

Abstract
Rationale
Superficial erosion currently causes up to a third of acute coronary syndromes (ACS), yet we lack understanding of its mechanisms. Thrombi due to superficial intimal erosion characteristically complicate matrix-rich atheromata in regions of flow perturbation.
Objective
This study tested in vivo the involvement of disturbed flow, and of neutrophils, hyaluronan, and TLR2 ligation in superficial intimal injury, a process implicated in superficial erosion.
Methods and Results
: In mouse carotid arteries with established intimal lesions tailored to resemble the substrate of human eroded plaques, acute flow perturbation promoted downstream endothelial cell (EC) activation, neutrophil accumulation, EC death and desquamation, and mural thrombosis. Neutrophil loss-of-function limited these findings. TLR2 agonism activated luminal ECs, and deficiency of this innate immune receptor decreased intimal neutrophil adherence in regions of local flow disturbance, reducing EC injury and local thrombosis (p<0.05).
Conclusions
These results implicate flow disturbance, neutrophils, and TLR2 signaling as mechanisms that contribute to superficial erosion, a cause of ACS of likely growing importance in the statin era.
Keywords: Superficial erosion, Endothelium, Neutrophils, Disturbed Flow, Acute coronary syndromes
Subject Terms: Vascular Biology, Cardiovascular Disease, Atherosclerosis, Thrombosis
INTRODUCTION
Rupture of an atheromatous plaque with a thin fibrous cap has received much attention as a cause of ACS1, 2. Abundant human and experimental findings indicate that lipid-lowering, in particular statin treatment, mitigates this mechanism of plaque disruption3. Current clinical data show a shift in the characteristics of plaques associated with rupture (lipid and macrophage rich) concomitant with increased statin use, smoking cessation, and other reductions in risk factors. Human atheromata today contain less lipid and fewer macrophages than just a decade ago4, 5. But even with the best current medical and interventional therapy, the residual burden of recurrent events post-ACS remains unacceptable6. Indeed, ruptured thin-capped atheromata may now cause fewer ACS7 and superficial erosion appears on the rise. Yet, mechanisms involved in superficial erosion have received scant attention and remain a major knowledge gap.
Thrombi associated with superficial erosion generally overlie fibrous rather than lipid-rich plaques. In contrast to plaques with ruptured fibrous caps, eroded plaques contain few macrophages but abundant smooth muscle cells (SMCs). In stark contrast with collagen-poor ruptured plaques, eroded atheromata characteristically contain abundant type III collagen8, 9, glycosaminoglycans, and proteoglycans10. Eroded plaques may localize preferentially in regions of low shear stress, exhibit impaired endothelial anti-thrombotic and atheroprotective functions11–13, and loss of EC14, 15. Markers of EC apoptosis increase downstream of obstructive atherosclerotic plaques in humans, sites of disturbed flow16. Oscillatory wall shear stress (WSS) favors EC death17–19 in stenotic arteries with expanded intimas in rabbits, promoting thrombosis20. EC apoptosis activates thrombin and platelet adhesion in vitro21, 22 as well as local EC desquamation and thrombosis in vivo23.
Zones of flow perturbation have high EC Toll-like receptor 2 (TLR2) expression in mouse and human atheromata24, 25, and loss of TLR2 function limits murine atherogenesis26. Hyaluronan (HA), a glycosaminoglycan prominent in human eroded plaques can ligate TLR210, 27. We and others recently provided in vitro data supporting the involvement of TLR2 and HA in EC activation28 associated with the release of the neutrophil chemoattractant IL-8 and augmented leukocyte adhesion molecules (e.g. VCAM-1 and E-Selectin). TLR2 expression further correlates with the number of apoptotic luminal ECs in human plaques with characteristics of superficial erosion and with neutrophils and neutrophil extracellular traps (NETs)28. Patients with ACS due to superficial erosion (vs. rupture) have higher concentrations of circulating myeloperoxidase29, a neutrophil enzyme linked with EC death and tissue factor expression30. Interaction with neutrophils alters many functions of cultured EC28, 31.
Thus, flow perturbation may comprise a “first hit” leading to chronic endothelial activation, propensity to slough, and impaired ability to repair small intimal breaches, setting the stage for an enhanced response to a “second hit” such as local recruitment of neutrophils, able to augment EC apoptosis and propagate desquamation28. Such unrepaired rents in the endothelial monolayer could then trigger platelet entrapment, activation, and thrombin generation, fostering clot formation. These hypotheses, derived from in vitro observations, require exploration in vivo, as cell culture experiments cannot fully mimic either the flow conditions or the abnormal intimal substrate associated with human superficial erosion.
METHODS
See online methods.
RESULTS
Thrombi due to superficial erosion typically complicate plaques rich in hyaluronan, collagen, and SMC, but with scant macrophages, often located at sites of human coronary arteries surrounding stenoses, regions of disturbed flow. We sought clues regarding the mechanisms which might participate in endothelial loss in humans through study of atheromata harvested at carotid artery surgery. We used morphologic criteria to classify different categories of plaques from specimens in our collection with well-preserved intimal surfaces (Figure 1). A group of plaques with a fibrous SMC-rich and macrophage-poor appearance was further subdivided into those with an “erosion-prone” morphology (high apoptotic EC content, n=7) or a “stable fibrous” morphology (low apoptotic EC content, n=10). A third group of fibrous plaques harbored a non-obstructive mural thrombus (“eroded” morphology, n=8) (Online Figure IA). We compared these fibrous lesions to a group of atheromata with features associated fibrous cap rupture: few SMC, many macrophages, and large lipid cores, (n=11). Fibrous plaques with many apoptotic EC or those with mural thrombi displayed much greater accumulation of neutrophils near the luminal endothelium than those with few apoptotic EC (“stable fibrous”) or the thin-capped lipid- and macrophage-laden lesions (Figure 1A through D). Mural thrombi contained abundant neutrophils (CD66b+ and neutrophil elastase [NE]+, Figure 1C and D) as previously described32. In the fibrous plaques, regions of high neutrophil content co-localized with disruption of the EC monolayer (Figure 1B and C). In contrast, thin-capped atheromatous lesions generally had an intact endothelial lining, and contained few if any neutrophils. Inspection of the deeper portions of plaques served as control for neutrophil content (Figure 1E). Further studies localized neutrophils in representative human atheromata with characteristics of superficial erosion. Double staining for MPO and CD61 showed neutrophils (MPO+) adjacent to but separate from platelets (CD61+). MPO/NE staining colocalized MPO with NE (Online Figure IB). Thus, neutrophil accumulation appears much more abundant in human lesions with erosion-prone characteristics than in those with features associated with fibrous cap rupture. Immunofluorescent staining showed a preferential accumulation of TUNEL+ cells in the intima, often co-localizing with CD31+ ECs, in erosion-prone plaques as compared to rupture-prone plaques (Figure 1F and Online Figure IC). Apoptotic ECs localized preferentially in the vicinity of NE+ neutrophils (Online Figure IC).
Figure 1. Neutrophils associate with the erosion-prone plaque morphology in humans.
