

Abstract
Polymer strip films have emerged as a robust platform for poorly water-soluble drug delivery. However, the common conception is that films cannot exceed low drug loadings, mainly due to poor drug stability, slow release, and film brittleness. This study explores the ability to achieve high loadings of poorly water-soluble drug nanoparticles in strip films while retaining good mechanical properties and enhanced dissolution rate. Aqueous suspensions containing up to 30 wt% griseofulvin nanoparticles were prepared via wet stirred media milling and incorporated into hydroxypropyl methylcellulose (HPMC) films. Griseofulvin loading in films was adjusted to be between 9–49 wt% in HPMC-E15 films and 30–73 wt% in HPMC-E4M films by varying the mixing ratio of HPMC solution-to-griseofulvin suspension. All films exhibited good content uniformity and nanoparticle redispersibility up to 50 wt% griseofulvin, while E4M films above 50 wt% griseofulvin had slightly worse content uniformity and poor nanoparticle redispersibility. Increasing drug loading in films generally required more time to achieve 100% release during dissolution, although polymer–drug clusters dispersed from E4M films above 50 wt% griseofulvin, resulting in similar dissolution profiles. While all films exhibited good tensile strength, a significant decrease in percent elongation was observed above 40–50 wt% GF, resulting in brittle films.
Keywords: pharmaceutical films, drug nanoparticles, drug loading, dissolution rate, mechanical properties, biodegradable polymers, physical stability
Graphical Abstract
1. Introduction
With an increasing abundance of new chemical entities in pharmaceutical development classified as poorly water-soluble, there has been an increasing demand for new and innovative drug delivery forms. One such format that has gained recent attention for its robustness and versatility is polymer strip films thanks to faster disintegration and dissolution in the oral cavity, improved patient compliance (Dixit and Puthli, 2009), and ease of continuous manufacturing (Hoffmann et al., 2011). Although best known for ease of incorporation of water-soluble drugs (Dixit and Puthli, 2009), particularly among films currently on the market (Borges et al., 2015a), recent work has explored a variety of means of incorporating poorly water-soluble drug into polymer strip films. One such method, hot melt extrusion, involves melting polymer, plasticizer, and drug together, and pumping the molten mixture through a die (Prodduturi et al., 2005). Another method, solvent casting, involves dissolving the poorly water-soluble drug in an organic solvent, mixing with polymer solution, and removing the solvent upon drying (Kumar et al., 2014; Visser et al., 2015). Various particle engineering techniques have also been employed to improve poorly water-soluble drug dissolution rate for incorporation into strip films. These include wet stirred media milling (WSMM) (Davé et al., 2014; Krull et al., 2017; Krull et al., 2016a; Krull et al., 2016b; Krull et al., 2015; Sievens-Figueroa et al., 2012a; Susarla et al., 2015; Susarla et al., 2013) and high pressure homogenization (Lai et al., 2015; Shen et al., 2013) to reduce drug particle size, as well as liquid antisolvent precipitation (Beck et al., 2013) and melt emulsification (Bhakay et al., 2016) to produce drug nanoparticles via bottom-up approach. However, one perceived limitation shared by all means of incorporating poorly water-soluble drug into films is the existence of an inherent limitation on drug loading (Borges et al., 2015b; Dixit and Puthli, 2009; Hoffmann et al., 2011).
While high drug loading in films is already difficult to achieve due to their small size, the means of incorporating drug into films can impose even greater limitations. Since a traditional strip film dosage only contains a fraction of the total mass of a typical tablet or pill, the amount of drug that can be conceivably loaded into a traditional strip film dosage cannot practically match the dosages available in tablets and capsules. In addition, the standard techniques by which poorly water-soluble drugs are incorporated into strip films further restrict the amount of drug that can be loaded into a film dosage, typically up to 30 wt%. In hot melt extruded films, the dissolution rate enhancement of the drug hinges on the stability of the amorphous solid dispersion during cooling and storage. This becomes increasingly difficult with increasing drug loading, resulting in uncontrolled crystallization if the drug loading is too high (Serajuddin, 1999). Similarly, organic solvent casting relies on the stability of drug in the amorphous form, specifically after the solvent in which the drug is dissolved is driven out of the film during drying. If the drug loading in solvent cast films is too high, they too will experience uncontrolled drug crystallization, resulting in a loss of drug dissolution rate enhancement (Hoffmann et al., 2011). Coupled with a drug delivery platform that is already limited in total mass per unit dosage, these additional restrictions on drug loading have made achieving even moderate dosages in strip films a significant challenge.
Inclusion of engineered drug particles into strip films over the last few years has revealed the potential for higher drug loading in films, although the subject has yet to be fully explored. The majority of work in this area has either focused on establishing a means of particle engineering that is suitable for incorporation into the film platform (Beck et al., 2013; Bhakay et al., 2016; Krull et al., 2015; Prodduturi et al., 2005; Sievens-Figueroa et al., 2012a; Visser et al., 2015) or the impact of critical material attributes (CMAs) on their performance (Krull et al., 2016a; Krull et al., 2016b; Susarla et al., 2015). In most cases, drug loading in films was kept below 20 wt% with none above 30 wt%, presumably to avoid potential confounding influences of high drug loading on other experimental factors under investigation. Woertz and Kleinebudde (2015) claimed to have prepared “acceptable films” with as high as 50 mg of homogenized ibuprofen in a 6 cm2 dosage, but neglected to demonstrate acceptable mechanical properties or dissolution rates for their films. Steiner et al. (2016) demonstrated HPMC films with up to 45 wt% WSMM naproxen nanoparticles that exhibited “good” mechanical properties, above which films became too stiff for practical application, although they did not investigate dissolution rate. Krull et al. (2017) mentioned the potential for higher drug loading (40 wt%) in films containing WSMM griseofulvin nanoparticles prepared with high molecular weight polymer in passing when investigating the impact of polymer molecular weight. However, to this point, no study has been able to demonstrate good mechanical properties and enhanced poorly water-soluble drug dissolution rate of strip films with high (> 40 wt%) drug loading.
The objective of this work is to investigate the effect of high loadings of poorly water-soluble drugs on polymer strip films. Aqueous suspensions of griseofulvin nanoparticles were mixed with hydroxypropyl methylcellulose solution, cast and dried to form griseofulvin nanoparticle-loaded films. These films were then characterized for drug nanoparticle redispersibility, drug content and uniformity, film mechanical properties, drug dissolution rate, moisture content, and stability. Through this work, the ability to achieve high loadings of poorly water-soluble drug nanoparticles in strip films while retaining good mechanical properties and enhanced dissolution rate is explored.
