

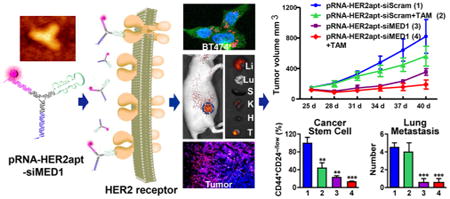
Abstract
Most breast cancers express estrogen receptor (ER) α, and the antiestrogen drug tamoxifen has been widely used for their treatment. Unfortunately, up to half of all ERα-positive tumors have intrinsic or acquired endocrine therapy resistance. Our recent studies revealed that the ER coactivator Mediator Subunit 1 (MED1) plays a critical role in tamoxifen resistance through cross-talk with HER2. Herein, we assembled a three-way junction (3-WJ) pRNA–HER2apt–siMED1 nanoparticle to target HER2-overexpressing human breast cancer via an HER2 RNA aptamer to silence MED1 expression. We found that these ultracompact RNA nanoparticles are very stable under RNase A, serum, and 8 M urea conditions. These nanoparticles specifically bound to HER2-overexpressing breast cancer cells, efficiently depleted MED1 expression, and significantly decreased ERα-mediated gene transcription, whereas point mutations of the HER2 RNA aptamer on these nanoparticles abolished such functions. The RNA nanoparticles not only reduced the growth, metastasis, and mammosphere formation of the HER2-overexpressing breast cancer cells but also sensitized them to tamoxifen treatment. These biosafe nanoparticles efficiently targeted and penetrated into HER2-overexpressing tumors after systemic administration in orthotopic xenograft mouse models. In addition to their ability to greatly inhibit tumor growth and metastasis, these nanoparticles also led to a dramatic reduction in the stem cell content of breast tumors when combined with tamoxifen treatment in vivo. Overall, we have generated multifunctional RNA nanoparticles that specifically targeted HER2-overexpressing human breast cancer, silenced MED1, and overcame tamoxifen resistance.
Keywords: MED1, HER2 RNA aptamer, pRNA of phi29 DNA packaging motor, tamoxifen resistance, breast cancer
Graphical abstract
Breast cancer has continually been one of the leading malignant cancers threatening global human health. Approximately 75% of patients express estrogen receptor alpha (ERα), which belongs to the steroid nuclear receptor family and has been well documented in promoting breast carcinogenesis in an estrogen-dependent manner.1–3 Hence, tamoxifen, an antagonist of ERα, has been widely used as a first-line adjuvant endocrine therapy to treat both premenopausal and postmenopausal ERα-positive breast cancer patients.4 Unfortunately, about half of these patients have intrinsic or acquired tamoxifen resistance, which significantly limits the clinical outcome of tamoxifen treatment.5 Recent research has provided deeper insight into the molecular mechanisms involved in tamoxifen resistance.6 Most notably, overexpression of HER2 has been shown to be one of the major mechanisms associated with tamoxifen resistance by enhancing ERα functions.7–9 Moreover, recent studies have further revealed the key role of the crosstalk between HER2 and ERα transcriptional coactivator Mediator Subunit 1 (MED1) in tamoxifen resistance.10,11
As a transcriptional coactivator of ERα, MED1 is associated with a subpopulation of the TRAP/mediator complex and directly interacts with ERα via its two classical LxxLL motifs to facilitate target gene expression through the recruitment of RNA polymerase II and general transcriptional machinery.12–14 Interestingly, the MED1 gene is located in close proximity to the HER2 gene within the HER2 amplicon on chromosome 17q12 and coamplifies with HER2 in almost all cases.10,15 Our recent studies have further confirmed that MED1 protein level highly correlated with HER2 status in human breast cancer by tissue microarray analyses.11 Importantly, MED1 can be phosphorylated and activated by an HER2 signaling pathway, while knockdown of MED1 by small interference RNA (siRNA) significantly sensitized HER2-overexpressing ERα positive breast cancer cells to tamoxifen treatment.11 Significantly, clinical data further indicated that MED1 overexpression strongly correlates with endocrine therapy resistance in ERα-positive breast cancer patients.16,17
