified that change the ratio of antisense/sense transcription at divergent promoters.
Transcription units are also marked by conserved 5′ to 3′ profiles of chromatin modifications. Thus, histone H3K4me3 is usually localized in a peak at 5′ ends of genes (Schneider et al., 2004) whereas H3K36me3, that is implicated in control of elongation and pre-mRNA splicing (Luco et al., 2011; Teves et al., 2014) is localized within genes, but biased toward their 3′ ends (Wagner and Carpenter, 2012)
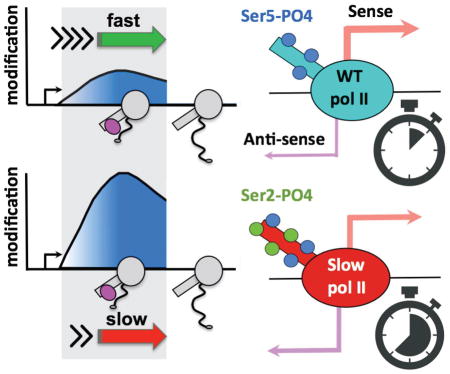
Despite the presumed functional importance of properly localized CTD and histone modifications within genes, it is not well understood how the 5′ to 3′ profiles of these modifications are set up and maintained. For example, they may be established by mechanisms that monitor position within a transcription unit or the time since elongation started. CTD Ser5-P and Ser2-P help to position H3K4me3 and K36me3, respectively because the Set1 H3K4 methyltransferase interacts with Ser5-P CTD (Ng et al., 2003) and the SET2 H3K36 methyltransferases interact with Ser2-P CTD (Kizer et al., 2005; Sun et al., 2005; Yoh et al., 2008) (C. Ebmeier, D. Taatjes, D.L.B, et al., in preparation). While CTD Ser2-P and H3K36me3 both increase 5′ to 3′ across transcribed genes, their ChIP profiles are distinct. Another possibility, not mutually exclusive with position sensing, is that the dwell-time spent by the transcription elongation complex (TEC) within the “target zone” where it is associated with a modifier, can modulate the deposition or removal of a post-translational modification (PTM, Fig. 1). The “dwell-time” principle is similar to that originally proposed for relating elongation rate with co-transcriptional splicing (Aebi and Weissman, 1987). In contrast to position sensing, this model predicts that if a modification reaction occurs on a similar time scale to pol II dwell time in the relevant “target zone”, then the extent of modification will be sensitive to elongation rate. Slow or fast elongation will increase or decrease the dwell-time of the TEC within the “target zone” and can thereby affect the extent of PTM deposition (or removal) (Fig. 1). If, on the other hand, the modification reaction is rapid relative to pol II dwell time in the relevant region of the gene, then PTM deposition will be unaffected by elongation rate. Interestingly fast elongation rates correlate with promoter distal localization of H3K36me3 in mouse ESCs (Jonkers et al., 2014) but whether establishment of the 5′ to 3′ profile of this mark is kinetically coupled to transcription elongation remains to be tested. Elongation rate has also been linked to the extent of pol II CTD S2 phosphorylation in yeast where the rat1-1 mutation accelerated transcription and increased CTD S2 phosphorylation (Jimeno-González et al., 2014). Whether there is a direct effect of elongation rate on CTD S2 phosphorylation is not known. In this report, we tested the dwell-time model by asking whether slow transcription elongation can affect the conserved 5′ to 3′ profiles of CTD and histone modifications. The results demonstrate that kinetic coupling with transcription elongation is an important determinant of where CTD Ser2-P and H3K36me3 are positioned within genes. Unexpectedly, genes enriched for CTD Ser2-P at their 5′ ends in slow pol II mutants have an elevated ratio of divergent antisense/sense transcription. The implication of these results is that pol II dwell time, and hence elongation rate, governs the function of pol II associated histone and CTD modifiers as well as the polarity of transcription at divergent promoters.
Figure 1. The “dwell-time in the target zone” model.

The “dwell-time in the target zone” model for establishment of profiles of co-transcriptionally deposited post-translational modifications (PTM). Diagram shows pol II (grey) that is associated with a post-translational modifier (pink) in the “target zone” (shaded). Y axis shows the amount of a PTM deposited by a “writer” of modifications in the “target zone” when transcription is fast (green arrow, short dwell-time) or slow (red arrow, long dwell-time). Note that activity of an “eraser” of PTMs associated with the TEC could also be affected by dwell-time.
Results
Elongation rate is a determinant of co-transcriptional histone H3K36me3
To investigate whether slow elongation rate affects co-transcriptional deposition of PTM’s, we employed HEK293 cell lines expressing inducible α-amanitin resistant (Amr) mutants of Rpb1 (Fong et al., 2014). Average in vivo elongation rates measured by Gro-seq at time points after release from a DRB block were approximately 1.7 kb/min for the WT Amr pol II, and 0.5 kb/min for the funnel domain mutant R749H (Fong et al., 2014). ChIP was conducted in cells induced with doxycycline to express the WT or mutant Amr large subunits, and then treated with α-amanitin for 42 hr to inhibit endogenous pol II.
Histone H3K36me3 is a co-transcriptionally deposited chromatin signature of expressed genes (Li et al., 2005; Vojnic et al., 2006). We tested whether pol II elongation rate influences establishment of the 5′ to 3′ profile of this mark. Anti-H3K36me3 ChIP-seq showed that the slow R749H mutant repositioned this modification toward the 5′ ends of many genes relative to WT pol II (Fig. 2, S1). This effect is evident in metaplots of over 5000 well expressed genes separated by over 2kb and from inspection of individual genes in replicate experiments (Fig. 2, S1A–D). “Early” H3K36me3 within the 5′ regions of genes in the slow mutant is widespread and highly significant (Fig. 2A, p<10−20 Welch’s two sample t-test). Slow transcription did not shift total histone H3 occupancy toward 5′ ends as measured by ChIP with antibody against the H3 C-terminus (Fig. S2A). Slow transcription also did not shift H3K36me3 toward 3′ ends, which occurs when splicing is inhibited (de Almeida et al., 2011; Kim et al., 2011). Therefore, the 5′ shift in H3K36me3 with slow transcription is not accounted for by indirect effects of nucleosome repositioning or splicing. In contrast to H3K36me3, there was no widespread effect of slow pol II on localization of H3K4me3, the chromatin signature of transcription start sites (Fig. S2B, C). We conclude that H3K36me3 localization across many human genes is sensitive to slow transcription, supporting the model that pol II dwell-time specifically modulates establishment of the 5′ to 3′ profile of this chromatin mark.