2. Materials and methods
2.1 Materials
Griseofulvin (GF; Letco Medical, Decatur, AL) was used as a model Biopharmaceutics Classification System (BCS) class II drug. Hydroxypropyl methylcellulose (HPMC; Methocel E15 Premium LV, MW ~14 kDa, and Methocel E4M Premium, MW ~88 kDa; The Dow Chemical Company, Midland, MI) served as the film-forming polymer. HPMC-E15LV also served as a nanoparticle stabilizer in suspension, along with the surfactant sodium dodecyl sulfate (SDS; Fisher Scientific, Pittsburgh, PA). Glycerin (Sigma–Aldrich, St. Louis, MO) was used as a film plasticizer. GF particle size reduction was performed by wet stirred media milling (WSMM) according to Section 2.2.1. All other materials were used without further processing.
2.2 Preparation methods
2.2.1 Preparation of GF nanosuspensions
GF nanosuspensions were prepared via wet stirred media milling (WSMM) using a Netzsch mill (Microcer, Fine particle technology LLC, Exton, PA). The 80 ml milling chamber was lined with zirconia. A peristaltic pump was used to recirculate the suspension between a holding tank and the milling chamber. A 200 μm screen was used to confine the yttrium-stabilized zirconia beads (430 μm median size; Zirmil Y, Saint Gobain ZirPro, Mountainside, NJ) inside the milling chamber while allowing passage of the drug suspension.
The methods and stabilizer concentrations used were selected according to previous optimization studies (Bilgili and Afolabi, 2012; Monteiro et al., 2013). Feed suspensions consisted of 10%, 20%, or 30% GF dispersed in a stabilizer solution of 2.5% HPMC-E15LV and 0.5% SDS (all w/w wrt water). The feed suspensions were prepared by adding stabilizer and GF to 200 ml batches of deionized water using an overhead shear mixer (Cat# 14-503, Fisher Scientific, Pittsburgh, PA) running at a fixed speed of 300 rpm. 1 g of SDS was added over 5 min and left stirring for 10 min. Then, 5 g of HPMC-E15LV was added over 15 min and left mixing for 30 min. Finally, respective amounts of GF (20 g, 40 g, and 60 g for 10%, 20%, and 30% suspensions, respectively) were added over 15 min and left mixing for 30 min. The feed suspensions were poured into the holding tank and milled under identical conditions: 50 ml bead loading, 126 ml/min suspension flow rate, and 3200 rpm stirrer (rotor) speed, corresponding to a tip speed of 11.7 m/s. The milling chamber and holding tank were both equipped with a cooling system (model number M1-.25A-11HFX, Advantage Engineering, Greenwood, IN) to keep the suspension below 35 °C in the holding tank. All three suspensions were milled for 120 min.
2.2.2 Preparation of polymer solutions and film precursor suspensions
Formulations for film-forming polymer solutions and film precursor suspensions are listed in Table 1. Polymer and plasticizer concentrations for polymer solutions were selected in such a way that all resulting film precursor formulations would be castable but not spread during drying (target viscosity range of 5000–12,000 cP). This was accomplished using a viscosity matching approach for polymer solution preparation outlined by Krull et al. (2017) for E15 and E4M polymer solutions, represented by 0’s in Table 1. Polymer solutions were prepared by adding the appropriate amounts of HPMC and plasticizer to water at 90 °C, after which the resulting suspension was allowed to cool to room temperature under continuous magnetic stirring to form a polymer solution. Polymer solutions were mixed with GF nanosuspensions using a Thinky ARE-310 planetary centrifugal mixer (Thinky, Laguna Hills, CA) at 2000 rpm for 30 s, followed by 7 min of deaeration at 2200 rpm, to form film precursor suspensions. If bubbles were still present in the precursor suspension after mixing, the precursor was left overnight to settle before casting.
Table 1.
Composition of polymer and film precursor suspensions.
| Formulation | HPMC grade | wt% GF in nanosuspension (wrt water) | wt% GF | wt% HPMC | wt% Glycerin | wt% SDS | wt% water | Polymer-to-nanosuspension mixing ratio (w/w) | Target GF loading in film (mg/cm2) |
|---|---|---|---|---|---|---|---|---|---|
| 15-0.0 | E15 | N/A | N/A | 15.0% | 5.0% | N/A | 80.0% | N/A | N/A |
| 15-1.2 | E15 | 10% | 1.9% | 12.2% | 3.9% | 0.10% | 81.8% | 3.6 | 1.2 |
| 15-2.5 | E15 | 10% | 3.5% | 9.9% | 3.0% | 0.18% | 83.4% | 1.5 | 2.5 |
| 15-3.3 | E15 | 20% | 4.1% | 11.8% | 3.8% | 0.11% | 80.3% | 3.0 | 3.3 |
| 15-5.4 | E15 | 20% | 6.2% | 10.1% | 3.1% | 0.17% | 80.5% | 1.6 | 5.4 |
| 15-6.0 | E15 | 30% | 6.8% | 11.2% | 3.5% | 0.13% | 78.6% | 2.3 | 6.0 |
| 15-8.6 | E15 | 30% | 9.4% | 9.7% | 2.9% | 0.18% | 78.0% | 1.4 | 8.6 |
| 4M-0.0 | E4M | N/A | N/A | 3.3% | 1.1% | N/A | 95.6% | N/A | N/A |
| 4M-1.2 | E4M | 10% | 1.9% | 3.1% | 0.9% | 0.10% | 94.1% | 3.6 | 1.2 |
| 4M-2.5 | E4M | 10% | 3.5% | 2.9% | 0.7% | 0.18% | 92.8% | 1.5 | 2.5 |
| 4M-3.3 | E4M | 20% | 4.1% | 3.0% | 0.8% | 0.11% | 92.0% | 3.0 | 3.3 |
| 4M-5.4 | E4M | 20% | 6.2% | 2.9% | 0.7% | 0.17% | 90.1% | 1.6 | 5.4 |
| 4M-6.0 | E4M | 30% | 6.8% | 3.0% | 0.8% | 0.13% | 89.5% | 2.3 | 6.0 |
Drug loadings in film precursor suspensions were adjusted by varying the drug concentration in WSMM nanosuspensions and polymer-to-nanosuspension mixing ratios (Table 1). This combination of factors was necessary in order to ensure the resulting film precursor suspensions were within the castable viscosity range. E15 formulations were designed to achieve the following target GF loadings in their respective dry films: 10 wt% for 15-1.2, 20 wt% for 15-2.5 and 15-3.3, 35 wt% for 15-5.4 and 15-6.0, and 50 wt% for 15-8.6. Similar drug loadings for 15-2.5/15-3.3 and 15-5.4/15-6.0 were chosen to observe the effects, if any, of adding GF nanoparticles from WSMM suspensions with different drug concentrations. Polymer-to-nanosuspension ratios were kept the same across both E15 and E4M formulations for ease of comparison. 4M–8.6 produced films that were too brittle for testing and were excluded from analysis.
