

Abstract
Drug design and discovery remains a popular topic of study to many students interested in visible, real-world applications of the chemical sciences. It is important that laboratory experiments detailing the early stages of drug discovery incorporate both compound design and an exploration of ligand/receptor interactions. Molecular modeling is widely employed in research endeavors seeking to predict the activity of potential compounds prior to synthesis and can therefore be used to illustrate these concepts. The following activity therefore details the use of AutoDock to predict the binding affinity and docked pose of a series of CDK2 inhibitors. Students can then compare their docking output to experimentally determined inhibitory activities and crystal structures. Finally, the AutoDock workflow detailed in this activity can be used in research settings, provided the receptor crystal structure is known.
Keywords: Upper-Division Undergraduate, Graduate Education/Research, Computational Chemistry, Computer-Based Learning, Drugs/Pharmaceuticals, Medicinal Chemistry, Biochemistry, Chemoinformatics, Inquiry-Based/Discovery Learning
Graphical abstract
INTRODUCTION
The topic of drug discovery is popular among undergraduate and graduate students, as the efficacy of novel therapeutics affords a socially visible application of the chemical sciences. As a result, medicinal chemistry courses often yield high enrollment at the university level. According to a recent Chem. Eng. News article,1 representatives from target (pharmaceutical) employers are not necessarily looking for students to have passed a dedicated medicinal chemistry course; rather, these employers are looking for well-rounded scientists with strong backgrounds in organic chemistry, biochemistry, and structural and cellular biology. It is therefore paramount that courses align learning objectives with the fundamental understanding of both the theory and application of various aspects of drug design and discovery.
Computational methods have been developed to predict the binding affinity of proposed ligands prior to synthesis and screening against the intended biochemical target and are currently a seminal tool in the medicinal chemist’s toolbox.2–6 Many specific software packages have been developed, some of which are open source for educational purposes. These software packages afford the opportunity to provide students with a skill that can be used for research applications outside of the traditional laboratory setting. One such software package, AutoDock, was developed at the Scripps Research Institute in the 1990s and has found wide application in both research and instructional settings.7 Although tutorials detailing the general AutoDock workflow exist,8–15 most do not demonstrate how the software can be utilized to reinforce early stage drug discovery methodologies for medicinal chemistry students. The graphical user interface of AutoDock provides the opportunity for students to view proteins in three dimensions, allowing for a greater understanding of tertiary structures. Additionally, students can view surface representations of protein crystal structures to demonstrate that finite ligand binding regions exist. This activity is therefore meant to act as supplementary material for medicinal chemistry courses (either as a homework assignment or a stand-alone laboratory activity) to aid students’ understanding of the mechanisms involved in protein–ligand interactions and provides the unique opportunity for students to explore protein structures in three dimensions. The following experiment therefore details both fragment growth hit modification and the general AutoDock workflow to train students on the use of the software and early stage drug discovery efforts. Students will dock a series of published cyclin-dependent kinase type-2 (CDK2) inhibitors16 and can compare their docking output to both actual inhibitory activities and bound crystallographic orientations. This macromolecular target was selected for utilization in the following study because the inhibitors represent a series that has been optimized according to fragment growth methodology, and the actual inhibitory activities and crystal structures with the target are known.