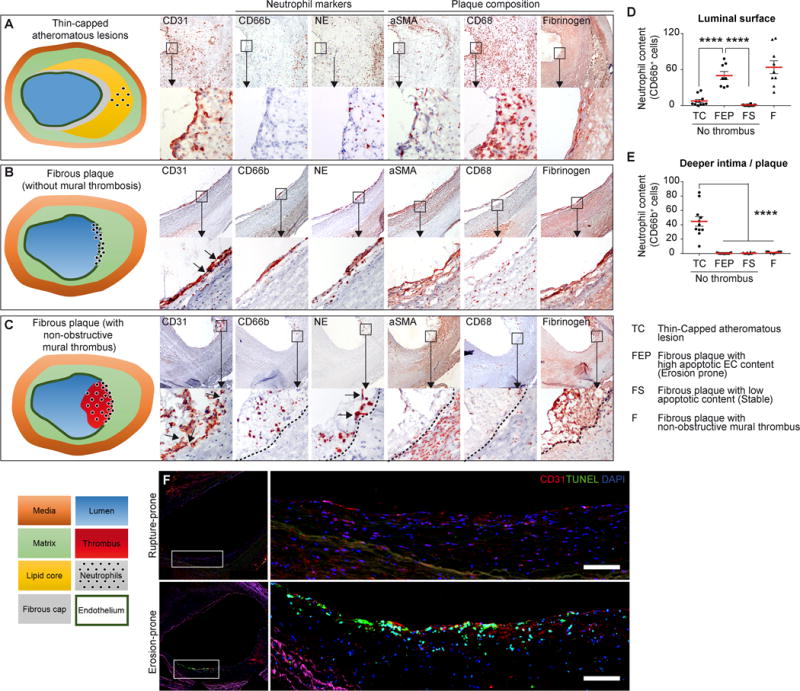
Human plaques with morphologies classified as thin-capped atheromatous lesions (n=11), fibrous without mural thrombosis (representing erosion prone or healed or resorbing erosive thrombus) (B), or fibrous plaques with non-obstructive mural thrombi (C), n=25, underwent serial cross sectional analysis (×40 magnification) for ECs/platelets (CD31), neutrophils (CD66b) and neutrophil elastase (NE), and other cell types (smooth muscle cells, αSMA+) and macrophages (CD68+), and thrombus (Fibrinogen). Insets show higher magnification (×100). CD66+ immunostaining quantified neutrophils located either on the luminal surface (D) or in the deeper intima / plaque (E). High content of apoptotic cells correlates with an erosion-prone morphology (F). Scale bars, 300μm. Data are expressed as mean ± s.e.m. ****p<0.0001. Mann–Whitney U test.
This study aimed to explore experimentally in vivo the mechanisms that underlie these findings in human plaques related to erosion vs. rupture. To this end, we first tailored arteries to harbor matrix-rich fibrous intimas by injury followed by healing. We then subjected these arteries with preformed fibrous intimal lesions to flow perturbation induced by partial stenosis. The initial injury impaired EC permeability at day 0, and the EC layer recovered after a 4 week healing period (Figure 2A and Online Figure IIA) with local neointima formation (Figure 2A right panel, and Online Figure IIB). The intima/wall (media + adventitia) ratio increased significantly post injury compared to uninjured left common carotid arteries (LCCA, Online Figure IIB and S2C, p<0.01), but the media-to-wall ratio did not change significantly (p=0.2). Post injury, LCCAs developed an SMC-rich neointima, with few macrophages, lacking a lipid core, and rich in HA (Figure 2A). This approach replicates key features of human plaques associated with superficial erosion9 (Figure 1). Post injury, arterial extracts contained significantly more mRNA encoding type I and type III collagen precursors and hyaluronan synthase 2, and decreased type I hyaluronidase mRNA, compatible with a slant toward HA accumulation (Figure 2B). E-Selectin (p<0.001) and VCAM-1 (p<0.0001) expression remained elevated 4 weeks after injury, indicating persistent endothelial activation at these sites, as we previously observed in injured arteries in rabbits (Figure 2C, Online Figure IID)33. Human saphenous vein ECs (HSVECs) cultured on a gelatin substrate supplemented with various molecular weight fractions of HA showed increased EC responses to a 5 kDa fraction of HA (Figure 2D). Furthermore, HSVECs cultured on a 5kDa HA-enriched coating showed concentration-dependent activation (Figure 2E). Thus, regional electric injury induces SMC and HA-rich intimal hyperplasia, and exposure of the basal surface of EC to low molecular weight (LMW)-HA can chronically activate EC.
Figure 2. Creation of intimal lesions for experimental probing of mechanisms related to superficial erosion in mice.
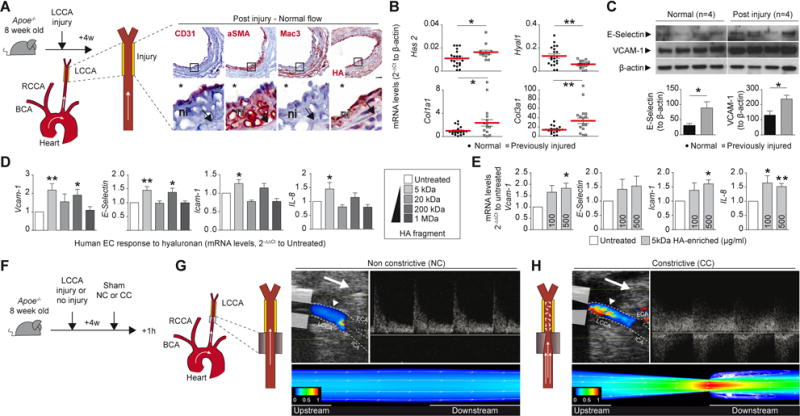
(A) Experimental protocol showing carotid electric injury (yellow segment) performed at day 0 and followed in 4 weeks by completed endothelial reconstitution. Composition of the neointima in post-injured arteries in ECs (CD31), vascular smooth muscle cells (SMA), macrophages (Mac3), and hyaluronan (HA). The insets show higher magnification views for each representative image. Carotid lumen (star) and internal elastic laminae (arrow) are shown. ni: neointima. The arrow represents flow orientation. LCCA: left common carotid injury. QPCR analysis was performed on normal vs previously injured carotid for the expression of extracellular matrix related mRNAs (B). Data are expressed in 2−ΔCt to β-actin. Each dot represents data from one animal. Mann–Whitney U test. (C) Western blot and relative quantification showing overexpression of E-Selectin and VCAM-1 in normal vs previously-injured arteries, n=4. Quantitative PCR on lysates from human saphenous vein ECs cultured 48h on native gelatin coating (untreated) vs coating enriched with hyaluronan (HA) of various molecular weights (D) or vs 100 or 500μg/ml of 5kDa HA-enriched coating (E). Data are expressed in 2−ΔΔCt to untreated condition, mean ± s.e.m, n=6 per group, *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Paired t-test (F,G,H) Placement of a cone-shape polyethylene cuff around the LCCA in mice and secured by a circumferential suture. Effect of non-constrictive control cuff (g) or constrictive cuff placement on flow dynamics in carotid arteries (H). Doppler-coupled ultrasonography showing velocity patterns along the downstream flow (left, arrow) and the velocity peak downstream of the cuff (right, arrow) measured at 1.5 mm downstream of the cuff (arrowhead). Computational fluid dynamics using finite volume analysis verified the extent of flow perturbation and recirculation. Longitudinal cross-sectional simulation of the effect of a non-constrictive cuff (G) or a constrictive cuff (H) showing a normalized axial velocity contour with superimposed velocity vectors (G,H). LCCA: left common carotid artery, LCCA: left common carotid artery, ECA: external carotid artery, ICA: internal carotid artery, NC: non-constrictive cuff, CC: constrictive cuff.
In vivo flow perturbation
Placement of cone-shaped polyethylene cuffs around the adventitia of the LCCA modulated downstream flow (Figure 2F through H and Supplementary video I). Placement of a non-constrictive control cuff did not alter the flow velocity pattern (Figure 2G, NC), but entailed the same operative manipulations and adventitial disturbance as the constrictive cuff (CC, Figure 2H). At peak systole (t = 0.1 ms), computational flow dynamic (CFD) analysis after NC placement showed a low variability in the velocity distribution of LCCA, and unidirectional vectors along the length of the vessel (Figure 2G). In contrast, CC placement yielded a high variability in the velocity distribution downstream, producing multidirectional vectors (Online Figure IIG). The highest velocity occurred at the narrowest end of the cuff and recirculation zones occurred further downstream (Online Figure IIG). At peak systole, the LCCA with NC presented a low evenly distributed wall shear stress (WSS) profile (Online Figure IIH), whereas a high variability in the WSS distribution accompanied the CC placement, with increasing values observed along the cuff and lowest values immediately downstream (Online Figure III). Time-averaged WSS plots obtained at sequential locations (Online Figure IIJ) showed no significant changes along the carotid in NC condition or proximal to the CC (site 1). Distal to the CC mural WSS greatly increased at the narrowest cuff site (2), became negative with increased amplitude slightly downstream from cuff (site 3), and oscillated from positive to negative values further downstream from the cuff (site 4). These data show that flow restriction in carotid arteries produces oscillatory shear stress downstream, permitting controlled and characterized experimental manipulation of local hydrodynamics in normal arteries or those with tailored fibrous intimal hyperplasia.