2.2.3 Preparation of GF nanoparticle-laden films
9–10 g of film precursor suspension was manually cast onto a stainless steel substrate with a doctor blade (Elcometer, MI) at a fixed 1000 μm thickness. Wet films were then dried in the convective zone of a Lab-Cast Model TC-71LC Tape Caster (HED International, NJ) in batch mode at 50 °C under laminar air flow for a period of 1 h (Davé et al., 2014). The films were peeled from the substrate after drying and stored in individual sealed plastic bags for characterization.
2.3 Characterization methods
2.3.1 Particle size measurements
GF particle size distributions were measured both in suspension immediately after milling and following redispersion from films utilizing a Coulter LS 13320 Laser Diffraction Particle Size Analyzer (Beckman Coulter, Miami, FL) employing a polarized intensity differential scattering (PIDS) obscuration water optical model. The PIDS was maintained between 40–50% and obscuration was maintained below 8%. Mie scattering theory was used to calculate particle size distributions. Suspension samples for particle size after milling were prepared by removing a 1.2 ml sample from the holding tank of the mill, dispersing the sample into 4 ml of stabilizer solution (2.5% HPMC-E15LV and 0.5% SDS, both w/w wrt water) via pipette, and vortex mixing for 1 min at 1500 rpm.
In order to evaluate the ability to recover GF nanoparticles from films upon delivery, redispersion samples to determine GF particle size after incorporation into dry films were prepared by dispersing three circular film punches ~0.7 cm2 in area into 3 ml of deionized water, followed by 3–5 min of vortex mixing at 1500 rpm. These samples were then subjected to the same particle size analysis as the milled suspension samples.
2.3.2 GF content and uniformity in films
Ten circular samples ~0.7 cm2 in area were removed from random points throughout each film and dissolved in 250 ml of 5.4 mg/ml SDS solution under continuous stirring for a minimum of 3 h. Despite being roughly 1/10th the size of a traditional film dosage, this smaller size was used to help elucidate differences in uniformity between film formulations. The UV absorbance of each sample at the wavelength of maximum absorbance for GF (291 nm) was then measured using a Thermo Scientific Evolution 300 UV-Vis spectrophotometer (Thermo Fisher Scientific Inc., MA). The concentrations of the respective samples were calculated via calibration curve. Content uniformity results are expressed as average GF weight per unit area of film and average weight percentage of GF in the film over ten samples.
2.3.3 Film mechanical properties
Film mechanical properties were measured using a TA-XT Plus Texture Analyzer (Stable Microsystems, UK). Rectangular film strips with dimensions of 50 mm × 15 mm were held between two grips and stretched at a constant speed of 1 mm/s until the point of tensile failure. Tensile strength (TS), yield strength (YS), Young’s modulus (YM), and percent elongation at break (EB) were computed from the resulting stress vs. strain data. Data represents an average of four strips.
2.3.4 Dissolution
Dissolution experiments were performed using a flow-through cell dissolution apparatus (USP IV; Sotax, Switzerland) with cells of 22.6 mm internal diameter and 0.2 μm Pall HT Tuffryn filters (Sievens-Figueroa et al., 2012b). Dissolution samples were automatically measured in-line every two minutes by the same Thermo Scientific Evolution 300 UV-Vis spectrophotometer (Thermo Fisher Scientific Inc., MA, USA) used for determination of drug content and uniformity in films (Section 2.3.2). One circular film sample ~0.7 cm2 in area was horizontally secured within the 5 g of 1 mm glass beads at the bottom of each cell (3 g below the film and 2 g above). 100 ml of 5.4 mg/ml SDS solution was circulated through each cell at a flow rate of 16 ml/min and a constant temperature of 37 ± 0.5 °C. These dissolution protocols were selected for their discriminatory power demonstrated in previous work between films containing nanoparticles, films containing microparticles, and compacted powders (Beck et al., 2013; Bhakay et al., 2016; Sievens-Figueroa et al., 2012b). Dissolution results are reported as percentage GF released as a function of time for an average of six samples from each film. Percentage GF released was calculated based on the expected drug loading in each sample from the drug content assessment performed for each film formulation (Section 2.3.2), taking into consideration the weight of each individual film sample.
2.3.5 Thermogravimetric analysis (TGA)
TGA was performed using a TGA/DSC1/SF STARe system (Mettler Toledo, Inc., Columbus, OH, USA). A ~4 mg film sample was placed in a ceramic crucible and heated under nitrogen flow from 25 °C to 150 °C at a constant rate of 10 °C/min, held at 150 °C for 15 min, heated to 250 °C at a rate of 10 °C/min, and finally cooled back to 25 °C at a rate of −10 °C/min.
2.3.6 X-ray diffraction (XRD)
The crystallinity GF in dried GF suspensions before and after milling as well as select films was assessed using X-ray diffraction (XRD; PANalytical, Westborough, MA, USA), using Cu Kα radiation (λ=1.5406 Å). The samples were scanned for 2θ ranging from 5° to 40° at a scan rate of 0.165 s−1.
2.3.7 Long-term stability of films
Films designated for long-term stability study were stored in a MicroClimate benchtop climate chamber (Cincinnati Sub-Zero, Cincinnati, OH) at 40 °C and 75% relative humidity (RH). Following 3 and 6 months of storage under these conditions, films were subjected to redispersion and dissolution tests as outlined in Sections 2.3.1 and 2.3.4, respectively.