Overview of Indazole-Based CDK2 Inhibitors
The indazole series of CDK2 inhibitors (Figure 1) was discovered through the high-throughput screening of fragment molecules against the receptor.16 The potency of the compounds against the CDK2 target was measured as the inhibitory concentration resulting in a reduction of activity by 50% compared to an uninhibited control (IC50). Thus, the difference in the relative IC50 values between two compounds suggests that the lower value corresponds to the more potent inhibitor, and a decrease in IC50 means an overall increase in potency. Indazole (1) was crystallized within the CDK2 active site, demonstrating the binding interactions and exposing a binding pocket near the CDK2 active site with exposed backbone carbonyl and amide NH groups oriented in the correct manner for exploitation. Using this information, an aniline amide was conjoined with the indazole fragment hit, and the resulting compound (N-phenyl-1H-indazole-3-carboxamide, 2) was found to have a significant increase in activity (62-fold)16 (Figure 1). This observed increase in activity was attributed to additional H-bonding contacts between the amide NH of 2 and the carbonyl of backbone residue Leu83. The aryl ring of 2 was proposed to be in close proximity to Asp86, and incorporation of a H-bond acceptor was suggested to promote additional contacts to the amide NH of the backbone residue. It was hypothesized that inclusion of a sulfonamide group at the para position could facilitate the desired interaction, and N-(4-sulfamoylphenyl)-1H-indazole-3-carboxamide (3) was synthesized and screened for activity16 (Figure 1). The compound was found to exhibit a slight increase in activity (4.5-fold) compared to that of 2. The binding mode of compound 3 was found to be as predicted in the cocrystal structure of the compound with the receptor, with additional H-bonding to the NH backbone amide bond of Asp86. To determine the necessity of the fused aryl system of the indazole fragment hit, N-phenyl-1H-pyrazole-3-carboxamide (4) was screened against the target and was found to exhibit a large decrease in activity (32-fold)16 in comparison to that of related compound 2 (Figure 1). This demonstrates the stabilizing energy associated with π–π stacking between the indazole aryl ring and the side chain of Phe80. The series of compounds demonstrates fragment growth, as original fragment 1 was astutely modified to accommodate additional H-bonding interactions with backbone residues close in proximity to the inhibitor; thus, a species exhibiting a drastic increase in activity (280-fold) was discovered.
Figure 1.
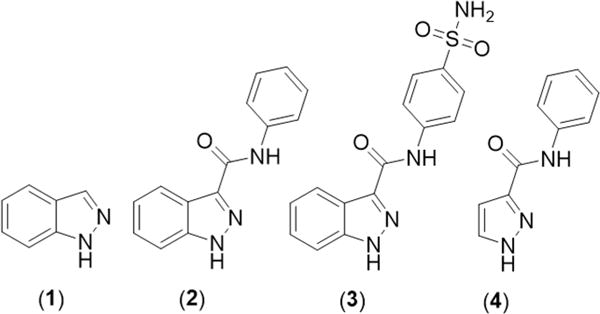
Structures of the CDK2 inhibitors utilized in the docking study. This series of inhibitors was designed by first identifying fragment 1 and employing fragment growth to arrive at CDK2 inhibitor 3.16
The activity described in the supplementary file details the use of AutoDock to simulate the binding poses and interactions of this series of inhibitors against the target. Students will draw compounds 1–4 with molecular graphics software (ChemSketch or ChemDraw) and will import these structures into AutoDock. The biochemical target (CDK2) will then be imported into AutoDock and can be prepared for docking by removing all nonpolar hydrogen atoms and any water molecules crystallized with the protein structure. After manually setting the region of the protein to be utilized for the docking run, students will dock this series of compounds. The docking output can then be compared to the actual crystal structures of compounds 1, 3, and 4, and students can visualize how the growth of the original fragment hit 1 into inhibitor 3 leads to an increase in potency via additional binding interactions between the protein and ligand.
RESULTS AND DISCUSSION
AutoDock uses pregenerated grid maps of the receptor to dock compounds into the binding site. Crystallographic receptor files deposited within the protein databank (PDB) are imported into AutoDock and must be modified prior to docking. Bound ligand, water, buffer, or other fragments must be removed from the receptor as these may interfere with docking runs; additionally, polar hydrogen atoms must be added to each residue, and Gasteiger charges must be assigned to every atom within the receptor. The user then selects the docking region (typically the known or predicted binding pocket) and generates a number of maps corresponding to each atom type found within the ligand to be docked. The ligand is then docked, and the output docking poses and scores are tabulated and can be visualized within the receptor binding pocket.