Flow perturbation promotes neutrophil recruitment and thrombus formation in previously injured arteries
Mice underwent placement of CC (n=45) or a NC (n=15) on either a normal or previously injured LCCA (Figure 3A and B, normal, n=19; post injury, n= 52). Control animals underwent a sham procedure (n=11). Neither NC placement nor sham procedure altered significantly intimal leukocyte accumulation downstream, demonstrating that neither the cuff material nor the surgical manipulations evoked acute inflammation. Neutrophils (Ly6G+ cells) did however adhere to the arterial intima distal to the CC, much more in previously injured than in normal LCCA (Figure 3B, p<0.001). Further experiments extended the histological observations by flow cytometric analysis of luminal eluates of downstream arterial segments from previously injured arteries: segments of arteries experiencing flow perturbation had significantly increased CD45+Ly6G+ cells (CC, Figure 3C and D, p<0.001). Ly6C+ monocytes also increased in the CC group, albeit 10 fold less than did neutrophils, illustrating the order-of-magnitude preponderance of neutrophils in this acute response (Figure 3C and D, p<0.05). Ly6G+ cells lined sites of flow disturbance in LCCA (Figure 3E, downstream, and Online Figure IVC and IVE). In contrast, upstream areas had few leukocytes. LCCA subjected to more prolonged flow perturbation (6h) still exhibited substantial Ly6G+ cell accumulation, although the cell number declined two fold compared to 1h (Online Figure IV, p<0.001). Flow disturbance caused Ly6G+ cell accumulation for up to 3 weeks (data not shown) compatible with chronic intimal disturbance. The adherent cells exhibited ultrastructural characteristics of neutrophils with segmented nuclei and plentiful cytoplasmic granules (Figure 3F). The CC groups displayed increased thrombus formation (Table II, p<0.05 and p<0.01), and fibrin-rich intraluminal thrombi in LCCAs following 1 h of flow perturbation (Figure 3G and H). Arterial segments subjected to flow perturbation accumulated intimal fibrinogen (Figure 3I). Moreover, circulating D-dimer, a product of fibrin degradation, increased in the serum of mice from CC group after 1h of flow disturbance (Figure 3J). Finally, en face observation of the LCCA showed thrombus formation downstream of stenoses (Online Figure IVD). These results demonstrate that flow perturbation provokes local neutrophil and thrombus accumulation in arteries with fibrous intimal hyperplasia.
Figure 3. Flow perturbation promotes neutrophil recruitment and thrombus formation.
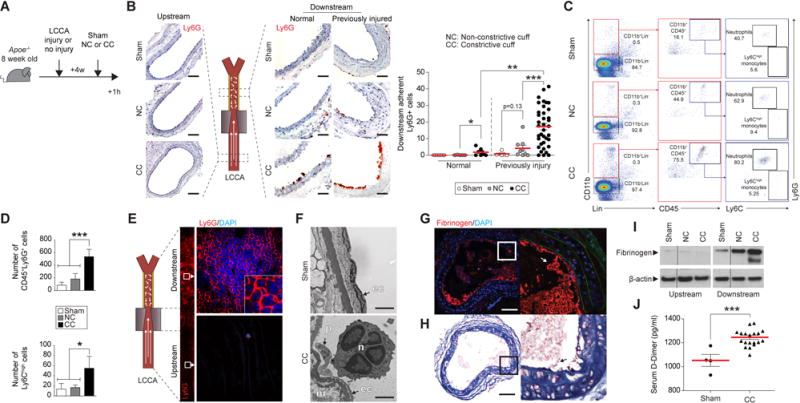
(A) Experimental protocol and time points studied. (B) Ly6G immunohistochemical staining of LCCA downstream segments shows the recruitment of neutrophils 1h after sham procedure (Sham, upper panel, n=9), placement of non-constrictive cuff (NC, intermediate panel, n=14) or constrictive cuff (CC, lower panels, n=46) either on normal (center) or post injured arteries (right). Uninjured upstream segments are shown on the left. LCCA: left common carotid artery. The graph shows assessment of the number of adherent neutrophils to the intima. Scale bars: 30 μm (C) Flow cytometry performed on post injured arteries after enzymatic digestion 1h after sham procedure (top), placement of a NC (middle) or a CC (bottom). n=5 per group. Viable neutrophils were identified as Lin−CD45+CD11b+Ly6G+ and viable Ly6Chigh monocytes were identified as CD45+CD11b+Ly6C+. (D) Flow cytometric quantification of neutrophils and Ly6Chigh monocyte. *p<0.05, ***p<0.001. (E) Representative en face Ly6G immunofluorescent staining performed on previously injured LCCA subjected to flow perturbation (n=3). Segment downstream from the cuff (CC) is shown in higher magnification. (F) Electron microscopic images show the intima of previously injured arteries after sham procedure (top) or CC placement (bottom). Scale bars, 2.5 nm. Immunofluorescent staining for fibrinogen (G) or Carstairs’ staining (H) performed on a selection of post injured LCCA that have formed intraluminal thrombus 1h after flow perturbation. Scale bars, 100 μm (I) Immunoblotting showing elevated fibrinogen downstream of CC. Normalization, β-actin. (J) Quantification by ELISA of circulating D-dimer concentrations in mice subjected to sham (n=4) vs flow perturbation (CC, n=19). Data are expressed as mean ± s.e.m. Mann–Whitney U test.
Flow-mediated neutrophil recruitment promotes endothelial injury
Sites of fatal superficial erosion complicating human atheromata show EC loss and neutrophil accumulation28. We therefore tested here the hypothesis that neutrophils contribute critically to loss of intimal EC in circumstances implicated in erosion. We examined the effect of 6h of continuous flow perturbation on luminal endothelium in arteries with fibrous thickened intimas (Figure 4A). In comparison to controls, in the CC group VCAM-1 and E-Selectin protein increased in extracts of downstream arterial segments (Figure 4B). Flow perturbation also promoted disturbed EC barrier function (Figure 4C, p<0.01) and apoptosis (Figure 4D). Further immunostaining using an anti-cleaved-caspase-3 antibody buttressed these findings (Online Figure IXB). Locally, ECs showed ultrastructural features of dying cells including blebbing and vacuolization (Figure 4E). Flow perturbation significantly decreased endothelial continuity (p<0.05), altered EC morphology, and led to EC desquamation (Figure 4F and 4H, p<0.05). Discontinuity in the intimal endothelium correlated with the number of firmly adherent neutrophils (Figure 4G). Numerous neutrophils also congregated with activated EC or co-localized with patches of intimal denudation (Figure 4I).
Figure 4. Flow-mediated neutrophil recruitment potentiates endothelial cell dysfunction and loss.