2.3.8 Statistical analysis
Basic calculations were performed using Microsoft Excel® (Microsoft Office 2010). Analysis of variance (ANOVA) and Tukey pairwise comparisons (two-tailed t-tests) were performed using Minitab® (Minitab 17.3.1). Results for viscosity, mechanical properties, and dissolution profiles are expressed as mean ± SD (standard deviation) while content uniformity results are expressed as mean with RSD% (relative standard deviation). Dissolution profiles were compared using similarity and difference factors (Boateng et al., 2012; Costa and Lobo, 2001).
3. Results and discussion
3.1 Drug particle size after milling and redispersed from films
Since poorly water-soluble drug particles naturally exhibit slow dissolution rates, it is critical that the WSMM nanoparticles do not aggregate upon incorporation into or delivery from films as their enhanced dissolution rate would be lost. In order to investigate the ability of the film format to stabilize and deliver poorly water soluble drug nanoparticles, even with high drug loading, films were tested for nanoparticle redispersibility in water, and the size distributions of the GF particles redispersed from films were compared to those of the nanosuspensions from which the GF nanoparticles were originally taken. Redispersion tests were performed on all films on the day of preparation, as well as after 3 and 6 months of storage under stress conditions (40 °C, 75% RH) to assess long-term stability (stability results discussed in Section 3.7). Particle size distribution results, reported as 10%, 50% (median), and 90% passing sizes (d10, d50, and d90, respectively), are shown in Fig. 1, organized by the WSMM suspension from which the films were prepared. Exact median GF particle sizes are listed in Supplementary Material.
Fig. 1.
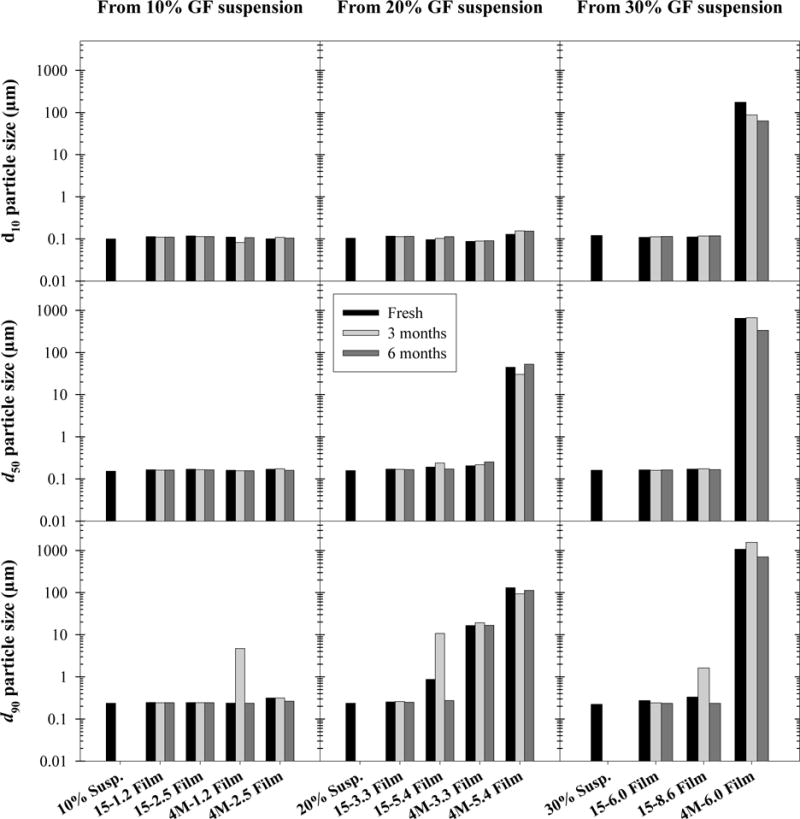
d10, d50 and d90 of GF nanoparticles in suspension and redispersed from HPMC-E15 and E4M films of varying API loading after 0, 3, and 6 months’ storage at 40 °C and 75% RH.
The size of the original GF nanoparticles (d50 between 150–165 nm) was recovered from all HPMC-E15 films with all d50 values < 200 nm. The same nanoparticle redispersibility was observed in HPMC-E4M films up to about 50 wt% drug loading (4M-2.5), above which original nanoparticle size could no longer be immediately recovered upon redispersion in water. In fact, coarse drug–polymer agglomerates or clusters were visible by the naked eye during redispersion tests for 4M-3.3, 44-5.4, and 4M-6.0 films (d50 values of 205 nm, 44.7 μm and 652 μm, respectively), appearing as undissolved film clusters after the film had physically broken apart during vortex mixing, rather than appearing to grow from suspended particles. However, as seen in the cross sectional SEM images (see Supplementary Material), the GF nanoparticles were finely dispersed and stabilized within all films, including E4M films and films with high drug loading. Therefore, it is likely that the existence and slow dissolution of these drug–polymer agglomerates was due to interactions between the GF nanoparticles and HPMC-E4M during redispersion, rather than GF nanoparticle aggregation during film formation. Although by a different mechanism (flocculation), similar behavior was reported by Wong and Bodmeier (1996) in ethyl cellulose dispersions with HPMC. They observed flocculation at lower HPMC concentrations in dispersions with higher molecular weight HPMC and higher poorly water-soluble solids content (ethyl cellulose), similar to how drug–polymer agglomerates were only observed upon redispersion from high molecular weight HPMC films (E4M) above a certain GF loading (> 50 wt%) in this study. That said, sub-200 nm GF particles were successfully recovered from all films with GF loadings as high as 50 wt%, demonstrating the ability to physically stabilize and deliver poorly water-soluble drug nanoparticles in polymer films with high drug loading.
3.2 GF content and uniformity in films
Content uniformity tests were performed on all films in order to assess their drug content and uniformity. These results, described here and shown in Supplementary Material, are expressed in terms of film thickness, mass of GF per unit area, and GF loading by wt%. In spite of using a small sample size (roughly 1/10th the size of a typical dose) to better elucidate potential differences between films, all HPMC-E15 films exhibited very good uniformity in terms of wt% GF (< 1% RSD) and mg GF/cm2 (most ~3% RSD). HPMC-E4M films, on the other hand, exhibited greater variability for both (~2% RSD and ~5% RSD, respectively), due in part to their decreased thickness. These results demonstrate the ability to achieve good content uniformity in films with high drug nanoparticle loading. Although all films were cast to be 1000 μm thick before drying, precursor suspensions had varying water concentrations (Table 1), the majority of which was lost upon drying. These differences resulted in thinner films for precursors with higher starting water concentrations, as they had less solids content (Krull et al., 2017). Consequently, although E15 and E4M precursors of the same number (e.g. 15-1.2 and 4M-1.2) contained roughly the same concentration of GF nanoparticles before drying, greater mass loss during drying drove up the percentage of GF remaining in the E4M films (despite still having the same loading by mass per unit area).