Figure 2. Slow elongation repositions H3K36me3 toward 5′ ends.

A. Metaplots of mean relative frequency of H3K36me3 ChIP signals at 5′ ends of human genes in WT (two independent experiments) and the R749H slow mutant. 100 bp bins are shown for the region from −1.5kb to +5.0 kb relative to the TSS for a set of well-expressed genes in HEK293 cells separated by 2kb (Brannan et al., 2012). Replicate experiments are shown in Fig. S1A–D. P-values (lower panel) were calculated using Welch’s two sample t-test. The horizontal dotted line indicates a p-value of 0.05.
B–E. UCSC genome browser shots of H3K36me3 ChIP signals in WT, and the R749H slow mutant. Note the 5′ shift in the profiles of H3K36me3 in the R749H slow mutant (arrows).
Slow pol II effects on CTD Ser2 phosphorylation and dwell time at promoters
We asked whether the 5′ shift of H3K36me3 caused by slow transcription was associated with altered CTD Ser2 phosphorylation because in yeast, this chromatin mark is affected by the CTD Ser2 kinase, Ctk1 (Xiao et al., 2006). Anti-CTD Ser-2P ChIP-seq revealed that in WT cells, as expected, this modification is predominantly located at 3′ ends, but surprisingly, in the R749H slow mutant, Ser2-P was strongly shifted toward 5′ ends (Fig. 3A). To confirm this effect we tested an independent slow mutant in the trigger loop (H1108Y) that severely inhibits transcription activity (Fong et al., 2014; Kaplan et al., 2008). ChIP signals in the H1108Y mutant are much lower than for R749H, but remarkably, on many genes, a novel peak of Ser2 phosphorylated pol II appeared at the TSS in both slow mutants (Fig. 3D–I, S3A, D–F, arrows). The effect of slow transcription on Ser2-P pol II is quite general as shown in a metaplot of ChIP signals in over 5000 well-expressed genes (Fig. 3A, B) and is specific to slow transcription as it was not observed in a fast trigger loop mutant (E1126G, Fig. S3A, B). The latter mutant had little or no effect on ChIP profiles of any PTMs in our experiments, possibly because it has a relatively modest effect on in vivo elongation rate (Fong et al., 2014). We identified a minimum of 829 genes where Ser2-P ChIP signals were significantly elevated (FDR<0.05, t-test) between −300 and +500 bp relative to the TSS in replicate experiments with the R749H mutant (Table S1). Interestingly, H3K36me3 shifted toward the 5′ ends of these genes, but not in a subset of genes where 5′ Ser2-P was unaffected by slow transcription (Fig. S1E, F).
Figure 3. Slow pol II prolongs dwell-time and elevates CTD Ser2 phosphorylation near start sites.

A. Metaplots of mean relative frequency of CTD Ser2-P ChIP signals for over 5000 genes in cells expressing WT Amr pol II and the R749H and H1108Y slow mutants. 100 base bins are shown in flanking regions from −1.5kb to +0.5kb relative to the TSS and from −0.5kb to +3.5kb relative to the poly (A) site. Gene body regions between +500 relative to the TSS and −500 relative to the poly (A) site are divided into 20 variable length bins. Note the peaks of Ser2-P at the TSS in both slow mutants (red arrow). Biological replicates are shown in Fig. S3.
B. Metaplots of Ser2-P ChIP signals normalized to total pol II (log2) in cells expressing WT, R749H and H1108Y pol II. Note elevated Ser2-P/total pol II in 5′ regions in both slow mutants.
C. Prolonged dwell time of R749H pol II relative to WT at promoters. Total pol II occupancy was measured by ChIP in cells treated with α-amanitin (42hr) and then with the initiation inhibitor triptolide (10 μM) for 0, 10, 20, 40 and 80 minutes. Q-PCR values were normalized to the tRNA MET gene as an internal control (see START Methods) and to t=0. SEM is shown for >5 PCR reactions from two biological replicates.
D–I. Anti-Pol II CTD Ser2-P ChIP-seq shows novel 5′ peaks (arrows) in the slow R749H and H1108Y pol II mutants. UCSC genome browser screen shots are shown. See also Fig. S3.
To verify that the effect of slow transcription on 5′ Ser2-P was due to enhanced phosphorylation rather than a gross re-localization of pol II, we normalized Ser2-P ChIP signals to total pol II. Average ChIP read density profiles for ~5000 well-separated genes (Fig. S3C) show that the characteristic peaks of paused pol II at transcription start sites are similar in WT and slow mutants and that relative pol II density within gene bodies is higher in the slow mutants as predicted by a mathematical model of elongation (Ehrensberger et al., 2013). Normalization to total pol II shows that the slow mutants enhanced the extent of Ser2 phosphorylation at 5′ ends of genes (Fig. 3B, S3B) consistent with the model that this modification is governed by pol II dwell-time near the TSS. The widespread repositioning of Ser2-P within genes in the R749H slow mutant is specific to this modification, and was not observed for other CTD phosphorylations on Ser5, Ser7 or Tyr1 that normally peak at 5′ ends (Fig. S4). These kinase reactions may therefore be too rapid on most genes to be affected by pol II dwell time. It remains possible that pol II dynamics affect phosphorylation of Ser5, Ser7, or Tyr1 residues on a specific minority of genes, however. Together these results suggest that in slow pol II mutants a distinct population of pol II phosphorylated on Ser2 as well as other residues within the CTD heptads accumulates at start sites. In this regard it is noteworthy that the 3E10 anti-Ser2-P monoclonal antibody (Chapman et al., 2007) we used for ChIP detects both Ser2-P and Ser2-P+Ser5-P doubly phosphorylated heptads. In summary, the results in Figure 3 reveal an unexpected plasticity in the CTD Ser2 phosphorylation profile. The canonical profile of 5′ low to 3′ high Ser2-P is radically changed in two slow pol II mutants that specifically enhance levels of this modification at 5′ ends.
To investigate whether slow pol II kinetics affects dwell time at start sites, we used ChIP to measure pol II occupancy at time points following inhibition of transcription initiation with triptolide (Chen et al., 2015; Henriques et al., 2013) that blocks the TFIIH-associated XPB helicase (Titov et al., 2011). For these experiments we compared WT Amr pol II with the R749H mutant that is much more active than H1108Y. The results in Figure 3C show that at four promoter regions tested, occupancy by the R749H mutant decays more slowly than WT in the presence of triptolide, as expected if the mutant polymerase has a longer dwell time at the 5′ ends of these genes. This result suggests that longer dwell time permits CTD Ser2 hyperphosphorylation at promoters.