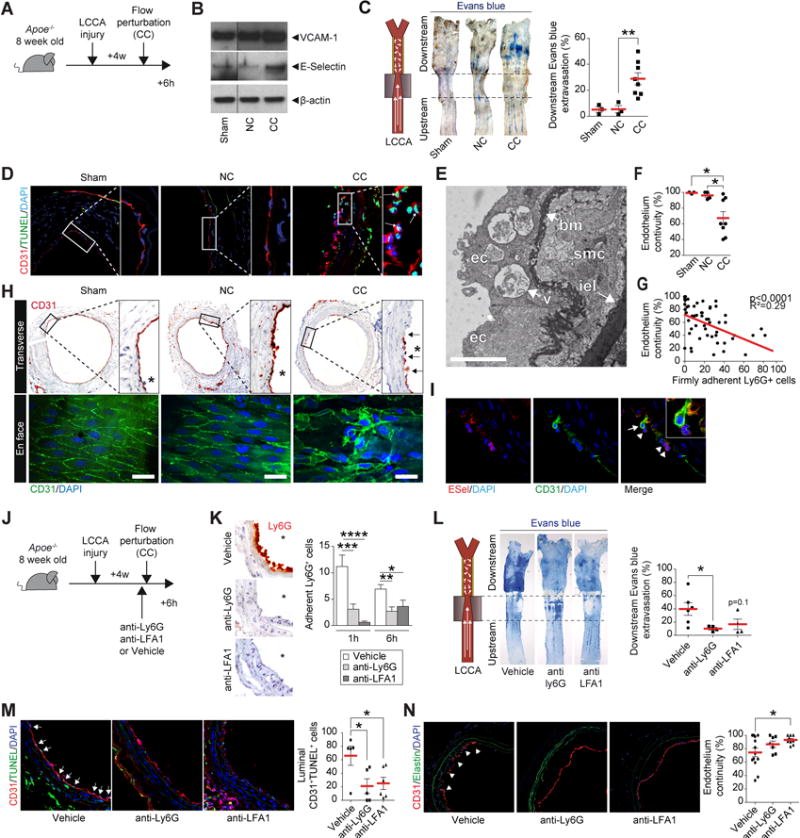
(A) Experimental protocol and time points studied: LCCA were subjected to injury and 4 weeks later to 6h of flow perturbation (CC). Sham, NC or CC arteries probed for the expression of VCAM-1 and E-Selectin by western blot (B) or for Evans blue extravasation (C). Normalization, β-actin. (D) Immunofluorescent staining for endothelium (CD31), apoptosis (TUNEL) and DNA (DAPI) in cross sections of Sham, NC, or CC arteries. Each image shows bigger magnification on the right. Arrows show the presence of luminal apoptotic ECs. (E) Electron microscopy image showing ECs subjected to flow perturbation in previously injured arteries. ec: endothelial cell, bm: basement membrane, smc: smooth muscle cell, iel: internal elastic laminae, v: vacuole. Scale bars, 2μm (F) Quantification of endothelial continuity. (G) Negative correlation between endothelial continuity and the number of adherent Ly6G+ neutrophils (p<0.0001, R2=0.29). Immunohistochemical staining for CD31 (top) or en face visualization in immunofluorescence of EC (CD31) and DNA (DAPI) (bottom) in downstream arteries after sham procedure (left, n=3), placement of a non-constrictive cuff (middle, n=4) or constrictive cuff (right, n= 8). Scale bars: 20μm. Arrows show patches of EC denudation. Semi-quantitative assessment of endothelial continuity cross sections CD31 immunohistochemistry is shown in graph (right). (I) Immunofluorescent staining for E-Selectin, EC (CD31) and DNA (DAPI) show EC undergoing detachment (arrow), in contact with neutrophils (arrowhead) in arteries subjected to flow perturbation (CC). (J) Experimental protocol and time points showing LCCA subjected to injury, followed 4 weeks later by systemic injections of neutralizing Ly6G antibody, LFA-1 antibody, or vehicle before flow perturbation (CC). (K) Ly6G staining in imunohistochemistry shows neutrophil recruitment after 1h of flow perturbation in LCCA subjected to flow in groups receiving vehicle, anti-ly6G, or anti-LFA1. The graph shows quantification of adherent neutrophils in each group, after either 1 or 6h of flow perturbation. (L) LCCA from group vehicle, anti-Ly6G, and anti-LFA-1 probed for endothelial permeability using Evans blue intravital staining after 6h of flow perturbation. (M) Immunofluorescent staining for endothelium (CD31, red), early apoptosis (green), and DNA (DAPI, blue) shows luminal endothelial apoptotic cells (arrows) in group vehicle, anti-Ly6G, or anti-LFA-1 after 6h of flow perturbation. (N) Immunofluorescent staining for endothelium (CD31, red), elastin auto fluorescence (green), and DNA (DAPI, blue) shows endothelial continuity in group vehicle, anti-Ly6G, or anti-LFA-1 after 6h of flow perturbation. Data are expressed as mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. Mann–Whitney U test.
Further experiments assessed the causal relationship between neutrophil presence and endothelial dysfunction/loss (Figure 4J). Anti-Ly6G blocking antibody treatment 24h before cuffing induced neutropenia (N=11). Alternatively, administration of an anti-LFA-1 blocking antibody 1h before the experiment aimed to reduce neutrophil recruitment. Either treatment abolished the recruitment of Ly6G+ neutrophils in the LCCA following 1 or 6h of flow perturbation (Figure 4K). After 6h of flow perturbation, either anti-neutrophil treatment likewise limited endothelial permeability in the arterial segments subjected to disturbed flow (Figure 4L), decreased the number of luminal CD31+TUNEL+ apoptotic EC (Figure 4M, p<0.05) and preserved endothelial continuity (Figure 4N, p<0.05). These results identify neutrophils as effectors of EC injury, death, and detachment under circumstances associated with thrombosis due to superficial erosion.
Endogenous TLR2 activates EC and promotes neutrophil chemoattraction
Further experiments tested the hypothesis that TLR2 participates in EC activation and potentiates neutrophil chemoattraction or leukocyte adhesion in arteries that share features of eroded human atheromata. HSVEC incubated with the TLR2 agonist Pam3CSK4 (Pam3) or with vehicle showed time-dependent increases in VCAM-1, ICAM-1, E-Selectin and IL-8 (Figure 5A). Six hours after Pam3 or vehicle administration i.p. to Apoe−/− or Apoe−/−Tlr2−/− mice, luminal eluates as well as extracts of the remaining aorta wall (intima excluded) underwent RNA isolation and QPCR analysis (Figure 5B). Arterial luminal eluates from Pam3-treated Apoe−/− mice showed significantly increased Vcam-1, E-Selectin, Cxcl-1, Cxcl-2, and Cxcl-5 mRNAs (Figure 5C). Aortic tunica media extracts from Pam3-treated Apoe−/− mice had increased concentrations of the chemoattractants Cxcl-1, Cxcl-2 and Cxcl-5 mRNAs but not messages that encode the endothelial adhesion molecules Vcam-1 or E-Selectin. Pam3 did not exert these actions in Apoe−/−Tlr2−/− animals, indicating that these arterial responses to Pam3 depend onTLR2. The Bax/Bcl2 mRNA ratio also increased in these intimal extracts, implicating TLR2 in endothelial apoptosis in vivo (Figure 5D). Pam3 administration augmented both VCAM-1 and E-Selectin protein in aortas isolated from Apoe−/− mice, but not in Apoe−/−Tlr2−/− animals (Figure 5E), and selectively activated luminal EC in vivo (Figure 5F). Circulating CXCL-1 also increased in Pam3-treated Apoe−/− but not Apoe−/−Tlr2−/− animals (Figure 5G). Together, these results indicate that TLR2 activation leads to the overexpression of neutrophil chemoattractants by both EC and mesenchymal cells, while it increases EC expression of leukocyte adhesion molecules, and augments luminal EC apoptosis.
Figure 5. TLR2 mediates endothelial cell activation in vivo.