Since the discovery and establishment of gene silencing by RNA interference at the turn of the century,18,19 many siRNA-based drugs have entered clinical trials with a number of them recently showing promising outcomes.20–22 However, developing the siRNA delivery systems with strong stability, specific tumor targeting ability, and low toxicity remains a major challenge. Recently, three-way junction (3-WJ) pRNA nanoparticles, which are derived from the RNA of bacteriophage phi29 DNA packaging motor, show promise as a highly desirable in vivo siRNA delivery system.23,24 Using RNA nanotechnology, Phi29 pRNA has been utilized to “bottom-up” assemble a variety of dimers, trimers, hexamers, tetramers, and higher order oligomers with controllable stoichiometry. The extending arms of pRNA structures could be intelligently replaced with siRNAs, miRNAs, riboswitches, and RNA aptamers and conjugated with fluorescent probes or other moieties to construct multifunctional pRNA nanoparticles.24 Notably, the 2′-fluoro modification of RNA bases rendered the pRNA nanoparticles ultrastable and resistant to RNase exposure.25 Moreover, after systemic administration, the pRNA nanoparticles demonstrated a favorable pharmacokinetic profile with a highly prolonged half-life and excellent biosafety in mice.26 Importantly, these pRNA nanoparticles have been applied to specifically target a variety of human tumors and tested for cancer therapy.24,27–30
In the current study, we exploited the 3-WJ pRNA structure to construct AlexaFluor647-labeled multifunctional pRNA–HER2apt–siMED1 nanoparticles bearing an HER2-targeting RNA aptamer and two different MED1 siRNAs to silence MED1 expression in HER2-overexpressing ERα-positive breast cancer cells. The pRNA–HER2apt–siMED1 nanoparticles exhibited a high Tm value and excellent stability upon exposure to 8 M urea, RNase A, and serum. Importantly, these nanoparticles could specifically target HER2-overexpressing breast cancer to silence MED1 expression both in vitro and in orthotopic xenograft mouse models. We further examined the antibreast cancer activities of these pRNA–HER2apt–siMED1 nanoparticles and dissected the underlying molecular mechanisms. Overall, our work has generated highly promising pRNA–HER2apt–siMED1 nanoparticles that could specifically deliver MED1 siRNAs to HER2-overexpressing human breast cancer and overcome tamoxifen resistance.
Results and Discussion
Generation and Characterization of 3-WJ pRNA–HER2apt–siMED1 Nanoparticles
Utilizing the three-way junction (3-WJ) of Phi29 pRNA as the core unit, we constructed a self-assembled double-strand pRNA nanoparticle bearing an HER2-targeting RNA aptamer and two different MED1 siRNAs for in vitro and in vivo delivery (termed pRNA–HER2apt–siMED1, Figure 1A). In the search for an HER2 aptamer suitable for delivering pRNA–HER2apt–siMED1 into HER2-overexpressing breast cancer cells, we tested several published HER2 RNA aptamers31,32 (Figure S1A,B) and found that the B3 aptamer could target HER2-overexpressing BT474 cells and knockdown MED1 expression with the highest efficiency (Figure S1C).
Figure 1.
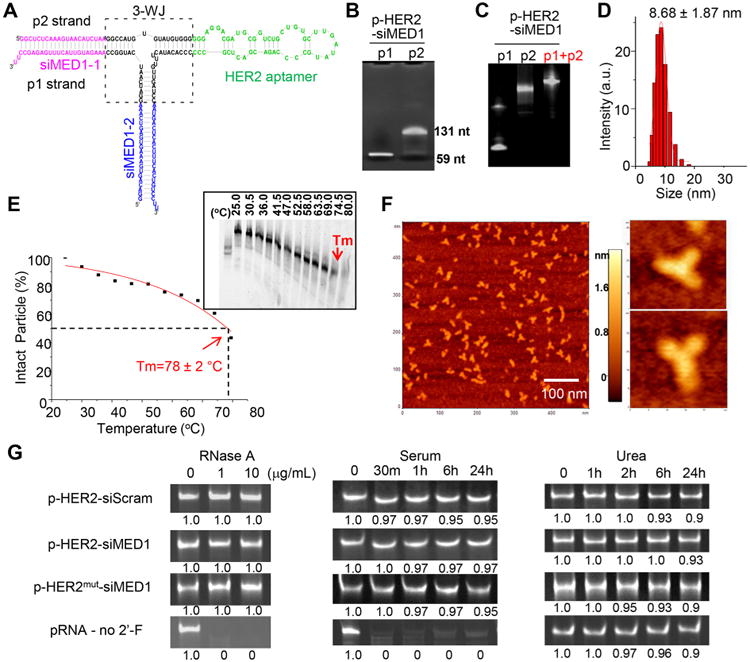
Construction and characterization of pRNA–HER2apt–siMED1 nanoparticles. (A) Scheme of the pRNA–HER2apt–siMED1 (p-HER2-siMED1) structure. (B) p1 and p2 strands of pRNA–HER2apt–siMED1were transcribed using an in vitro RNA transcription system and separated in 8% denatured PAGE gel. (C) pRNA–HER2apt–siMED1 nanoparticles were generated by annealing equal molar of strands p1 and p2 and subjected to 8% native PAGE gel electrophoresis. (D) DLS assay of hydrodynamic size of pRNA–HER2apt–siMED1 nanoparticle. (E) Tm value of pRNA–HER2apt–siMED1 nanoparticle determined by TGGE assay. (F) Atomic force microscopy (AFM) images of pRNA–HER2apt–siMED1 nanoparticles. (G) Stability of control unmodified and 2′-F-modified pRNA nanoparticles was examined by 8% native PAGE gel electrophoresis after RNase A, 10% FBS-supplemented DMEM medium, and 8 M urea treatments for the indicated time at 37 °C.