A conserved effect of elongation rate on CTD Ser2-P in yeast
We tested whether the effect of elongation rate on Ser2 phosphorylation was conserved in budding yeast using the slow rpb2-10 mutant (Powell and Reines, 1996; Scafe et al., 1990). Replicate ChIP-seq experiments in rpb2-10 and isogenic WT strains revealed premature Ser2 CTD phosphorylation on most protein coding genes after normalization to total pol II (Fig. 4A) as well as non-coding snoRNA and U snRNA genes (Fig. 4B, E, H). Overlaid ChIP-seq profiles reveal that the positions where Ser2-P comes up at 5′ ends (red arrows, Fig. 4C–H) and where it declines at 3′ ends (grey arrows) are both shifted upstream when transcription is slow. A similar 5′ shift in Ser2-P pol II occurred on some genes in the slow N488D mutant within the Rpb1 catalytic center (Fig. 4F–H), but the effect was modest relative to rpb2-10. In summary, these experiments show that slow pol II elongation causes a similar 5′ re-localization of CTD Ser2-P in human and yeast cells. We conclude that kinetic coupling of Ser2 phosphorylation with transcription elongation is conserved across a wide evolutionary distance and is therefore likely to be of functional importance.
Figure 4. Slow elongation repositions CTD Ser2-P toward 5′ ends in yeast.

A. Metaplots of mean ChIP signals for Ser2-P normalized to total pol II (log2) in biological replicate ChIP-seq experiments with isogenic WT (DY103) and slow rpb2-10 mutant (DY105) strains at over 3000 ORFs >1kb long. 100 base bins are shown in 5′ and 3′ flanking regions and gene body regions are divided into 20 variable length bins. Note the 5′ shift in Ser2-P in rpb2-10 relative to WT (arrow).
B. Metaplots of mean relative frequency of Ser2-P ChIP signals at intergenic snoRNAs in WT and rpb2-10. Note the 5′ shift in Ser2-P in rpb2-10 (arrow).
C–E. 5′ shifting of Ser2-P (arrows) in the slow rpb2-10 mutant relative to isogenic WT. UCSC genome browser screen shots of anti-Ser2-P ChIP signals are shown for overlaid tracks of WT (grey) and rpb2-10 (red).
F–H. 5′ shifting of Ser2-P (arrows) in the slow rpb2-10 and rpb1 N488D (GRY3038) mutants (red) relative to isogenic WT strains (grey).
5′ Ser2-P associated with reduced transcriptional directionality and H3K4me2 re-positioning
To investigate the potential functional significance of Ser2 hyperphosphorylation at 5′ ends of genes, we conducted total nascent RNA sequencing in WT and slow pol II mutants by a modification of NET-seq (Churchman and Weissman, 2011) called anti-pol II tNET-seq in which total nascent RNA immunoprecipitated with anti-pol II antibody is sequenced. RNA pol II was immunoprecipitated from DNAse I digested nuclei (Fig. S5A) expressing WT or mutant Amr pol II followed by RNA isolation, random priming of cDNA and construction of strand-specific sequencing libraries (Fig. 5A, S5B). As expected for nascent transcripts anti-pol II tNET-seq reads for WT Amr HEK293 cells were enriched for intron reads relative to mRNA-seq of the same cells (Fong et al 2014) by 14.8 fold on average (n=31677 introns). Comparison of tNET-seq reads with published 3′ end NET-seq (Mayer et al., 2015; Nojima et al., 2015) shows that they are also enriched for, 3′ flanking sequences, divergent antisense transcripts, and enhancer RNAs, as well as being depleted for tRNAs and processed snoRNAs relative to chromatin associated RNA (Fig. S6). Nascent RNA sequencing in triplicate revealed a remarkable stimulation of divergent antisense transcription in both the R749H and H1108Y mutants that is evident from inspection of individual genes and metaplots of many genes (Fig. 5, 6). We defined the Divergence Index, DI, as a measure of divergent antisense transcription relative to sense transcription from each promoter region: DI= (divergent antisense reads in region −5kb to −1 relative to TSS/sense reads in region +1 to polyA site)×100. DI was significantly increased (FDR < .05) at close to 1000 genes in each of the slow mutants relative to WT pol II (Fig. S7A, B). Furthermore enhanced DI in the slow pol II mutants is common to genes with high and low levels of sense transcription (Fig. S7A, B). A small subset of genes with increased divergent transcription in the R749H mutant (35/1034; Table S1) were previously found to have convergent transcription across their start sites (Mayer et al., 2015). We also confirmed that there is an absolute increase in antisense transcription in the R749H slow mutant by QRT-PCR of nascent transcripts at 9 of 10 genes where it was detected by tNET-seq (Fig. S7C). Remarkably the antisense divergent transcription elicited by the slow mutants is elevated near the TSS consistent with more initiation and also often extended >20kb (Figs. 5F, S8A). In contrast anti-pol II tNET-seq does not reveal extensive transcription downstream of genes encoding mRNAs or snRNAs (Fig. S7D) consistent with previous anti-pol II ChIP-seq experiments (Fong et al., 2015). The apparent failure of divergent antisense transcripts to terminate early is not accounted for by a low frequency of polyA sites however (Fig. S7E). Increased divergent antisense transcription in the slow mutants was associated with reduced levels of sense transcription (Fig. 5D, E, 6A, S8B). Because the R749H and H1108Y substitutions are in different domains and modulate elongation by different mechanisms, the results in Figures 5, 6 strongly suggest that slow kinetics, rather than some other property of these mutants, reduces the polarity of transcription at many promoters.
Figure 5. Elevated divergent antisense transcription in a slow pol II funnel domain mutant.

A. Anti-pol II total Nascent Elongating Transcript sequencing (anti-pol II tNET-seq). Isolated nuclei are digested with DNAse I, lysed and used to immunoprecipitate RNA pol II (blue circles) along with nascent transcripts (red). Precipitated RNA was processed into random primed strand-specific libraries (see STAR Methods).
B, C. Integrated genomics viewer (IGV) screen shots of anti Ser2-P ChIP-seq and sense and antisense anti-pol II tNET-seq reads. Note that high 5′ CTD-Ser2-P in R749H (blue arrows) is associated with increased divergent antisense transcription (red arrows). Sense and antisense tNET-seq tracks are scaled equivalently for WT and R749H samples to accurately reflect changes in the amount of divergent versus sense transcription. Read numbers are not normalized, and differ between experiments.