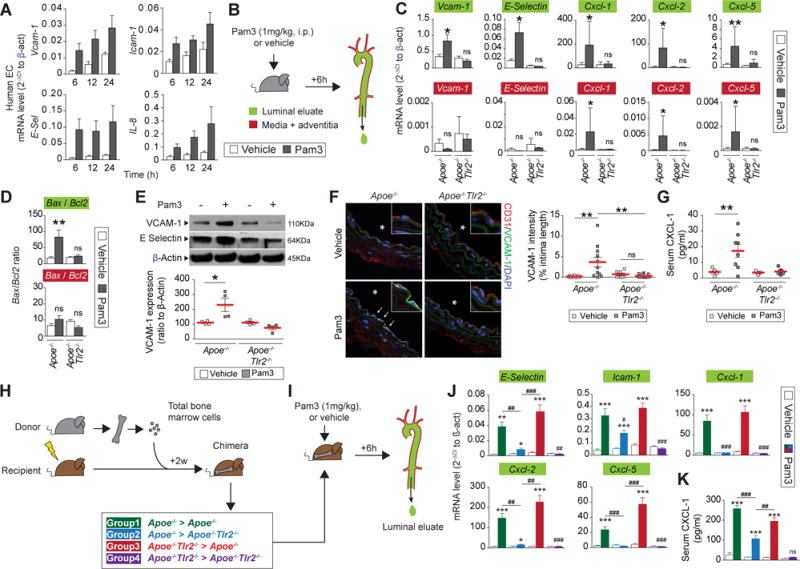
(A) Quantitative PCR on lysates from human saphenous vein ECs 6, 12 or 24h after Pam3csk4 incubation, n=6 per condition. (B) Experimental protocol and time points used in the study. (C,D) Quantitative PCR performed on aortic luminal eluates (top, green) or media-adventitia lysates (bottom, red) isolated 6h after Pam3csk4 (Pam3, grey bars) or vehicle (white bars) injection in either Apoe−/− or Apoe−/−Tlr2−/− mice, n=6 per condition. (c) mRNA expression of adhesion molecules and neutrophil chemoattractants. (D) mRNA expression ratio between Bax and BCl2. (E) Immunoblotting for VCAM-1, E-Selectin performed on aorta after Pam3 (grey bars) or vehicle (white bars) injection in either Apoe−/− or Apoe−/−Tlr2−/− mice. Normalization used β-actin. The graph shows quantification of VCAM-1 expression (below). (F) Immunofluorescent staining for endothelium (CD31), VCAM-1, and DNA (DAPI) in LCCA isolated from Apoe−/− (left) or Apoe−/−Tlr2−/− mice (right) 6h after injection of vehicle (top) or Pam3 (bottom). The stars indicate the lumen, the arrows show EC overexpressing VCAM-1. The graph (right) depicts semi-quantitatively VCAM-1 immunopositivity in luminal EC expressed as % of intima length. (G) Quantification by ELISA of circulating CXCL-1 levels. (H) Generation of four groups of chimeric mice and experimental protocol of the study (I). Quantitative PCR performed on luminal eluate (J) and quantification by ELISA of CXCL-1 serum levels (K) isolated from mice from each group, 6h after vehicle (white bars) or Pam3 injection (colored bars), n=5–6 per condition. Data are expressed as mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001, Pam3 vs vehicle. ## p<0.01, ### p<0.001, comparison of selected groups, paired t-test.
Testing the hypothesis that TLR2 of arterial origin participates in the recruitment of neutrophils used chimeric mice constructed by transplantation of either Apoe−/− or Apoe−/−Tlr2−/− bone marrow to lethally irradiated Apoe−/− or Apoe−/−Tlr2−/− recipients. Mice from each of the 4 groups thus generated (Figure 5H) received Pam3 or vehicle and underwent analysis after 6h (Figure 5I). Eluates from aortic luminal extracts furnished RNA for QPCR analysis (Figure 5I). As expected, Pam3-injection strongly activated luminal cells in mice expressing TLR2 in both intrinsic arterial and bone-marrow derived cells (Group 1, Figure 5J) but not in Tlr2−/− mice (Group 4). Animals lacking TLR2 in bone marrow-derived cells alone (Group 3) retained responsiveness to Pam3, to an extent comparable with mice from Group 1. In contrast, mice lacking TLR2 in intrinsic arterial cells, but reconstituted with bone marrow from TLR2 sufficient animals (Group 2), showed a much lower activation than Group 1 mice. While Pam3 increased circulating CXCL-1 in Groups 1, 2, and 3, Group 2 showed significantly lower blood concentration of CXCL-1 compared to Groups 1 and 3 (Figure 5K, p<0.001). Pre-treatment with Pam3 strongly increased TLR2 expression in luminal ECs (Online Figure VIII. Immunofluorescent colocalization by confocal microscopy reveals that ECs express most arterial TLR2, although adherent neutrophils and some smooth muscle cells also show limited positivity. A 3D reconstruction revealed that TLR2 localizes mainly on the basal surface of ECs in contact with the basement membrane (Supplementary video I). In contrast, the apical surface of EC did not containTLR2. These results demonstrate that TLR2 expressed by intrinsic arterial cells participates prominently in intimal cell activation and supports the hypothesis that endothelial TLR2 promotes neutrophil recruitment in the context of superficial erosion.
TLR2 participates in neutrophil recruitment, EC death, and dysfunction in arteries with fibrous intimal hyperplasia in response to flow disturbance
Areas of disturbed arterial flow and mouse and human atheromata exhibit overexpression of TLR2. These observations and the ability of TLR2 ligation to activate ECs suggested the hypothesis that TLR2 participates in the recruitment of intimal neutrophils. Eight-week-old Apoe−/− or Apoe−/−Tlr2−/− mice first underwent LCCA injury (Figure 6A). LCCA with fibrous intimal thickening in Apoe−/− versus Apoe−/−Tlr2−/− showed no significant differences in internal or external diameter, or in the intima-to-wall ratio (p=0.2, Figure 6B and C). Tlr2 deficiency in normal or previously injured arteries did not affect the arterial expression of the atherothrombosis-related genes evaluated (Online Figure VC). Other mice underwent LCCA injury followed 4 weeks later by flow perturbation (Online Figure VID). While local neutrophil number did not differ between groups after 1h (Online Figure VD), after 6 h the distal arterial segments of Apoe−/−Tlr2−/− contained significantly fewer Ly6G+ neutrophils than Apoe−/− (Figure 6E, p<0.01). Furthermore, Apoe−/−Tlr2−/− mice showed less disruption in endothelial continuity in arterial segments subjected to flow perturbation (Figure 6F), as well as decreased permeability (Figure 6G). Apoe−/−Tlr2−/− mice had reduced local thrombus formation after 1 or 6h of flow perturbation (Table II), and diminished circulating D-Dimer concentrations after 6h of flow perturbation (Online Figure IIIE). These results support the participation of TLR2 in the recruitment of neutrophils, in EC dysfunction/loss, and in subsequent thrombosis in the context of superficial erosion (Figure 6H).
Figure 6. Flow perturbation promotes neutrophil recruitment through TLR2.
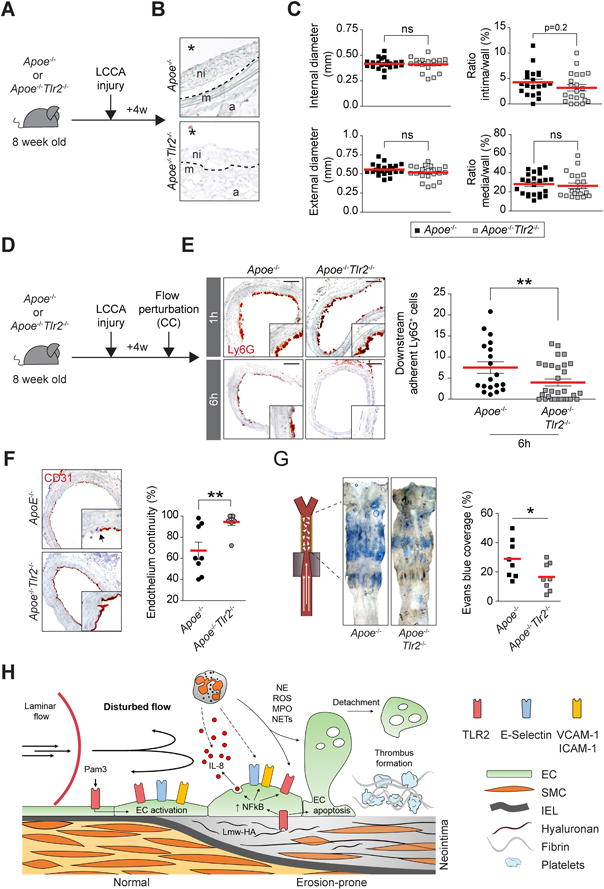
(A) Experimental timeline. (B) H&E staining shows neointima formation after LCCA injury in Apoe−/− and Apoe−/−Tlr2−/− mice. ni: neointima, m: media, a: adventitia. The star indicates the lumen. (C) Morphometric analysis showing measurement of internal (top, left) and external diameters (bottom, left), and the ratio intima/wall (top, right) and media/wall (bottom, right). (D) Experimental protocol involving LCCA injury followed by flow perturbation (CC) in Apoe−/− and Apoe−/−Tlr2−/− mice. (E) Ly6G immunohistochemistry staining of LCCA cross-sections showing the recruitment of neutrophils 1 or 6h after flow perturbation in Apoe−/− and Apoe−/−Tlr2−/− mice. The graph shows the semi-quantitative assessment of adherent neutrophils to the intima after 6h of flow perturbation. Scale bar: 60 μm (F) CD31 immunohistochemistry staining shows endothelium. The graph (right) shows assessment of endothelial continuity (G) LCCA isolated from Apoe−/− and Apoe−/−Tlr2−/− mice were probed for endothelial permeability using Evans blue intravital staining after 6h of flow perturbation. (H) Summary diagram of the main findings of this study. ROS: Reactive oxygen species. MPO: Myeloperoxidase, NETs: Neutrophils extracellular traps, Lmw-HA: Low molecular weight-hyaluronan. IEL: Internal elastic lamina. Data are expressed as mean ± s.e.m. *p<0.05, **p<0.01. Mann–Whitney U test.