Compounds 1–4 were docked against the CDK2 receptor, and the results for the most favorable docking pose of each were compared to the conformations revealed in the protein/ inhibitor complex crystal structures (PDB: 2VTA,16 2VTI,16 2VTL16). As shown in Figure 2, the output docking poses were found to closely agree with the published structures. The fundamental H-bonding to Glu81 carbonyl O and Leu83 NH amide present in the published crystal structure of 1 was also predicted by the docking software. Similarly, the output docking pose of 3 and 4 closely matched the interactions observed in the crystal structures. To quantitatively validate the docking output, predicted bond distances were measured with AutoDockTools and compared to distances observed in the crystal structures. All predicted bond distances were found to be within ±0.2 Å of the actual distances, with the exception of the sulfonyl H-bond to the Asp86 backbone amide observed for 3 (difference of 0.9 Å). The docked pose of 3 was observed to have a slightly twisted conformation when compared to the actual crystal structure. This conformation pushes the sulfonamide away from Asp86, resulting in the observed increase in predicted bond distance. Additionally, the sulfonamide is rotated ∼120° in the docking pose, resulting in H-bonding to Asp86 via the opposite sulfonyl O than what is observed within the crystal structure. The source of this discrepancy is currently unknown, although the increase in the number of allowed torsions for compound 3 as compared to compound 2 adds variability to the docking method, complicating the computation. However, the other bond distances are in good agreement with the experimentally determined values. In addition to predicted binding interactions, AutoDock also predicts binding affinities of each inhibitor. A comparison of the predicted and observed binding affinities can also be found it Table 1. The predicted binding affinities supplied by AutoDock for each docking pose were found to closely agree with the actual observed activities. Although the predicted affinities are in the form of the inhibitor–enzyme dissociation constant (Ki) and the actual activities are expressed as inhibitor concentration resulting in a 50% reduction in enzyme activity (IC50), the relative values should correlate.
Figure 2.
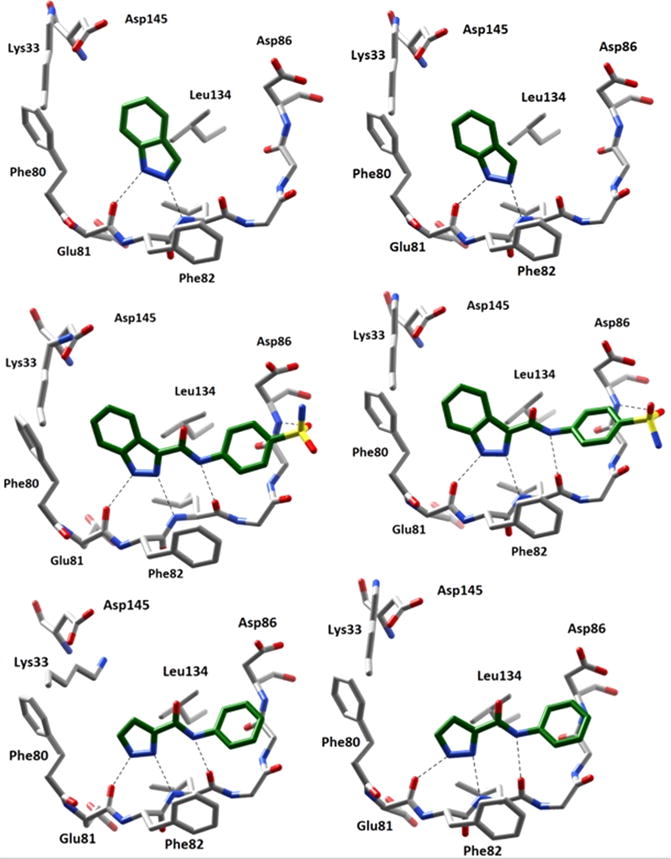
Comparison of crystalline and docked poses (left and right, respectively) of various CDK2 inhibitors. Dashed lines indicate H-bonding interactions between the ligand and receptor. The docking poses were found to closely resemble those suggested by crystal structures. Top: Compound 1 (PDB: 2VTA16). Middle: Compound 3 (PDB: 2VTI16). Bottom: Compound 4 (PDB: 2VTL16).
Table 1.
Comparison of Actual and Predicted Binding Affinities and Bond Distances
| compound | actual affinity (IC50)a | predicted affinity (Ki)a | actual bond distances (Å)b | predicted bond distances (Å) |
|---|---|---|---|---|
| 1 | 185 | 69.0 | NH-carbonyl (2.9) | NH-carbonyl (2.8) |
| N-amide (3.0) | N-amide (2.9) | |||
| 2 | 3 | 0.992 | c | NH-carbonyl (2.9) |
| N-amide (3.1) | ||||
| amide—carbonyl (2.7) | ||||
| 3 | 0.66 | 0.122 | NH-carbonyl (2.8) | NH-carbonyl (2.7) |
| N-amide (3.2) | N-amide (3.2) | |||
| amide—carbonyl (2.9) | amide—carbonyl (2.7) | |||
| SO-amide (3.0) | SO-amide (4.1) | |||
| 4 | 97 | 16.5 | NH-carbonyl (2.5) | NH-carbonyl (2.6) |
| N-amide (3.0) | N-amide (3.1) | |||
| amide—carbonyl (2.7) | amide—carbonyl (2.8) |
Affinities are expressed in units of μM.