D–E. Metaplots of mean anti-pol II tNET-seq sense and antisense reads/75bp bin from 3 replicate experiments in WT and R749H slow mutant in over 5000 well expressed genes separated by 2 kb (D) and in a subset of genes (Table S1) with elevated 5′ Ser2-P (E). Note increased divergent antisense transcription in R749H (arrows), which is more pronounced among genes with elevated 5′ Ser2-P.
F. Metaplots of mean anti-pol II tNET-seq antisense reads/200bp bin (n=3) at genes with 5′ Ser2 hyperphosphorylation in the R749H slow mutant (Table S1). Note that high level antisense transcription in the slow mutant extends many kilobases.
G. Metaplots as in D of H3K4me2 ChIP signals in WT and the R749H slow mutant. Note that slow transcription is associated with a shift in H3K4me2 upstream and downstream of the TSS (red arrows). Similar results were observed for genes with elevated DI in R749H. See Fig. S2D–G.
Figure 6. Elevated divergent antisense transcription in a slow pol II trigger loop mutant.

A, B. Metaplots of mean anti-pol II tNET-seq sense and antisense reads from WT (n=3) and H1108Y (n=2) slow mutant at over 4000 well expressed genes separated by 2 kb, and genes with 5′ Ser2 hyperphosphorylation (Table S1) as in Fig. 5D. Note increased divergent antisense transcription (red arrows) in H1108Y.
C–E. IGV screen shots of anti-pol II tNET-seq sense and antisense reads in WT and R749H and H1108Y slow mutants and in Fig. 5B. Note elevated antisense transcription relative to WT in both slow mutants (arrows).
We asked whether H3K4me2, a chromatin mark enriched on nucleosomes upstream of divergently transcribed promoters (Duttke et al., 2015), was affected by the R749H slow pol II mutant. Anti-H3K4me2 ChIP-seq revealed a global repositioning of this modification in the slow mutant relative to WT. Most notably, H3K4me2 was elevated at positions upstream of the TSS in the R749H mutant among well-expressed genes (Fig. 5G, S2E–G) and those with elevated DI in the R749H mutant (Fig. S2D). H3K4me2 was also reduced in the slow mutant at nucleosomes immediately downstream of start sites (Fig. 5G, S2D–G). In summary, the increased DI of transcription by the slow R749H mutant is associated with elevated H3K4me2 in a region that extends further upstream of the TSS than in WT cells. These observations show that, in contrast to H3K4 trimethylation, which is relatively insensitive to pol II dynamics (Fig. S2B, C), K4 dimethylation is elevated upstream of many start sites in a slow pol II mutant. The upstream repositioning of H3K4me2 is consistent with the previously reported correlation between this chromatin mark and divergent antisense transcription (Duttke et al., 2015).
We asked whether the effects of elongation rate on transcriptional polarity and CTD Ser2 phosphorylation are correlated. Metaplots of nascent RNA-seq reads for a subset of several hundred genes with high Ser2-P/total pol II at 5′ ends in the R749H mutant showed particularly high levels of antisense transcription in both slow mutants relative to WT (compare Figs. 5E, 6B with Fig. 5D, 6A). The H1108Y trigger loop mutant, which has a more severe effect on transcription activity, (Fong et al., 2014; Kaplan et al., 2008) also has a more pronounced effect on transcriptional polarity than the R749H funnel domain mutant. We examined transcriptional polarity at genes separated by >5kb that have elevated Ser2-P at their 5′ ends in the R749H mutant (selected from Table S1) and found a strong bias toward increased DI in the mutant relative to WT (Fig. 7A). Furthermore the genes with increased DI in the slow mutant are strongly enriched (P=2×10−63, ChiSq-test) among those with 5′ Ser2 hyperphosphorylation relative to all genes (Fig. 7B). CTD Ser2 hyperphosphorylation at 5′ ends in the slow R749H mutant therefore strongly correlates with reduced transcriptional polarity at divergent promoters. Consistent with this correlation, the two independently identified gene sets with high 5′ Ser2-P and high DI in R749H, have remarkably similar functional profiles as determined by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005). The top four functional categories enriched for both gene groups included: 1: “Regulated by KLF1”, 2: “Has Sp1 transcription factor binding site (G/C rich)” and 3: “Co-regulated by BRCA1” (Table S1). These gene groups have in common that their promoters are enriched for proximity to CpG islands (P-values for gene groups 1–3: 4.84454E-21; 5.26704E-77; 7.42022E-31, respectively Chisq). Interestingly proximity to CpG islands correlates with bidirectional transcription, and the binding of Klf/Sp family transcription factors correlates with directional chromatin opening (Core et al., 2008; Sherwood et al., 2014).
Figure 7. Divergent antisense transcription correlated with high 5′ CTD Ser2 phosphorylation.

A. Scatter plot comparing the divergence index (DI, see STAR Methods) between WT and R749H. Genes analyzed are separated by >5kb upstream and downstream and have elevated Ser2-P at their 5′ ends in R749H (Table S1) and those with altered values (n=3, FDR <.05, t-test) are shown as red and blue points. Note the majority of genes with increased 5′ Ser2-P show increased DI in the slow mutant (red dots).
B. Genes with high 5′ Ser2-P in the R749H slow mutant (n=344) are enriched for elevated (2X increase, FDR<.05, n=3) divergence index (DI) relative to all genes scored in the DI analysis (n= 2940, see STAR Methods). P-value calculated by ChiSq.
C. IGV screen shots of anti-Ser2-P tNET-seq sense and antisense nascent RNA reads as in Fig. 5B. Note increased divergent antisense transcription by Ser2 phosphorylated pol II in the R749H slow mutant relative to WT (arrows).
D, E. Metaplots of mean coverage of anti-Ser2-P tNET-seq sense and antisense reads as in Fig. 5D for a group of highly expressed genes separated by 2kb (C) and those with 5′ Ser2 hyperphosphorylation in R749H (Table S1). Note elevated levels of promoter proximal sense transcription (blue arrows) and divergent antisense transcription (red arrows) by Ser2 phosphorylated pol II in the slow R749H mutant.
F. Model showing WT and slow mutant pol II at the transcription start site. The slow mutant has increased dwell time (stopwatch), CTD Ser2 phosphorylation and divergent antisense transcription relative to WT.