To construct pRNA–HER2apt–siMED1 and control nanoparticles, two strands, p1 and p2, were transcribed from the DNA templates using an in vitro T7 promoter-controlled RNA transcription system33 (Figure 1B and Table S1). These two strands were then mixed in an equal molar ratio and annealed to generate uniform pRNA nanoparticles (Figure 1C). The hydrodynamic size of the pRNA nanoparticles was determined to be 8.68 ± 1.87 nm by dynamic light scattering (DLS) measurements (Figure 1D). The Tm value was determined to be 78 ± 2 °C by temperature gradient gel electrophoresis (TGGE) assay (Figure 1E), and the atomic force microscopy (AFM) images clearly demonstrated the formation of homogeneous three-way junction architecture of the pRNA nanoparticles as previously described28 (Figure 1F).
As shown in Figure 1G, the 2′-F-modified but not unmodified pRNA nanoparticles were resistant to 10 μg/mL RNase A treatment and were highly stable in 10% FBS-supplemented DMEM medium at 37 °C. Interestingly, both modified and unmodified pRNA nanoparticles maintained their structure in 8 M urea, reflecting the highly stable nature of the three-way junction. Next, the pRNA nanoparticles were digested with recombinant Dicer enzyme (Genlantis)34 to confirm the release of siRNAs as indicated by the generation of 21–23 bp small RNAs after digestion (Figure S1D). Taken together, these results indicated that the self-assembled pRNA– HER2apt–siMED1 nanoparticles formed uniformly sized ultracompact and stable structures capable of releasing siRNAs.
Specific Targeting of HER2-Overexpressing Breast Cancer Cells by pRNA–HER2apt–siMED1 Nanoparticles in Vitro and in Vivo
We next examined the targeting capabilities of the AlexaFluor 647 (AF647)-conjugated pRNA–HER2apt–siMED1 nanoparticles using HER2-overexpressing BT474 cells and control MDA-MB-231 cells (Figure S2A).35 We found that pRNA–HER2apt–siMED1 nanoparticles can bind and be readily internalized into BT474 but not MDA-MB-231 cells (Figure S2B,C and Figure 2A). However, mutating the HER2 aptamer in the stem region dramatically impaired the binding and internalization of the pRNA–HER2apt–siMED1 nanoparticles (Figure S2B and Figure 2A). Moreover, flow cytometric analyses further determined that pRNA–HER2apt– siMED1 nanoparticles were effectively taken up by BT474 cells rather than MDA-MB-231 cells after 3 h incubation at 37 °C, whereas HER2 aptamer mutation notably decreased the uptake efficiency (Figure 2B and Figure S2D). Together, these results demonstrated the specific and efficient targeting of pRNA–HER2apt–siMED1 nanoparticles into HER2-overexpressing breast cancer cells.
Figure 2.