DISCUSSION
Superficial erosion causes arterial thrombosis, and hence ACS, without plaque rupture, as disclosed by post mortem examination and optical coherence tomographic imaging in intact patients. Studies of human autopsy specimens do not permit dissection of the mechanisms that underlie this modus of thrombotic complication of atherosclerosis. This study extends in vivo our prior in vitro observations that implicated TLR2 signaling, engagement of this innate immune receptor by hyaluronan or other ligands, and the participation of neutrophils in aspects of superficial erosion.
Among several processes that may contribute to superficial erosion, flow perturbation can promote endothelial dysfunction and death. Yet, areas subjected solely to flow perturbation seldom develop superficial erosion, suggesting that flow disturbance alone does not suffice to trigger thrombosis. We therefore recently proposed a “two hit” schema for the pathogenesis of superficial erosion, a mechanism of coronary thrombosis apparently on the rise5, 28. Plaques that have precipitated thrombi due to erosion differ distinctly from the so-called rupture-prone plaque. These differences suggest striking divergences in the pathological mechanisms and support the concept that plaque composition influences the mode of thrombotic complication. This in vivo study used an experimental approach designed to mimic certain characteristics of plaques that have caused thrombosis due to superficial erosion. We created in mouse arteries expanded intimas enriched in SMC and a glycosaminoglycan-rich extracellular matrix with few inflammatory cells, which recapitulates some key features of the substrate associated with eroded plaques in humans8, 9. We used ApoE-deficient mice consuming a chow rather than atherogenic diet to avoid producing plaques overloaded with lipids and macrophage foam cells, characteristics of ruptured rather than eroded lesions34. The use of electrical injury to stimulate intimal expansion proved more reproducible in extensive pilot experiments than endovascular intervention. The resultant lesions accumulated HA, and exhibited altered HA turnover, as indicated by reciprocal changes in the concentrations of mRNAs that encode enzymes involved in HA production and degradation. Heightened expression of E-Selectin and VCAM-1 in these arteries indicated sustained chronic EC activation, findings concordant with our prior work on injured rabbit arteries33. Other studies have shown HA accumulation during neointimal hyperplasia in association with the migration and the proliferation of SMC, and that intimal HA may accentuate atherosclerosis35, 36. In vitro, a culture substrate enriched with low molecular weight HA caused low-level activation of human ECs, consistent with the notion that intimal HA could pave the way for thrombotic complication due to superficial erosion. Prior in vitro studies have seldom aimed to replicate the hydrodynamic conditions that prevail downstream of arterial stenosis in humans. The current in vivo findings show that flow disturbance sets the stage for thrombosis, in arteries with fibrous intimal thickening.
Disturbed flow and low-shear stress can activate arterial ECs through NF-κB37, 38 and also rapidly augment P-Selectin and IL-8 elaboration non-transcriptionally by triggering their translocation to the EC luminal surface39. Experimental stenosis in veins promotes the accumulation of neutrophils that participate in P-Selectin-dependent initiation/amplification of thrombosis40. Here, arterial ECs exposed to flow perturbation displayed markers of activation and apoptosis acutely and developed patches of endothelial desquamation. Oscillatory shear stress can induce EC apoptosis through various pathways including p53 and protein kinase C-ζ (PKC-ζ)18, 41. Apoptosis could in turn exacerbate EC detachment and promote thrombosis.
Substantial neutrophil accumulation distal to stenosis occurred in the arteries with fibrous intimal expansion. Interruption of neutrophil trafficking protected EC from activation, apoptosis, and detachment. These observations support the local recruitment of neutrophils in eroded plaques and their role in extending injury28, 42, whether or not these leukocytes participate in plaque formation43. Neutrophil arrival at sites of early erosion could thus amplify and propagate conditions that promote local thrombosis, in accord with our proposed “two hit” scheme. Granulocytes can harm EC in many ways44, including by producing proteinases that sever the tethers of the ECs to the basement membrane favoring desquamation45. Neutrophils strongly activate cultured ECs, disturb their ability to adhere to the basement membrane, and disturb EC morphology28, 44. Neutrophil elastase can degrade basement membrane constituents46, enhance EC injury47, favor anoikis and apoptosis48, and concentrations of this enzyme increase in patients with myocardial infarction49. Neutrophils also contain abundant myeloperoxidase (MPO), an enzyme that produces hypochlorous acid, an inducer of EC apoptosis and tissue factor production30. While a subset of recruited cells after flow perturbation express MPO in this study, MPO does not necessarily colocalize with Ly6G+ neutrophils. Rather, in mice, macrophages contain considerable MPO, an example of the discrepancy between murine and human innate immunity. Dying granulocytes also generate neutrophil extracellular traps (NETs), structures that can induce EC death and dysfunction50, 51. NETs could also promote EC detachment as they contain MMP-9, a metalloproteinase involved in the degradation of the basement membrane type IV collagen and able to activate endothelial pro-MMP-251. We previously reported the presence of NETs and neutrophil elastase at the surface of human plaques resembling those implicated in superficial erosion28. NETs contain tissue factor procoagulant and can further furnish scaffolds for coronary thrombi in culprit lesions of ACS52, 53.
This study demonstrates in vivo that TLR2 participates in EC activation and amplifies the recruitment of neutrophils after flow perturbation. ECs express TLR2 that participates in experimental atherosclerosis. TLR2 also localizes in human atheromata with characteristics of those that have caused thrombosis due to erosion28. Previous studies showed that low molecular weight fractions of HA can activate TLR2 signaling through a NF-κB dependent pathway27. Thus, HA could act as an endogenous agonist of TLR2 and contribute to neutrophil recruitment in the setting of disturbed flow, as supported by the present findings. The use of chimeric mice indicates that TLR2 expressed by intrinsic arterial cells rather than leukocytes mediates the release of neutrophil chemoattractants and the production of endothelial adhesion molecules. In addition to TLR2, HA can also bind various receptors including CD44 or ICAM-1. Flow perturbation could also activate alternative signaling (i.e. by integrins) that may synergize with constitutive TLR2 signaling driven by HA binding. Defining the roles of these and other potential pathways will require further study.
The lack of experimental tools in vivo for studying superficial erosion has constituted a considerable hindrance, and contributes to the knowledge gap in the field. The experimental approach used here permitted us to test hypotheses in vivo regarding the pathogenesis of superficial erosion that emerged from in vitro or descriptive observations made by our laboratory and others. This in vivo approach permitted testing of focused mechanistic hypotheses related to the pathophysiology of superficial erosion. The short time course of the experiments presented here represents one of several limitations to the ready extrapolation of our results to a human disease that plays out over decades. Yet, these findings furnish some early and novel insights into the mechanisms of superficial erosion, a major gap area in our understanding of the thrombotic complications of human atherosclerosis. This quest has become increasingly clinically compelling, as current therapies have made substantial inroads in reducing plaque rupture, and as superficial erosion has emerged as a considerable continuing contributor to residual risk in the current era5, 54, 55.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Superficial erosion involves discontinuity in the intimal endothelium and thrombus formation without plaque rupture.