To test directly whether divergently transcribing pol II in a slow mutant is actually Ser2 phosphorylated, we conducted tNET-seq with anti-CTD Ser2-P antibody. As expected for anti-Ser2-P tNET-seq was enriched for reads over introns, 3′ flanking regions and enhancers (Fig. S5C), and shows greater relative signals at 3′ ends of long genes than anti-total pol II tNET-seq (Fig. 7C, S5D). This experiment revealed much higher levels of antisense nascent transcripts associated with Ser2-P pol II in the R749H slow mutant than WT (red arrows Fig. 7D, E, S8C), showing that divergently transcribing transcription complexes are indeed Ser2 phosphorylated. The enrichment of Ser2-P on divergently transcribing slow mutant pol II is further confirmed by the elevated ratio of antisense Ser2-P/total pol II tNET-seq signal relative to WT (Fig. S8D). Promoter-proximal anti-Ser2-P tNET-seq signal in the sense direction was also higher in the slow mutant relative to WT (Fig. 7D, E blue arrows). The latter result is consistent with anti-CTD Ser2-P ChIP (Fig. 3) and suggests that Ser2 hyperphosphorylated pol II at 5′ ends is active in both sense and antisense transcription.
Discussion
In this report we investigated how conserved 5′ to 3′ profiles of pol II CTD and histone PTMs are established, by asking how they are affected by transcription elongation rate. We postulated that if the dwell time spent by pol II and associated modification factors in particular regions of a gene is important, then elongation rate would influence how 5′ to 3′ profiles of PTMs take shape (Fig. 1). On the other hand, if position within the transcription unit were the primary determinant of how the profile of a PTM is established, then elongation rate would have little effect. In support of the “dwell time model”, our results revealed genome-wide effects of slow elongation on 5′ to 3′ profiles of specific histone and CTD modifications. The salient results are:
H3K36me3, a co-transcriptionally deposited chromatin mark associated with the elongation phase of the transcription cycle, is repositioned toward 5′ ends (Fig. 2) when transcription is slow.
Slow pol II has a prolonged dwell time at human promoters and caused CTD Ser2-P to be re-positioned toward 5′ ends of genes in human and yeast (Fig. 3, 4, S3). Moreover novel peaks of Ser2 phosphorylation appeared at the 5′ ends of many human genes.
Ser2 hyperphosphorylated slow pol II is correlated with reduced transcriptional polarity at bidirectional promoters. Slow mutants displayed strongly enhanced, divergent antisense transcription that extended far upstream of genes and slightly reduced sense transcription than WT pol II (Fig. 5–7, S7).
H3K36 trimethylation is catalyzed by SETD2 that associates with Ser2 phosphorylated pol II (Kizer et al., 2005; Sun et al., 2005; Yoh et al., 2008) and our results show that this process is kinetically coupled to transcription elongation. Slow transcription widely altered H3K36me3 profiles along genes by shifting the position of this modification in a promoter-proximal direction and this shift correlated with 5′ Ser2 hyperphosphorylation (Fig. 2, S1). These results are therefore consistent with a mechanism where remodeling of the H3K36me3 profile when transcription is slow is caused by longer pol II dwell time and Ser2 hyperphosphorylation at 5′ ends, both of which could promote methylation by pol II-associated SETD2. Note however that there is not a precise coincidence between novel peaks of Ser2-P at the TSS in the slow mutant and peaks of H3K36me3. It was previously reported that genes with slower elongation rates have more promoter proximally localized H3K36me3 in mouse ESCs (Jonkers et al., 2014). Our results strongly suggest that this correlation is the result of a kinetic coupling mechanism where pol II dwell time within genes helps determine how H3K36me3 is deposited. Kinetic coupling is not a universal property of co-transcriptionally deposited chromatin modifications however, since H3K4me3 is relatively unaffected by slow elongation (Fig. S2B, C).
Pol II CTD phosphorylation on Ser2 residues is a highly conserved property of pol II at 3′ ends of genes. Two slow mutants of human pol II in the trigger loop and funnel domain (R749H and H1108Y) caused widespread repositioning of CTD Ser2-P toward 5′ ends of genes (Fig. 3, S3, Table S1). The effect of these mutants on CTD phosphorylation is likely caused by slow kinetics rather than some other phenotype as they lie in distinct domains of the large subunit and inhibit elongation by different mechanisms. Moreover, the slow pol II R749H mutant persists at start sites for longer than WT pol II (Fig. 3C). These results are therefore consistent with the model that Ser2 hyperphosphorylation at start sites results from a longer window of opportunity for deposition of this modification by Cdk9 and/or Cdk12. On the other hand Tyr1, Ser5, or Ser7 phosphorylations are relatively insensitive to slow kinetics (Fig. S4), presumably because pol II dwell time is not limiting for these modification reactions. Pol II with high Ser2-P at start sites may constitute a novel isoform that also has high Ser5-P and other phosphorylations. Interestingly, premature Ser2 phosphorylation near 5′ ends was previously reported in cells knocked down of the pol II-associated FUS protein (Schwartz et al., 2012). It is possible that this effect of FUS depletion also results from prolonged pol II dwell time at promoter regions. Remarkably, slow pol II mutants in budding yeast also shifted the profile of Ser2-P toward 5′ ends (Fig. 4). The conservation of elongation rate effects on Ser2-P profiles over a wide evolutionary distance implies that kinetic coupling of Ser2 phosphorylation with transcription is of fundamental functional importance.
It is not known whether the sensitivity of specific PTMs to transcription elongation rate has functional significance. PTM profile shifting could be a homeostatic mechanism that compensates for the effects of slow elongation. For example, flexible Ser2-P and H3K36me3 deposition might help to buffer the effects of variations in elongation rate on co-transcriptional mRNA processing. This idea is consistent with the modulation of mRNA 3′ end formation and splicing by CTD Ser2-P and H3K36me3 (Ahn et al., 2004; Schwartz et al., 2012), (Luco et al., 2011; Luco et al., 2010). We speculate that natural variation in elongation rates, which range from < 0.5kb/min to >5kb/min, (Fuchs et al., 2014; Jonkers et al., 2014) is sufficient to cause changes in co-transcriptional deposition of PTM’s on the CTD and histones by virtue of its effects on dwell time of the TEC.