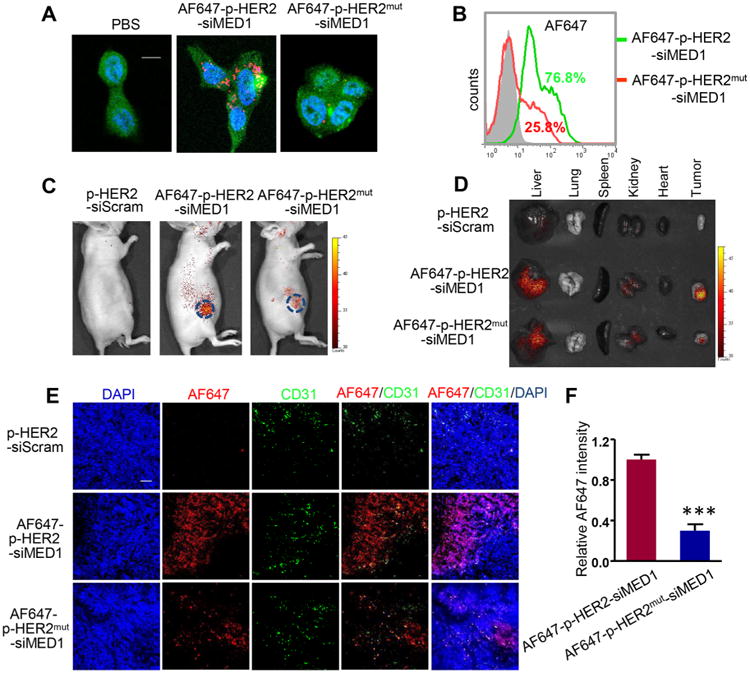
pRNA–HER2apt–siMED1 nanoparticles specifically targeted BT474 cells in vitro and in vivo. (A) Confocal microscopy analyses of the internalization of AF647-labeled control and pRNA–HER2apt–siMED1 nanoparticles by BT474 cells. Scale bar: 10 μm. (B) Flow cytometry assays of the cellular uptake of AF647-labeled control and pRNA–HER2apt–siMED1 nanoparticles by BT474 cells. (C) IVIS Lumina live imaging of BT474 orthotopic xenograft mice 24 h after i.v. injection of indicated AF647-labeled pRNA nanoparticles (10 mg/kg). (D) Major organs and tumors of above mice were excised and imaged for AF647 fluorescence. (E) Frozen tumor sections were examined for localization of AF647-labeled pRNA nanoparticles (red) using confocal microscopy. The blood vessels were stained with anti-CD31 primary antibody and Alexa488-conjugated secondary antibody (green). The nuclei were stained with DAPI (blue). Scale bar: 50 μm. (F) The fluorescence intensity of AF647-labeled pRNA nanoparticles (red) in frozen tumor sections was quantified with Image-pro Plus software.
To examine the in vivo siRNA delivery effects of pRNA–HER2apt–siMED1 nanoparticles, we utilized an orthotopic xenograft mouse model by implanting luciferase-overexpressing BT474 cells into the fourth mammary fat pad of the nude mice. The overexpression of HER2 in both BT474 cells and xenograft tumors was confirmed by Western blot analyses (Figure S2A,E). The live animal imaging demonstrated that AF647-conjugated pRNA–HER2apt–siMED1 nanoparticles but not HER2 aptamer mutant nanoparticles were strongly accumulated in the area of the xenograft tumor after systemic administration (Figure 2C). Further in vivo biodistribution analyses confirmed the predominant accumulation of wild type but not HER2 mutant aptamer-containing nanoparticles in the xenograft tumors, while similar low levels of residual signals were detected in liver and kidney in both groups (Figure 2D). Importantly, confocal microscopic analyses of frozen tumor sections indicated that pRNA–HER2apt–siMED1 nanoparticles very effectively penetrated to tumor cells, while a majority of HER2 aptamer mutant nanoparticles remained in the microvessels (stained with an anti-CD31 antibody) as indicated by their localizations (Figure 2E,F). These results indicated that pRNA–HER2apt–siMED1 nanoparticles could specifically target HER2-overexpressing breast cancer both in vitro and in orthotopic xenograft mouse models.