3.3 Film mechanical properties
TS, YS, YM, and EB were measured for each film, with the exception of 4M-6.0 which was too brittle for texture analysis. As can be seen in Table 2, films generally exhibited TS between 35–45 MPa and YS between 30–40 MPa, demonstrating good mechanical strength regardless of GF nanoparticle loading. There was no observable trend in YM for E15 films with increasing GF nanoparticle loading, while YM appeared to increase significantly in E4M films prepared by suspensions with lower polymer-to-nanosuspension mixing ratios (Table 1), indicating increasing stiffness. EB decreased dramatically with increasing drug nanoparticle loading above ~40 wt% GF. It is no surprise that one of the greatest challenges posed by high drug loading in films is simultaneously maintaining good mechanical properties for manufacture and patient compliance. Typically, film-forming polymer mass must be reduced to account for higher drug loading, reducing the integrity of the resulting film matrix. Although there are currently no fixed guidelines regarding acceptable mechanical properties of pharmaceutical films (Hoffmann et al., 2011), the brittleness of high drug loading films which exhibited EB < 5% was readily apparent as they were being handled. Steiner et al. (2016) also observed brittleness in poorly water-soluble drug nanoparticle-loaded films with similarly high drug loading using a lower molecular weight grade of HPMC as film-forming polymer. Although it may be possible to push the maximum drug particle loading in films above 45–50 wt% while maintaining acceptable film mechanical properties with more thorough formulation development, such as modulation of plasticizer content (Krull et al., 2016b), these results suggest that film brittleness is a major hurdle to overcome to achieve higher drug particle loadings in strip films for practical use.
Table 2.
Mechanical properties of films with different GF loadings.
| Formulation* | Tensile strength (MPa) | Yield strength (MPa) | Young’s modulus (GPa) | Elongation at break (%) |
|---|---|---|---|---|
| 15-1.2 | 38.6 ± 0.8bc | 28.9 ± 1.7c | 2.38 ± 0.05d | 19.6 ± 1.3a |
| 15-2.5 | 40.7 ± 0.1abc | 30.8 ± 1.6bc | 2.61 ± 0.16d | 18.8 ± 1.6ab |
| 15-3.3 | 38.0 ± 0.3bc | 32.3 ± 3.3bc | 2.27 ± 0.21d | 18.5 ± 3.6ab |
| 15-5.4 | 34.5 ± 2.7c | 31.0 ± 1.2c | 1.99 ± 0.40d | 13.9 ± 4.3b |
| 15-6.0 | 41.7 ± 1.9ab | 41.7 ± 1.9a | 2.45 ± 0.24d | 6.1 ± 0.7c |
| 15-8.6 | 37.7 ± 7.1abc | 37.7 ± 7.1abc | 2.67 ± 0.12cd | 2.4 ± 0.7c |
| 4M-1.2 | 46.0 ± 3.0a | 35.0 ± 1.2abc | 3.63 ± 0.62bc | 13.7 ± 1.2b |
| 4M-2.5 | 38.3 ± 4.5bc | 38.3 ± 4.5ab | 4.80 ± 0.48a | 1.4 ± 0.1c |
| 4M-3.3 | 38.0 ± 2.1bc | 38.0 ± 2.1ab | 2.43 ± 0.59d | 4.2 ± 1.1c |
| 4M-5.4 | 42.2 ± 4.6abc | 36.1 ± 8.0abc | 4.04 ± 0.42ab | 2.5 ± 0.3c |
4M-6.0 not listed as it was impossible to cut strips due to film brittleness
Values in each column that do not share a superscript are statistically different
When comparing directly between HPMC molecular weights for films with the same drug loading by mass (e.g. 15-1.2 and 4M-1.2), the observed trends are in line with those presented in previous work which also incorporated a polymer solution viscosity matching approach (Krull et al., 2017). There was no statistical difference between the TS or YS of E15 and E4M films for most drug loadings (with the exception of TS for 1.9 films). However, YM was generally lower and EB was generally higher for E15 than for E4M, suggesting that E4M films were more brittle than E15 films. As in the previous study, it is worth noting that E4M polymer solution was prepared using a lower concentration of HPMC to match the viscosity of E15 polymer solution to ensure the precursor was castable. As a result, the lower HPMC concentration (and, consequently, higher GF nanoparticle loading by wt%) in E4M films likely confounded the influence of polymer MW on film mechanical properties.
3.4 Dissolution
Dissolution curves for fresh films are shown in Fig. 2, separated by HPMC molecular weight grade. The extent of drug release relative to the estimated drug content established in Section 3.2 decreased slightly with increasing GF loading, likely due to increasing deviation from sink conditions. There was a clear increasing trend in total release time for both HPMC-E15 and HPMC-E4M films with increasing drug nanoparticle loading up to ~50 wt% GF. Additional time required to achieve 100% drug release may be partially attributed to other factors, including increasing film thickness (Krull et al., 2016a) and varying polymer concentration (Krull et al., 2017) with increasing drug loading (Table 1). Above 50 wt% GF, however, the dissolution profiles for E4M films 4M-3.3, 4M-5.4, and 4M-6.0 exhibited statistically similar release rates according to similarity and difference factors (Boateng et al., 2012; Costa and Lobo, 2001) (see Supplementary material). These films also exhibited poor GF nanoparticle redispersibility (Section 3.1) and polymer–drug agglomerates were visible upon film disintegration during dissolution testing similar to those observed in redispersion tests. It appears likely that, above 50 wt% drug nanoparticle loading, the dissolution of these polymer–drug agglomerates became the controlling drug release mechanism, particularly for high molecular weight polymers, resulting in similar release rates for all E4M films with high drug loading. When directly comparing drug release rates of E15 and E4M films with similar drug loading by mass (e.g. 15-1.2 and 4M-1.2), the higher molecular weight E4M films appear to release more slowly than their E15 counterparts for all drug loadings, in line with the findings of Krull et al. (2017).