Our experiments uncovered an unexpected functional link between Ser2 hyperphosphorylation at 5′ ends and transcriptional polarity. Mammalian promoters frequently give rise to transcription in both the sense and divergent antisense directions. We found that pol II kinetics can have a major impact on the balance between sense and divergent antisense transcription. Genes with high levels of Ser2-P at their 5′ ends in slow pol II mutants are strongly enriched for a high divergence index associated with elevated antisense transcription and modestly reduced sense transcription as measured by nascent RNA sequencing (Fig. 5E, F, 6B). Divergent transcription from promoters by slow pol II also extends further than WT; often persisting for more than 20kb (Fig. 5F, S8A, C). This result suggests that the slow mutant is relatively resistant to early termination that is typical of these transcription units (Almada et al., 2013; Ntini et al., 2013). The apparent insensitivity to termination signals of slow pol II initiating at divergent antisense promoters does not apply at conventional mRNA and snRNA genes, however (Fong et al., 2015) (Fig. S7D). Increased pol II dwell time at promoters and/or increased divergent antisense transcription in the slow mutant might cause the repositioning of H3K4 dimethylation by pol II-associated K4 methyltransferases, resulting in elevated levels of this mark upstream, and reduced levels downstream, of start sites (Fig. 5G, S2D–G).
How might slow pol II dynamics alter transcriptional polarity? We speculate that longer dwell time at the TSS might favor successful initiation events in the antisense direction that normally fail at an early step perhaps due to abortive initiation. There is precedent for pol II activity mutants affecting transcription initiation in yeast where slow and fast mutants in the trigger loop alter start site selection (Kaplan et al., 2012). It is also possible that enhanced H3K4 dimethylation of upstream nucleosomes by Set1 complexes associated with slow pol II (Fig. 5G) might facilitate divergent transcription. Another possibility is that slow transcription dynamics and/or Ser2 hyperphosphorylation make divergently transcribing polymerases more resistant to mechanisms that normally terminate anti-sense transcription before it extends very far. For example if slow transcription by Ser2 hyperphosphorylated pol II favored recruitment of U1 snRNP upstream of genes, then polyA site recognition and coupled termination of divergent transcription is expected to be inhibited (Almada et al., 2013; Berg et al., 2012; Ntini et al., 2013). In summary, our results show that pol II kinetics, and hence dwell time, is an important determinant of how specific CTD and histone modifications are established and how the decision between sense and antisense transcription is made at bidirectional promoters.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, David Bentley( david.bentley@ucdenver.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HUMAN CELL LINES
Flp-In-293 TREX cells (Female, Invitrogen) expressing inducible α-amanitin resistant WT and mutant Rpb1 with slow (R749H, H1108Y) and fast (E1126G) elongation rates have been described (Fong et al., 2014). The cells were maintained in DMEM media supplemented with 10% FBS, 200μg/ml hygromycin B, 6.5μg/ml blasticidin, and pen/strep. All experiments were performed after induction with 2.0 μg/ml doxycycline for 12–16 hr. and treatment with α-amanitin (2.5 μg/ml) for a further 42hr at which time all cell lines were viable and endogenous pol II is inactive.
YEAST STRAINS
Isogenic WT and rpb2-10 strains, DY103 and DY105 (Lennon et al., 1998), and WT and rpb1-N488D, GRY3031 and GRY3038 (Malagon et al., 2006) strains were kind gifts of D. Reines and J. Strathearn.
METHOD DETAILS
Pol II dwell time
HEK 293 Flp-in Rpb1 WTAmr and R749H Amr mutant cells were induced with doxycycline and treated with α-amanitin as described above then triptolide (10μM) or an equal volume of DMSO for 0–80 min. before cross-linking with formaldehyde and anti-pol II ChIP. Q-PCR ChIP signals from 2 replicate experiments for the GAPDH, FUS, MAT2B and RETSAT promoter regions were normalized to the tRNA MET gene. The latter was used as internal control because it has a prominent peak of pol II occupancy like some other pol III genes (Raha et al., 2010) that is resistant to triptolide over the time course of these experiments. Primer sequences are in Table S3.
ChIP-seq
ChIP of yeast and human extracts has been described (Kim et al., 2011; Schroeder et al., 2000). Enzymatic steps and size fractionation of libraries were done on AMPure XP SPRI beads (Beckman Coulter Genomics) and sequenced on the Illumina Hi-Seq platforms. Reads were mapped to the hg19 UCSC human genome (Feb. 2009) with Bowtie version 0.12.5 (Langmead et al., 2009)(Table S2). We generated bed and wig profiles using 50bp bins and 200bp windows assuming a 180bp fragment size shifting effect. Results were viewed with the UCSC genome browser and meta plots were generated using R. Except where noted metaplots show relative frequency of mean read counts per bin divided by the total number of aligned reads in all bins. The y-axis of these plots represents the proportion of counts contained in each bin and the area under each curve is equal. We used 100 base bins in the 5′ region from −1.5kb to +0.5kb (−0.5kb to +0.5kb in yeast) relative to the TSS, and 3′ region −0.5kb to +3.5kb (−0.5kb to +0.5kb in yeast) relative to the poly (A) site. Gene body regions between +500 relative to the TSS and −500 relative to the poly (A) site were divided into 20 variable length bins. Gene body regions between +500 relative to the TSS and −500 relative to the poly (A) site were divided into 20 variable length bins. Metaplots of human genes are from a list of 5507 well-expressed genes separated from their neighbors by >2kb (Brannan et al., 2012). Metaplots include all genes in common between the data sets for which a minimum ChIP signal (at least one read in each of the 5′, gene body and 3′ regions) was obtained. In Figure 2A Welch’s two sample t-test was used to generate p-values comparing each bin for the indicated datasets. The p-value for each bin was then plotted in R. Similar results were obtained using the Wilcoxon rank sum test (not shown). Yeast ORF gene coordinates are from (Nagalakshmi et al., 2008).
Ser2 hyperphosphorylated were identified by counting normalized reads in the regions −300 to +500 in Ser2 ChIP-seq/total pol II in two replicates using paired t-test. FDR values were calculated using Benjamini-Hochberg procedure. Those with FDR < .05 and >1.5 fold change are shown in Table S1.