Erosion-prone plaques have specific features and associate with the presence of neutrophils.
What New Information Does This Article Contribute?
Validation of an in vivo approach to recapitulate aspects of superficial erosion in mice to permit mechanistic explorations.
A demonstration that neutrophils contribute critically to arterial EC injury in regions of disturbed arterial flow.
Establishing a role for endothelial TLR2 in local neutrophil recruitment
Post-mortem pathological studies demonstrated the loss of endothelial cells at sites of superficial erosion of the culprit lesions of acute coronary syndromes, yet the mechanisms that drive this process remain elusive. We developed a new in vivo approach in mice that involves creating a chronic intimal lesion that recapitulates certain features associated with superficial erosion, followed by introduction of flow disturbance. We found that neutrophils selectively accumulate in the lumen of carotid arteries under these circumstances. This neutrophil recruitment locally disrupted the endothelial layer. TLR2 loss of function in arterial but not in bone-marrow-derived cells blunted these effects. These findings illustrate the utility of a novel experimental tool to study the mechanisms of arterial thrombi in the absence of plaque rupture.
Acknowledgments
We thank David Lynn, Marc Belanger, Mark MacMillan and Chelsea Swallom for providing administrative, technical, and editorial support throughout the project.
SOURCES OF FUNDING
This work was supported by grants from the National Heart, Lung and Blood institute (NIH-R01 HL080472) by the Leducq Foundation (Paris, France) and from the RRM charitable fund. G.F was supported by the Harold M. English Fellowship Fund from Harvard Medical School (Boston, USA), the Fondation Bettencourt Schueller (Neuilly-sur-Seine, France), and the Philippe Foundation (New York, USA and Paris, France). T.M. received support from by the Sarnoff Cardiovascular Research Foundation (Great Falls, USA). G.S. received financial support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior and the Lemann Foundation (Brazil). M.B.M. received support from the Lemann Foundation and the National Council for Scientific and Technologic Development (CNPq, Brazil). E.A was supported by grants from the National Heart, Lung and Blood institute (NIH-R01 HL114805 and NIH-R01 HL109506). We thank Pr. Ronglih Liao and Dr. Sudeshna Fisch from the Brigham and Women’s Hospital Cardiovascular Physiology Core for assistance in ultrasound visualization.
Nonstandard Abbreviations and Acronyms
- ACS
Acute coronary syndrome
- ApoE
Apolipoprotein E
- CC
Constrictive cuff
- CXCL
Chemokine (C-X-C motif) ligand
- EC
Endothelial cell
- HA
Hyaluronic acid
- HSVEC
Human saphena vein endothelial cell
- IL-8
Interleukin-8
- LCCA
Left common carotid artery
- MMP
Metalloproteinase
- MPO
Myeloperoxidase
- NC
Non-constrictive cuff
- NETs
Neutrophil extracellular traps
- NF-κB
Nuclear factor-kappa B
- PKC
Protein kinase C
- RCCA
Right common carotid artery
- SMC
Smooth muscle cell
- TLR2
Toll like receptor 2
- VCAM-1
Vascular cell adhesion molecule 1
- WSS
Wall shear stress
Footnotes
AUTHOR CONTRIBUTIONS
Designing research studies: GF, KC, PL. Conducting experiments GF, TM, GS, GSM, YT, ES. Acquiring data: GF, TM, GS, MS, GSM, AC, MBM, TQ. Analyzing data: GF, GS, MS, GS, MS. Advice on experimental design: MS, YC, GS, FS, MN, EA, KC, PL. Writing the manuscript: GF and PL.
DISCLOSURES
None.
References
- 1.Libby P. Mechanisms of acute coronary syndromes. The New England journal of medicine. 2013;369:883–884. doi: 10.1056/NEJMc1307806. [DOI] [PubMed] [Google Scholar]
- 2.Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circulation research. 2014;114:1852–1866. doi: 10.1161/CIRCRESAHA.114.302721. [DOI] [PubMed] [Google Scholar]
- 3.Libby P. How does lipid lowering prevent coronary events? New insights from human imaging trials. European heart journal. 2015;36:472–474. doi: 10.1093/eurheartj/ehu510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Lammeren GW, den Ruijter HM, Vrijenhoek JE, van der Laan SW, Velema E, de Vries JP, de Kleijn DP, Vink A, de Borst GJ, Moll FL, Bots ML, Pasterkamp G. Time-dependent changes in atherosclerotic plaque composition in patients undergoing carotid surgery. Circulation. 2014;129:2269–2276. doi: 10.1161/CIRCULATIONAHA.113.007603. [DOI] [PubMed] [Google Scholar]
- 5.Libby P, Pasterkamp G. Requiem for the ‘vulnerable plaque’. European heart journal. 2015;36:2984–2987. doi: 10.1093/eurheartj/ehv349. [DOI] [PubMed] [Google Scholar]
- 6.Jernberg T, Hasvold P, Henriksson M, Hjelm H, Thuresson M, Janzon M. Cardiovascular risk in post-myocardial infarction patients: Nationwide real world data demonstrate the importance of a long-term perspective. European heart journal. 2015;36:1163–1170. doi: 10.1093/eurheartj/ehu505. [DOI] [PubMed] [Google Scholar]
- 7.Jia H, Abtahian F, Aguirre AD, Lee S, Chia S, Lowe H, Kato K, Yonetsu T, Vergallo R, Hu S, Tian J, Lee H, Park SJ, Jang YS, Raffel OC, Mizuno K, Uemura S, Itoh T, Kakuta T, Choi SY, Dauerman HL, Prasad A, Toma C, McNulty I, Zhang S, Yu B, Fuster V, Narula J, Virmani R, Jang IK. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. Journal of the American College of Cardiology. 2013;62:1748–1758. doi: 10.1016/j.jacc.2013.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: A comprehensive morphological classification scheme for atherosclerotic lesions. Arteriosclerosis, thrombosis, and vascular biology. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 9.Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: The pathologists’ view. European heart journal. 2013;34:719–728. doi: 10.1093/eurheartj/ehs411. [DOI] [PubMed] [Google Scholar]
- 10.Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, Wight TN, Virmani R. Differential accumulation of proteoglycans and hyaluronan in culprit lesions: Insights into plaque erosion. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:1642–1648. doi: 10.1161/01.atv.0000034021.92658.4c. [DOI] [PubMed] [Google Scholar]
- 11.Chien S, Li S, Shyy YJ. Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension. 1998;31:162–169. doi: 10.1161/01.hyp.31.1.162. [DOI] [PubMed] [Google Scholar]
- 12.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: Molecular, cellular, and vascular behavior. Journal of the American College of Cardiology. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. [DOI] [PubMed] [Google Scholar]
- 13.Gimbrone MA, Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circulation research. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fry DL. Acute vascular endothelial changes associated with increased blood velocity gradients. Circulation research. 1968;22:165–197. doi: 10.1161/01.res.22.2.165. [DOI] [PubMed] [Google Scholar]
- 15.Joris I, Zand T, Majno G. Hydrodynamic injury of the endothelium in acute aortic stenosis. The American journal of pathology. 1982;106:394–408. [PMC free article] [PubMed] [Google Scholar]
- 16.Tricot O, Mallat Z, Heymes C, Belmin J, Leseche G, Tedgui A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation. 2000;101:2450–2453. doi: 10.1161/01.cir.101.21.2450. [DOI] [PubMed] [Google Scholar]
- 17.Chaudhury H, Zakkar M, Boyle J, Cuhlmann S, van der Heiden K, Luong le A, Davis J, Platt A, Mason JC, Krams R, Haskard DO, Clark AR, Evans PC. C-jun n-terminal kinase primes endothelial cells at atheroprone sites for apoptosis. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:546–553. doi: 10.1161/ATVBAHA.109.201368. [DOI] [PubMed] [Google Scholar]
- 18.Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, McClain C, Reinhart-King CA, King MR, Berk BC, Fujiwara K, Woo CH, Abe J. Pkczeta mediates disturbed flow-induced endothelial apoptosis via p53 sumoylation. J Cell Biol. 2011;193:867–884. doi: 10.1083/jcb.201010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Civelek M, Manduchi E, Riley RJ, Stoeckert CJ, Jr, Davies PF. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circulation research. 2009;105:453–461. doi: 10.1161/CIRCRESAHA.109.203711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sumi T, Yamashita A, Matsuda S, Goto S, Nishihira K, Furukoji E, Sugimura H, Kawahara H, Imamura T, Kitamura K, Tamura S, Asada Y. Disturbed blood flow induces erosive injury to smooth muscle cell-rich neointima and promotes thrombus formation in rabbit femoral arteries. J Thromb Haemost. 2010;8:1394–1402. doi: 10.1111/j.1538-7836.2010.03843.x. [DOI] [PubMed] [Google Scholar]