Fig. 2.
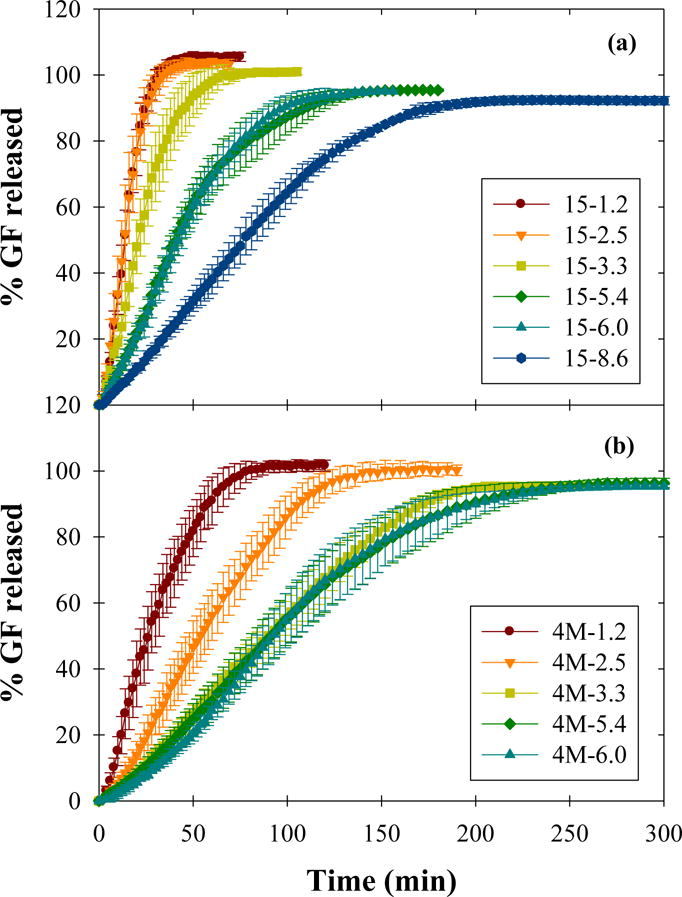
Dissolution profiles of HPMC (a) E15 and (b) E4M films with various GF nanoparticle loadings.
3.5 Thermogravimetric analysis (TGA)
Results from TGA are described here and TGA curves for E15 and E4M films can be found in Supplementary Material. All films exhibited significant mass loss up to 110 °C, most likely due to the loss of water. E15 films exhibited the greatest such loss on average (~6.5 wt%) compared to E4M films (~4.0 wt%) despite E15 precursor suspensions having less moisture content than E4M precursor suspensions when cast. In addition, films with higher GF nanoparticle loading generally exhibited less weight loss during this time, suggesting lower moisture content. This may be due to the fact that films with higher GF concentrations exhibit greater overall hydrophobicity, driving out additional moisture during drying, resulting in films with less moisture content. Films with higher GF nanoparticle loading also contained lower concentrations of hydrophilic HPMC, which is expected to retain some moisture even after the film dries, likely resulting in lower moisture content. The significant weight loss after holding at 150 °C for 15 min was most likely due to the loss of the plasticizer glycerin. Relative weight loss between film formulations during this time was generally in line with the plasticizer concentrations in the precursor suspensions (Table 1).
3.6 X-ray diffraction (XRD)
In order to monitor the crystallinity of GF throughout the WSMM and film manufacture processes, XRD diffractograms were measured for as-received GF and HPMC-E15 powder, dried 30% GF suspensions before and after milling, as well as 15-6.0 and 15-8.6 films (Fig. 3). As can be seen in the diffractograms, the characteristic peaks of crystalline GF were preserved both in suspension before and after milling as well as in the film samples in the absence of broad halos. These results suggest the stability of crystalline GF throughout the milling and film manufacture processes, even in high drug loading formulations.
Fig. 3.
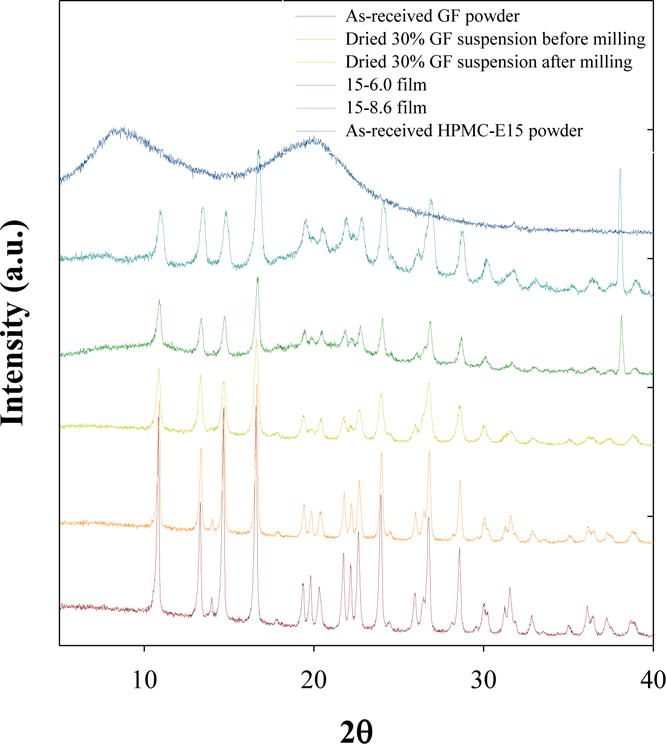
XRD diffractograms of as-received GF and HPMC-E15 powder, dried 30% GF suspensions before and after milling, 15-6.0 and 15-8.6 films.
3.7 Long-term stability of films
Redispersion and dissolution tests were performed for all film formulations over the course of six months’ storage at 40 °C and 75% RH to simulate the effects of long-term storage on the ability of the film format to preserve drug nanoparticle size and enhanced drug dissolution rate. First, the effect of long-term storage on the ability to recover GF nanoparticles from films via redispersion in water was assessed (Fig. 1). Aside from order of magnitude increases in the d90 of a select three films for 3 month samples only (4M-1.2, 15-5.4, 15-6.0), there was no observable impact of long-term storage on GF nanoparticle redispersibility from films. The ability to recover GF nanoparticles after 6 months of storage under stress conditions was demonstrated for all E15 films and E4M films with 50 wt% drug loading or less. Interestingly, long-term storage did not appear to influence the inability to immediately recover GF nanoparticles from E4M films with higher drug loading, as the particle size statistics for such films did not appear to deviate significantly after storage.