Anti-Pol II/anti Ser2-P total NetSeq - tNetseq
HEK 293 cells expressing WT or mutant RNA pol II were lysed in 300 mM sucrose, 10mM HEPES pH 7.3, 85mM KCl, 0.1 mM EDTA, 0.5% NP40, 0.5mM DTT. Nuclei (~5×106 per IP) were treated with DNase I (10–20 U) for 1hr at 4°C in 200μl 10 mM Tris pH 7.5, 10 mM CaCl2, 10 mM MnCl2, 1 μl RNAseOut. Nuclei were lysed by addition of an equal volume of 2X RIPA buffer and the resultant lysate was diluted 10X with IP buffer (20 mM Tris pH7.5, 100 mM KCl, 0.2 mM EDTA, 1% NP40, 1.0mM DTT, 20 μg/ml heparin, 10mM CaCl2, 10mM MnCl2). The diluted lysate was immunoprecipitated with rabbit anti-pan pol II CTD serum or affinity purified rabbit anti-CTD Ser2-P pre-bound to a 1:1 mix of magnetic protein A and protein G beads (Dynabeads) in the presence of 2U DNAse I and 1 μl RNAseOut. Beads were washed 3X in IP buffer, 1X IP buffer+200mM KCl and then 2X in IP buffer. RNA purified with Trizol Reagent from a total of ~1.5 × 107 cells was pooled and used as input in the KAPA RNA-seq kit (#07962169001) to create stand-specific random-primed libraries. Libraries were sequenced on the Illumina Hi-seq platform and resultant reads were mapped using Hisat2 (Kim et al., 2015) to the hg19 UCSC human genome. Aligned reads were viewed on the Integrated Genome Viewer (IGV) in either bam or bedgraph format. A custom python program was employed for metagene analysis of tNET-seq data. For different defined promoter regions, genes were aligned to the TSS and total coverage was calculated per gene using bedtools. Mean coverage is reported per bin and further normalized to total number of reads mapped to REFseq genes +/− 3kb of flanking sequence. Metaplots show well-expressed genes that are separated by >5kB from other genes (coding and non-coding).
Divergence Index, DI, was calculated as a measure of the polarity of transcription at bidirectional promoters from tNET-seq reads. DI = (divergent antisense reads (up to −5kB from TSS)/sense reads (TSS to TTS) *100. Genes selected for analysis of DI are separated from their neighbors by >5kb upstream and have at least 10 divergent antisense tNET-seq reads in region to −5kb and a mean coverage of 5 reads per base pair in the region from the TSS to the polyA site. Significant differences in DI were determined by t-test and FDRs were calculated using the Benjamini-Hochberg procedure.
Q RT-PCR of nascent transcripts
Quantitative RT-PCR was performed on two independent biological replicates of wild type and R749H nascent RNA purified by anti-pol II IP. Random primed cDNA was synthesized with SuperScript IV (Invitrogen) and amplified using primers (shown in Table S3) within 5kb upstream of the TSS in the indicated genes (Fig. S7C). Signal was normalized to the signal upstream of two control genes (MCRS1 and ACAD9) that showed no change in divergent antisense expression between wild type and R749H. The fold change in signal between wild type and R749H was calculated using the delta/delta Ct method by normalizing the signal to each control gene separately and then taking the average fold change calculated from each control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details including the number of replicates (n) are provided in Figure legends. Significance was defined by P values determined by using t-test, and ChiSq test and FDR values calculated using Benjamini-Hochberg procedure.
DATA AND SOFTWARE AVAILABILITY
The CHIP-Seq and tNET-seq datasets have been deposited in the GEO under GSE97827.
Supplementary Material
Acknowledgments
We thank T. Blumenthal, R. Davis, C. Kaplan and members of our lab for helpful discussions; B. Erickson for the metaplot analysis tool, D. Reines and J. Strathearn for yeast strains and D. Eick for antibody. We thank K. Diener, B. Gao and the UC Denver Sequencing facility for sequencing. This work was supported by NIH grants GM58613 and R35GM118051 to D.B. and by an American Cancer Society fellowship PF-15-188-01-RMC to T.S.
Footnotes
Author Contributions
N.F. T.S and D.B conceived the experiments. N.F. and T.S. performed all experiments. N.F., T.S., M.C. and R.S. performed informatics analysis. D. B. and T.S. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aebi M, Weissman C. Precision and orderliness in splicing. Trends in Genetics. 1987;3:102–107. [Google Scholar]
- Ahn SH, Kim M, Buratowski S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell. 2004;13:67–76. doi: 10.1016/s1097-2765(03)00492-1. [DOI] [PubMed] [Google Scholar]
- Almada AE, Wu X, Kriz AJ, Burge CB, Sharp PA. Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature. 2013;499:360–363. doi: 10.1038/nature12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15:163–175. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Kaida D, Zhang Z, Wan L, Dreyfuss G. U1 snRNP Determines mRNA Length and Regulates Isoform Expression. Cell. 2012;150:53–64. doi: 10.1016/j.cell.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol Cell. 2012;46:311–324. doi: 10.1016/j.molcel.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S. Progression through the RNA polymerase II CTD cycle. Mol Cell. 2009;36:541–546. doi: 10.1016/j.molcel.2009.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- Chen F, Gao X, Shilatifard A. Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev. 2015;29:39–47. doi: 10.1101/gad.246173.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Pai AA, Herudek J, Lubas M, Meola N, Järvelin AI, Andersson R, Pelechano V, Steinmetz LM, Jensen TH, et al. Principles for RNA metabolism and alternative transcription initiation within closely spaced promoters. Nat Genet. 2016;48:984–994. doi: 10.1038/ng.3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, Levezinho H, Gut M, Eick D, Gut I, et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol. 2011;18:977–983. doi: 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- Duttke SH, Lacadie SA, Ibrahim MM, Glass CK, Corcoran DL, Benner C, Heinz S, Kadonaga JT, Ohler U. Human promoters are intrinsically directional. Mol Cell. 2015;57:674–684. doi: 10.1016/j.molcel.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrensberger AH, Kelly GP, Svejstrup JQ. Mechanistic Interpretation of Promoter-Proximal Peaks and RNAPII Density Maps. Cell. 2013;154:713–715. doi: 10.1016/j.cell.2013.07.032. [DOI] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, Bentley DL. Effects of Transcription Elongation Rate and Xrn2 Exonuclease Activity on RNA Polymerase II Termination Suggest Widespread Kinetic Competition. Molecular Cell. 2015;60:256–267. doi: 10.1016/j.molcel.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, Bentley DL. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 2014;28:2663–2676. doi: 10.1101/gad.252106.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs G, Voichek Y, Benjamin S, Gilad S, Amit I, Oren M. 4sUDRB-seq: measuring genomewide transcriptional elongation rates and initiation frequencies within cells. Genome Biol. 2014;15:R69. doi: 10.1186/gb-2014-15-5-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Kim S, Espinosa J, Bentley DL. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol. 2008;15:71–78. doi: 10.1038/nsmb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidemann M, Hintermair C, Voss K, Eick D. Dynamic phosphorylation patterns of RNA polymerase II CTD during transcription. Biochim Biophys Acta. 2013;1829:55–62. doi: 10.1016/j.bbagrm.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, Adelman K. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell. 2013;52:517–528. doi: 10.1016/j.molcel.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimeno-González S, Schmid M, Malagon F, Haaning LL, Jensen TH. Rat1p maintains RNA polymerase II CTD phosphorylation balance. RNA. 2014;20:551–558. doi: 10.1261/rna.041129.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, Lis JT. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife. 2014;3:e02407. doi: 10.7554/eLife.02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Jin H, Zhang IL, Belyanin A. Dissection of Pol II trigger loop function and Pol II activity-dependent control of start site selection in vivo. PLoS Genetics. 2012;8:e1002627. doi: 10.1371/journal.pgen.1002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD, Larsson KM, Kornberg RD. The RNA polymerase II trigger loop functions in substrate selection and is directly targeted by alpha-amanitin. Molecular Cell. 2008;30:547–556. doi: 10.1016/j.molcel.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A. 2011;108:13564–13569. doi: 10.1073/pnas.1109475108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon JC, 3rd, Wind M, Saunders L, Hock MB, Reines D. Mutations in RNA polymerase II and elongation factor SII severely reduce mRNA levels in Saccharomyces cerevisiae. Mol Cell Biol. 1998;18:5771–5779. doi: 10.1128/mcb.18.10.5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Phatnani HP, Guan Z, Sage H, Greenleaf AL, Zhou P. Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc Natl Acad Sci U S A. 2005;102:17636–17641. doi: 10.1073/pnas.0506350102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Alló M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010;327:996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagon F, Kireeva ML, Shafer BK, Lubkowska L, Kashlev M, Strathern JN. Mutations in the Saccharomyces cerevisiae RPB1 gene conferring hypersensitivity to 6-azauracil. Genetics. 2006;172:2201–2209. doi: 10.1534/genetics.105.052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt S, Escalante-Chong R, Pho N, Wang J, Churchman LS, Springer M, Buratowski S. A Chromatin-Based Mechanism for Limiting Divergent Noncoding Transcription. Cell. 2014;157:1712–1723. doi: 10.1016/j.cell.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, Churchman LS. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell. 2015;161:541–554. doi: 10.1016/j.cell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science. 2012;336:1723–1725. doi: 10.1126/science.1219651. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso AR, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell. 2015;161:526–540. doi: 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntini E, Järvelin AI, Bornholdt J, Chen Y, Boyd M, Jørgensen M, Andersson R, Hoof I, Schein A, Andersen PR, et al. Polyadenylation site–induced decay of upstream transcripts enforces promoter directionality. Nat Struct Mol Biol. 2013;20:923–928. doi: 10.1038/nsmb.2640. [DOI] [PubMed] [Google Scholar]
- Powell W, Reines D. Mutations in the 2nd largest subunit of rna-polymerase-ii cause 6-azauracil sensitivity in yeast and increased transcriptional arrest in-vitro. Journal Of Biological Chemistry. 1996;271:6866–6873. doi: 10.1074/jbc.271.12.6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Almvig K, Christensen MS, Valen E, Mapendano CK, Sandelin A, Jensen TH. PROMoter uPstream Transcripts share characteristics with mRNAs and are produced upstream of all three major types of mammalian promoters. Nucleic Acids Res. doi: 10.1093/nar/gkr370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- Raha D, Wang Z, Moqtaderi Z, Wu L, Zhong G, Gerstein M, Struhl K, Snyder M. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc Natl Acad Sci U S A. 2010;107:3639–3644. doi: 10.1073/pnas.0911315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, Kellis M, Gymrek M, Issner R, Coyne M, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scafe C, Martin C, Nonet M, Podos S, Okamura S, Young RA. Conditional mutations occur predominantly in highly conserved residues of RNA polymerase II subunits. Mol Cell Biol. 1990;10:1270–1275. doi: 10.1128/mcb.10.3.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Schroeder SC, Schwer B, Shuman S, Bentley D. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 2000;14:2435–2440. doi: 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JC, Ebmeier CC, Podell ER, Heimiller J, Taatjes DJ, Cech TR. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012;26:2690–2695. doi: 10.1101/gad.204602.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scruggs BS, Gilchrist DA, Nechaev S, Muse GW, Burkholder A, Fargo DC, Adelman K. Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Molecular Cell. 2015:1–13. doi: 10.1016/j.molcel.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Hashimoto T, O’Donnell CW, Lewis S, Barkal AA, van Hoff JP, Karun V, Jaakkola T, Gifford DK. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat Biotechnol. 2014;32:171–178. doi: 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XJ, Wei J, Wu XY, Hu M, Wang L, Wang HH, Zhang QH, Chen SJ, Huang QH, Chen Z. Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J Biol Chem. 2005;280:35261–35271. doi: 10.1074/jbc.M504012200. [DOI] [PubMed] [Google Scholar]
- Teves SS, Weber CM, Henikoff S. Transcribing through the nucleosome. Trends Biochem Sci. 2014;39:577–586. doi: 10.1016/j.tibs.2014.10.004. [DOI] [PubMed] [Google Scholar]
- Titov DV, Gilman B, He QL, Bhat S, Low WK, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, et al. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nature chemical biology. 2011;7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojnic E, Simon B, Strahl BD, Sattler M, Cramer P. Structure and CTD binding of the Set2 SRI domain that couples histone H3 K36 methylation to transcription. J Biol Chem. 2006;281:13–15. doi: 10.1074/jbc.C500423200. [DOI] [PubMed] [Google Scholar]
- Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Shibata Y, Rao B, Laribee RN, O’Rourke R, Buck MJ, Greenblatt JF, Krogan NJ, Lieb JD, Strahl BD. The RNA Polymerase II Kinase Ctk1 Regulates Positioning of a 5′ Histone Methylation Boundary along Genes. Mol Cell Biol. 2006;27:721–731. doi: 10.1128/MCB.01628-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoh SM, Lucas JS, Jones KA. The Iws1:Spt6:CTD complex controls cotranscriptional mRNA biosynthesis and HYPB/Setd2-mediated histone H3K36 methylation. Genes Dev. 2008;22:3422–3434. doi: 10.1101/gad.1720008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Schroeder S, Fong N, Bentley DL. Altered nucleosome occupancy and histone H3K4 methylation in response to ‘transcriptional stress’. EMBO J. 2005;24:2379–2390. doi: 10.1038/sj.emboj.7600711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.