- 21.Bombeli T, Karsan A, Tait JF, Harlan JM. Apoptotic vascular endothelial cells become procoagulant. Blood. 1997;89:2429–2442. [PubMed] [Google Scholar]
- 22.Bombeli T, Schwartz BR, Harlan JM. Endothelial cells undergoing apoptosis become proadhesive for nonactivated platelets. Blood. 1999;93:3831–3838. [PubMed] [Google Scholar]
- 23.Durand E, Scoazec A, Lafont A, Boddaert J, Al Hajzen A, Addad F, Mirshahi M, Desnos M, Tedgui A, Mallat Z. In vivo induction of endothelial apoptosis leads to vessel thrombosis and endothelial denudation: A clue to the understanding of the mechanisms of thrombotic plaque erosion. Circulation. 2004;109:2503–2506. doi: 10.1161/01.CIR.0000130172.62481.90. [DOI] [PubMed] [Google Scholar]
- 24.Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation. 2002;105:1158–1161. [PubMed] [Google Scholar]
- 25.Mullick AE, Soldau K, Kiosses WB, Bell TA, 3rd, Tobias PS, Curtiss LK. Increased endothelial expression of toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med. 2008;205:373–383. doi: 10.1084/jem.20071096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by toll-like receptor 2. The Journal of clinical investigation. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging tlr2. Journal of immunology. 2006;177:1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 28.Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P. Tlr2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. European heart journal. 2015;36:1394–1404. doi: 10.1093/eurheartj/ehv044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrante G, Nakano M, Prati F, Niccoli G, Mallus MT, Ramazzotti V, Montone RA, Kolodgie FD, Virmani R, Crea F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: A clinicopathological study. Circulation. 2010;122:2505–2513. doi: 10.1161/CIRCULATIONAHA.110.955302. [DOI] [PubMed] [Google Scholar]
- 30.Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: Involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:1309–1314. doi: 10.1161/01.ATV.0000131784.50633.4f. [DOI] [PubMed] [Google Scholar]
- 31.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. Journal of immunology. 2011;187:538–552. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tavora FR, Ripple M, Li L, Burke AP. Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc Disord. 2009;9:27. doi: 10.1186/1471-2261-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka H, Sukhova GK, Swanson SJ, Clinton SK, Ganz P, Cybulsky MI, Libby P. Sustained activation of vascular cells and leukocytes in the rabbit aorta after balloon injury. Circulation. 1993;88:1788–1803. doi: 10.1161/01.cir.88.4.1788. [DOI] [PubMed] [Google Scholar]
- 34.Raman KG, Gandley RE, Rohland J, Zenati MS, Tzeng E. Early hypercholesterolemia contributes to vasomotor dysfunction and injury associated atherogenesis that can be inhibited by nitric oxide. Journal of vascular surgery. 2011;53:754–763. doi: 10.1016/j.jvs.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riessen R, Wight TN, Pastore C, Henley C, Isner JM. Distribution of hyaluronan during extracellular matrix remodeling in human restenotic arteries and balloon-injured rat carotid arteries. Circulation. 1996;93:1141–1147. doi: 10.1161/01.cir.93.6.1141. [DOI] [PubMed] [Google Scholar]
- 36.Evanko SP, Angello JC, Wight TN. Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 1999;19:1004–1013. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- 37.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The nf-kappa b signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fang Y, Shi C, Manduchi E, Civelek M, Davies PF. Microrna-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13450–13455. doi: 10.1073/pnas.1002120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hol J, Wilhelmsen L, Haraldsen G. The murine il-8 homologues kc, mip-2, and lix are found in endothelial cytoplasmic granules but not in weibel-palade bodies. J Leukoc Biol. 2010;87:501–508. doi: 10.1189/jlb.0809532. [DOI] [PubMed] [Google Scholar]
- 40.von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Magid R, Davies PF. Endothelial protein kinase c isoform identity and differential activity of pkczeta in an athero-susceptible region of porcine aorta. Circulation research. 2005;97:443–449. doi: 10.1161/01.RES.0000179767.37838.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–2900. doi: 10.1161/01.cir.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
- 43.Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circulation research. 2012;110:875–888. doi: 10.1161/CIRCRESAHA.111.257535. [DOI] [PubMed] [Google Scholar]
- 44.Harlan JM, Killen PD, Harker LA, Striker GE, Wright DG. Neutrophil-mediated endothelial injury in vitro mechanisms of cell detachment. The Journal of clinical investigation. 1981;68:1394–1403. doi: 10.1172/JCI110390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Libby P. Seeing and sampling the surface of the atherosclerotic plaque: Red or white can make blue. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:2275–2277. doi: 10.1161/ATVBAHA.116.308491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watanabe H, Hattori S, Katsuda S, Nakanishi I, Nagai Y. Human neutrophil elastase: Degradation of basement membrane components and immunolocalization in the tissue. J Biochem. 1990;108:753–759. doi: 10.1093/oxfordjournals.jbchem.a123277. [DOI] [PubMed] [Google Scholar]
- 47.Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. The American journal of physiology. 1998;275:H385–392. doi: 10.1152/ajpheart.1998.275.2.H385. [DOI] [PubMed] [Google Scholar]
- 48.Yang JJ, Kettritz R, Falk RJ, Jennette JC, Gaido ML. Apoptosis of endothelial cells induced by the neutrophil serine proteases proteinase 3 and elastase. The American journal of pathology. 1996;149:1617–1626. [PMC free article] [PubMed] [Google Scholar]
- 49.Dinerman JL, Mehta JL, Saldeen TG, Emerson S, Wallin R, Davda R, Davidson A. Increased neutrophil elastase release in unstable angina pectoris and acute myocardial infarction. Journal of the American College of Cardiology. 1990;15:1559–1563. doi: 10.1016/0735-1097(90)92826-n. [DOI] [PubMed] [Google Scholar]
- 50.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PloS one. 2012;7:e32366. doi: 10.1371/journal.pone.0032366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Annals of the rheumatic diseases. 2015;74:1417–1424. doi: 10.1136/annrheumdis-2013-204837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mangold A, Alias S, Scherz T, Hofbauer T, Jakowitsch J, Panzenbock A, Simon D, Laimer D, Bangert C, Kammerlander A, Mascherbauer J, Winter MP, Distelmaier K, Adlbrecht C, Preissner KT, Lang IM. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in st-elevation acute coronary syndrome are predictors of st-segment resolution and infarct size. Circulation research. 2015;116:1182–1192. doi: 10.1161/CIRCRESAHA.116.304944. [DOI] [PubMed] [Google Scholar]
- 53.Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, Ritis K. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. European heart journal. 2015;36:1405–1414. doi: 10.1093/eurheartj/ehv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jia H, Dai J, Hou J, Xing L, Ma L, Liu H, Xu M, Yao Y, Hu S, Yamamoto E, Lee H, Zhang S, Yu B, Jang IK. Effective anti-thrombotic therapy without stenting: Intravascular optical coherence tomography-based management in plaque erosion (the erosion study) European heart journal. 2016 doi: 10.1093/eurheartj/ehw381. [DOI] [PubMed] [Google Scholar]
- 55.Libby P. Superficial erosion and the precision management of acute coronary syndromes: Not one-size-fits-all. European heart journal. 2017 doi: 10.1093/eurheartj/ehw599. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.