Dissolution tests were also performed on films following 0, 3, and 6 months’ storage at 40 °C and 75% RH in order to investigate the effect of long-term storage on the drug dissolution rate from films (Fig. 4 and 5). Despite slight variations in extent of release due to differences in drug loading between film samples according to Section 3.2, four of six E15 film formulations exhibited similar rates of drug release after 3 and 6 months’ storage according to similarity and difference factors (see Supplementary material) while the remaining two formulations, 15-2.5 and 15-6.0, were not drastically different. This suggests that drug release rates from E15 films are relatively stable, even after long-term storage, for a wide range of drug nanoparticle loadings. On the other hand, while E4M films exhibited similar release profiles after 3 months’ storage, they appeared to release drug more quickly after 6 months of storage, particularly from high drug loading films for which polymer–drug agglomerates were observed during initial redispersion and dissolution tests. It is possible that prolonged exposure to high humidity weakened the interaction between HPMC-E4M and GF nanoparticles within the film, resulting in faster dissolution of the polymer–drug agglomerates. However, as seen in redispersion tests from the same films, the agglomerates do not dissolve immediately, requiring a short induction time (~10–20 min) before dissolving.
Fig. 4.
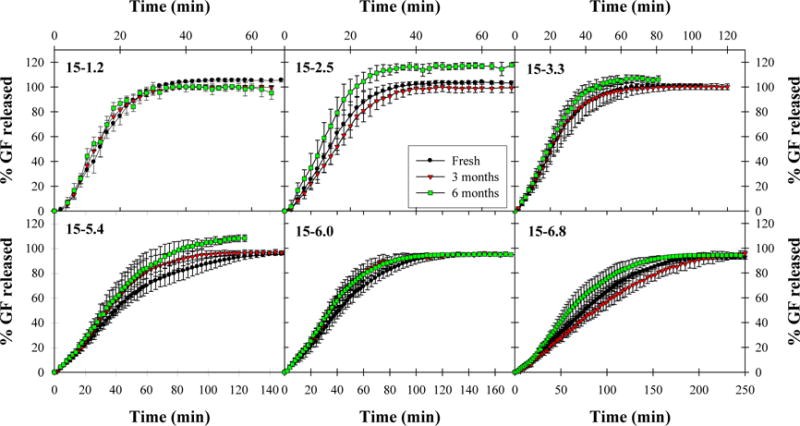
Comparison of dissolution profiles for E15 films immediately after film preparation, after 3 months of storage at 40 °C, 75% RH, and after 6 months of storage at 40 °C, 75% RH. Values are mean ± SD, n = 6.
Fig. 5.
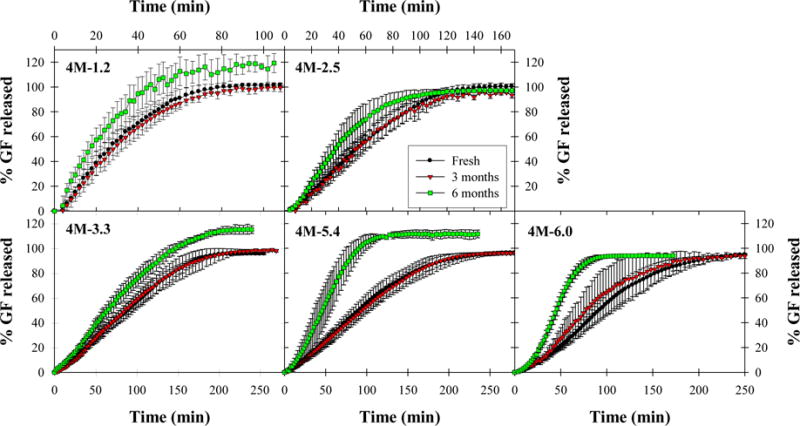
Comparison of dissolution profiles for E4M films immediately after film preparation, after 3 months of storage at 40 °C, 75% RH, and after 6 months of storage at 40 °C, 75% RH. Values are mean ± SD, n = 6.
4. Conclusions
The objective of this work was to investigate the capability of producing strip film dosages containing high loadings of poorly water-soluble drug nanoparticles with good mechanical properties that retain the enhanced dissolution rate of the drug nanoparticles. While drug loadings of 50 wt% and 73 wt% were achieved in HPMC-E15 and E4M films, respectively, films with drug loadings above 40–50 wt% were unacceptably brittle, imposing a practical drug loading limitation for films with good mechanical properties. In addition, undissolved polymer– drug agglomerates were observed upon redispersion and dissolution from E4M films with drug loadings above 50 wt%, severely limiting the ability to recover the embedded GF nanoparticles and retain their enhanced dissolution upon delivery. In spite of these challenges, all film formulations exhibited good content uniformity (6% RSD or less) and were able to preserve the size, morphology, and crystallinity of the GF nanoparticles. These results suggest that the greatest barriers to producing pharmaceutical films with high loadings of poorly water-soluble drug nanoparticles are overcoming poor film mechanical properties and ensuring recovery of the embedded drug nanoparticles, both of which can be conceivably overcome by further formulation development.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Science Foundation (NSF) in part through the ERC (EEC-0540855) award and from the National Institute of Health (NIH) NIH-U01 in part through award U01FD005521. We also thank Dr. R. Susarla for her valuable input on experimental design as well as N. Malik and P. Orbe for their assistance with preliminary studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beck C, et al. Effects of stabilizers on particle redispersion and dissolution from polymer strip films containing liquid antisolvent precipitated griseofulvin particles. Powder Technol. 2013;236:37–51. [Google Scholar]
- Bhakay A, et al. Incorporation of fenofibrate nanoparticles prepared by melt emulsification into polymeric films. J Pharm Innov. 2016;11(1):53–63. [Google Scholar]
- Bilgili E, Afolabi A. A combined microhydrodynamics-polymer adsorption analysis for elucidation of the roles of stabilizers in wet stirred media milling. Int J Pharm. 2012;439(1–2):193–206. doi: 10.1016/j.ijpharm.2012.09.040. [DOI] [PubMed] [Google Scholar]
- Boateng JS, et al. Comparison of the in vitro release characteristics of mucosal freeze-dried wafers and solvent-cast films containing an insoluble drug. Drug Dev Ind Pharm. 2012;38(1):47–54. doi: 10.3109/03639045.2011.590496. [DOI] [PubMed] [Google Scholar]
- Borges AF, et al. Oral films: Current status and future perspectives II — Intellectual property, technologies and market needs. J Control Release. 2015a;206:108–121. doi: 10.1016/j.jconrel.2015.03.012. [DOI] [PubMed] [Google Scholar]
- Borges AF, et al. Oral films: Current status and future perspectives: I — Galenical development and quality attributes. J Control Release. 2015b;206:1–19. doi: 10.1016/j.jconrel.2015.03.006. [DOI] [PubMed] [Google Scholar]
- Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13(2):123–133. doi: 10.1016/s0928-0987(01)00095-1. [DOI] [PubMed] [Google Scholar]
- Davé RN, et al. System and method for fabrication of uniform polymer films containing nano and micro particles via continuous drying process. PCT Application No PCT/US14/30506 Filed March. 2014;17:2014. [Google Scholar]
- Dixit RP, Puthli SP. Oral strip technology: Overview and future potential. J Control Release. 2009;139(2):94–107. doi: 10.1016/j.jconrel.2009.06.014. [DOI] [PubMed] [Google Scholar]
- Hoffmann EM, et al. Advances in orodispersible films for drug delivery. Expert Opin Drug Deliv. 2011;8(3):299–316. doi: 10.1517/17425247.2011.553217. [DOI] [PubMed] [Google Scholar]
- Krull SM, et al. Critical Material Attributes of Strip Films Loaded With Poorly Water-Soluble Drug Nanoparticles: II. Impact of Polymer Molecular Weight. J Pharm Sci. 2017;106(2):619–628. doi: 10.1016/j.xphs.2016.10.009. [DOI] [PubMed] [Google Scholar]
- Krull SM, et al. Preparation and characterization of fast dissolving pullulan films containing BCS class II drug nanoparticles for bioavailability enhancement. Drug Dev Ind Pharm. 2016a;42(7):1073–1085. doi: 10.3109/03639045.2015.1107094. [DOI] [PubMed] [Google Scholar]
- Krull SM, et al. Critical material attributes (CMAs) of strip films loaded with poorly water-soluble drug nanoparticles: I. Impact of plasticizer on film properties and dissolution. Eur J Pharm Sci. 2016b;92:146–155. doi: 10.1016/j.ejps.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krull SM, et al. Polymer strip films as a robust, surfactant-free platform for delivery of BCS Class II drug nanoparticles. Int J Pharm. 2015;489(1–2):45–57. doi: 10.1016/j.ijpharm.2015.04.034. [DOI] [PubMed] [Google Scholar]
- Kumar GP, et al. Polyvinylpyrrolidone oral films of enrofloxacin: Film characterization and drug release. Int J Pharm. 2014;471(1–2):146–152. doi: 10.1016/j.ijpharm.2014.05.033. [DOI] [PubMed] [Google Scholar]
- Lai F, et al. Maltodextrin fast dissolving films for quercetin nanocrystal delivery. A feasibility study Carbohydr Polym. 2015;121:217–223. doi: 10.1016/j.carbpol.2014.11.070. [DOI] [PubMed] [Google Scholar]
- Monteiro A, et al. Continuous production of drug nanoparticle suspensions via wet stirred media milling: A fresh look at the Rehbinder effect. Drug Dev Ind Pharm. 2013;39(2):266–283. doi: 10.3109/03639045.2012.676048. [DOI] [PubMed] [Google Scholar]
- Prodduturi S, et al. Solid‐state stability and characterization of hot‐melt extruded poly (ethylene oxide) films. J Pharm Sci. 2005;94(10):2232–2245. doi: 10.1002/jps.20437. [DOI] [PubMed] [Google Scholar]
- Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- Shen Bd, et al. Development and characterization of an orodispersible film containing drug nanoparticles. Eur J Pharm Biopharm. 2013;85(3, Part B):1348–1356. doi: 10.1016/j.ejpb.2013.09.019. [DOI] [PubMed] [Google Scholar]
- Sievens-Figueroa L, et al. Preparation and characterization of hydroxypropyl methyl cellulose films containing stable BCS Class II drug nanoparticles for pharmaceutical applications. Int J Pharm. 2012a;423(2):496–508. doi: 10.1016/j.ijpharm.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Sievens-Figueroa L, et al. Using USP I and USP IV for discriminating dissolution rates of nano- and microparticle-loaded pharmaceutical strip-films. AAPS PharmSciTech. 2012b;13(4):1473–1482. doi: 10.1208/s12249-012-9875-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner D, et al. Efficient production of nanoparticle-loaded orodispersible films by process integration in a stirred media mill. Int J Pharm. 2016;511(2):804–813. doi: 10.1016/j.ijpharm.2016.07.058. [DOI] [PubMed] [Google Scholar]
- Susarla R, et al. Novel use of superdisintegrants as viscosity enhancing agents in biocompatible polymer films containing griseofulvin nanoparticles. Powder Technol. 2015;285:25–33. [Google Scholar]
- Susarla R, et al. Fast drying of biocompatible polymer films loaded with poorly water-soluble drug nano-particles via low temperature forced convection. Int J Pharm. 2013;455(1–2):93–103. doi: 10.1016/j.ijpharm.2013.07.051. [DOI] [PubMed] [Google Scholar]
- Visser JC, et al. Orodispersible films in individualized pharmacotherapy: The development of a formulation for pharmacy preparations. Int J Pharm. 2015;478(1):155–163. doi: 10.1016/j.ijpharm.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Woertz C, Kleinebudde P. Development of orodispersible polymer films containing poorly water soluble active pharmaceutical ingredients with focus on different drug loadings and storage stability. Int J Pharm. 2015;493(1–2):134–145. doi: 10.1016/j.ijpharm.2015.07.032. [DOI] [PubMed] [Google Scholar]
- Wong D, Bodmeier R. Flocculation of an aqueous colloidal ethyl cellulose dispersion (Aquacoat®) with a water-soluble polymer, hydroxypropyl methylcellulose. Eur J Pharm Biopharm. 1996;42(1):12–15. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.