Bond distances were determined from published crystal structures; see ref 16.
Compound 2 was not crystallized with the target; thus, actual bond distances are unknown.
Student Outcomes
This activity was used to train students conducting independent research on the general workflow of AutoDock, and students then used the program to guide synthetic efforts for the research group. A total of 15 undergraduate and 5 graduate students have completed the activity to date. We recommend that students are introduced to topics including protein–ligand interactions, fragment-based drug discovery, and fragment modification techniques prior to administration of this activity. Typically, the activity requires about 6 h to complete while working individually. The overall pedagogical goals were not inherently different for students at various education levels, as the activity was primarily used to introduce students to the processes involved in in silico drug discovery efforts, beginning with the assumption that students have no prior experience with this topic. Because the programs used in this activity are all open source, students were able to work outside of the laboratory, and many used their personal computers. All students were able to successfully complete the exercise and obtain docking results similar to those shown in Figure 2 and Table 1.
However, most students encountered a number of problems when learning the AutoDock workflow, although all were based upon issues regarding software errors. For example, if too many commands are issued or if commands are issued in the incorrect order, the program is often unable to produce grid maps or dock compounds. The supplementary content supplied with this paper includes an example laboratory procedure with an introduction discussing fragment growth and the application of computational modeling in drug discovery, a detailed procedure outlining the generation of molecular structures, and subsequent docking with AutoDock, examples of postlab questions, and a troubleshooting section with solutions to the most common errors students have encountered while performing this experiment. Additionally, videos demonstrating this experiment and the AutoDock workflow can be found by following the links embedded in the supplementary content.
Further Considerations for Fragment Growth
While the typical workflow regarding fragment growth methodology is laid out in this activity, the types of molecules proven to be effective as therapeutic agents are not discussed. It may be difficult for students to determine which changes could be applied to a molecule to produce species of increased binding affinity or drug-likeness. Therefore, it would be useful to utilize this paper in conjunction with resources detailing various aspects of drug design, including favorable ligand–protein interactions, bioisosterism, pharmacokinetics and dynamics, and bioavailability. Encourage students to attempt to adhere to the Lipinski Rule-of-Five, as most small-molecule pharmaceuticals are compliant, and all of the properties involved can be determined from widely available software (notably, clogP determination from ChemDraw).
CONCLUSIONS
Drug discovery is an important, visible discipline of the chemical and biological sciences, and ligand/receptor interactions and fragment modification are key principles. It is therefore necessary that students understand these topics in courses or research endeavors concerning therapeutic development. Because molecular modeling is employed to predict the potency of prospective compounds, docking software and its application could be an excellent opportunity for practical instruction. We have therefore developed this activity to demonstrate molecular docking in the context of fragment growth, where known inhibitors are docked and the output is compared with experimental results. All students, at both the undergraduate and graduate levels, were able to apply the docking method for research applications, demonstrating students’ understanding following the completion of the activity. Because all of the software necessary to draw, convert, and dock ligands is open source for educational applications and runs on popular operating systems (MacOS, Windows, and Linux), the processes described herein are applicable to essentially any personal computer. Anyone with interest can therefore participate in drug discovery away from conventional laboratories.
Supplementary Material
Acknowledgments
We acknowledge Northern Illinois University and the National Institutes of Health under Grant 1R15AI113653-01 for supporting this work. Molecular graphics were generated with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). Structural images of chemicals used in both the paper and the Supporting Information were generated with ChemDraw under licensing to the Hagen group.
Footnotes
Supporting Information
The Supporting Information is available on the ACS Publications website at DOI: 10.1021/acs.jchemed.6b00555.
Printable instructions and videos detailing the use of AutoDockTools (PDF, DOCX)
Typical results (PDF, DOCX)
ORCID
Timothy J. Hagen: 0000-0003-1929-9121
Notes
The authors declare the following competing financial interest(s): TJH is a consultant to Mission Life Sciences.
References
- 1.Ainsworth SJ. Universities Tailor Programs to Meet the Pharmaceutical Industry’s Needs. Chem Eng News. 2014;92(21):65–69. [Google Scholar]
- 2.Joseph-McCarthy D, Campbell AJ, Kern G, Moustakas D. Fragment-Based Lead Discovery and Design. J Chem Inf Model. 2014;54(3):693–704. doi: 10.1021/ci400731w. [DOI] [PubMed] [Google Scholar]
- 3.Shang E, Yuan Y, Chen X, Liu Y, Pei J, Lai L. De Novo Design of Multitarget Ligands with an Iterative Fragment-Growing Strategy. J Chem Inf Model. 2014;54(4):1235–1241. doi: 10.1021/ci500021v. [DOI] [PubMed] [Google Scholar]
- 4.Chéron N, Jasty N, Shakhnovich EI. OpenGrowth: An Automated and Rational Algorithm for Finding New Protein Ligands. J Med Chem. 2016;59(9):4171–4188. doi: 10.1021/acs.jmedchem.5b00886. [DOI] [PubMed] [Google Scholar]
- 5.Meng X-Y, Zhang H-X, Mezei M, Cui M. Molecular Docking: A powerful approach for structure-based drug discovery. Curr Comput-Aided Drug Des. 2011;7(2):146–157. doi: 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Macalino S, Gosu V, Hong S, Choi S. Role of computer-aided drug design in modern drug discovery. Arch Pharmacal Res. 2015;38(9):1686–1701. doi: 10.1007/s12272-015-0640-5. [DOI] [PubMed] [Google Scholar]
- 7.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turk JA, Beckmann JD. Resources for an Investigative and Sustainable Undergraduate Medicinal Chemistry Research Program. J Chem Educ. 2013;90(1):137–139. [Google Scholar]
- 9.Sutch BT, Romero RM, Neamati N, Haworth IS. Integrated Teaching of Structure-Based Drug Design and Biopharmaceutics: A Computer-Based Approach. J Chem Educ. 2012;89(1):45–51. [Google Scholar]
- 10.Rodrigues RP, Andrade SF, Mantoani SP, Eifler-Lima VL, Silva VB, Kawano DF. Using Free Computational Resources To Illustrate the Drug Design Process in an Undergraduate Medicinal Chemistry Course. J Chem Educ. 2015;92(5):827–835. [Google Scholar]
- 11.Price GW, Gould PS, Marsh A. Use of Freely Available and Open Source Tools for In Silico Screening in Chemical Biology. J Chem Educ. 2014;91(4):602–604. [Google Scholar]
- 12.Tsai CS. Using Computer Applications and Online Resources To Teach and Learn Pharmaceutical Chemistry. J Chem Educ. 2007;84(12):2019–2023. [Google Scholar]
- 13.Jacob RB, Andersen T, McDougal OM. Accessible High-Throughput Virtual Screening Molecular Docking Software for Students and Educators. PLoS Comput Biol. 2012;8(5):e1002499. doi: 10.1371/journal.pcbi.1002499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mukherjee S, De S, Ghosh Z, Dasgupta S. A docking interaction study of the effect of critical mutations in ribonuclease a on protein-ligand binding. Biochem Mol Biol Educ. 2005;33(5):335–343. doi: 10.1002/bmb.2005.49403305335. [DOI] [PubMed] [Google Scholar]
- 15.Rudnitskaya A, Török B, Török M. Molecular docking of enzyme inhibitors. Biochem Mol Biol Educ. 2010;38(4):261–265. doi: 10.1002/bmb.20392. [DOI] [PubMed] [Google Scholar]
- 16.Wyatt PG, Woodhead AJ, Berdini V, Boulstridge JA, Carr MG, Cross DM, Davis DJ, Devine LA, Early TR, Feltell RE, Lewis EJ, McMenamin RL, Navarro EF, O’Brien MA, O’Reilly M, Reule M, Saxty G, Seavers LCA, Smith D-M, Squires MS, Trewartha G, Walker MT, Woolford AJA. Identification of N-(4-Piperidinyl)-4-(2,6-dichlorobenzoylamino)-1H-pyrazole-3-carboxamide (AT7519), a Novel Cyclin Dependent Kinase Inhibitor Using Fragment-Based X-Ray Crystallography and Structure Based Drug Design. J Med Chem. 2008;51(16):4986–4999. doi: 10.1021/jm800382h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.