Inhibition of Cell Growth and Metastatic Capabilities of HER2-Overexpressing Breast Cancer Cells by pRNA–HER2apt–siMED1 Nanoparticles in Vitro
Next, we examined the ability of pRNA–HER2apt–siMED1 nanoparticles to silence the expression of MED1 in BT474 and MDA-MB-231 human breast cancer cells. Using real-time PCR analyses, we found a significant depletion of nearly 70% of MED1 mRNA in BT474 cells after pRNA–HER2apt–siMED1 but not pRNA-HER2aptmut-siMED1 treatment (Figure 3A). Western blots further confirmed that MED1 protein level was greatly reduced by pRNA–HER2apt–siMED1 treatment in BT474 cells but not in MDA-MB-231 cells (Figure 3B and Figure S2F, top panel). As controls, transfection of wild type and mutant HER2 aptamer containing-nanoparticles with lipofectamine 2000 resulted in MED1 silencing in both BT474 and MDA-MB-231 cells (Figure 3B and Figure S2F, bottom). Moreover, the pRNA–HER2apt–siMED1 nanoparticles decreased MED1 protein level in BT474 cells in a dose-dependent manner (Figure S3A). In addition, we further confirmed the effect of pRNA–HER2apt–siMED1 treatment on MED1 silencing in two other HER2-overexpressing and tamoxifen-resistant breast cancer cell lines MCF-7/HER2 and MCF-7/TAM, respectively (Figure S4A,B).36,37 These data demonstrated that the pRNA–HER2apt–siMED1 nanoparticles could specifically and efficiently silence MED1 expression in HER2-overexpressing breast cancer cells.
Figure 3.
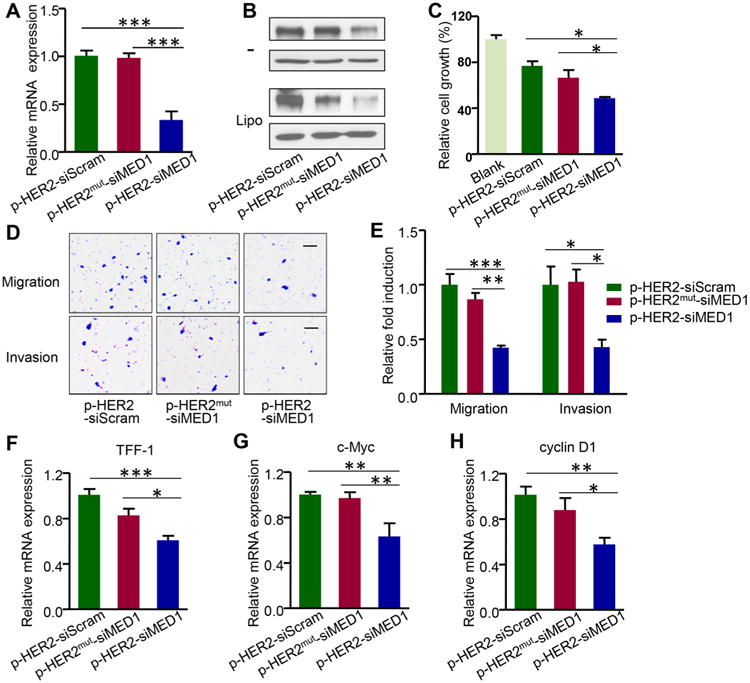
pRNA–HER2apt–siMED1 nanoparticles silenced MED1 expression and inhibited the cell growth and metastatic capabilities of HER2-positive breast cancer cells in vitro. (A) BT474 cells were incubated with 10 μg/mL control and pRNA–HER2apt–siMED1 nanoparticles for 48 h, and the MED1 mRNA level was determined by real-time PCR. (B) BT474 cells were incubated directly with (as indicated by –) or transfected with indicated pRNAs using lipofectamine 2000. At 48 h post treatment, MED1 protein levels were determined by Western blotting. (C–E) BT474 cells were treated with 10 μg/mL pRNA nanoparticles for 48 h and assayed for cell viability by MTT assay (C). Cells were treated as above and seeded for migration (D) and invasion (E) transwell assays. Scale bar: 50 μm. (F–H) After pRNA treatment for 48 h, the mRNA levels of ERα target genes TFF-1 (F), c-Myc (G), and cyclin D1 (H) in BT474 cells were determined by realtime PCR.
As MED1 plays a key role in ERα-dependent growth and metastasis of HER2-overexpressing breast cancer, we further examined the effect of pRNA–HER2apt–siMED1 nanoparticles on BT474 cell growth and metastatic capabilities. As shown by MTT assay, the cell growth was dose-dependently inhibited by pRNA–HER2apt–siMED1 but not pRNA–HER2aptmut–siMED1 treatment (Figure 3C and Figure S3B). Furthermore, transwell assays indicated that both migration and invasion abilities of BT474 cells were specifically suppressed by the treatment of pRNA–HER2apt–siMED1 but not pRNA– HER2aptmut–siMED1 nanoparticles (Figure 3D,E). Next, we investigated the mRNA expression levels of ERα-dependent genes using real-time PCR analyses and found the expression of well-known ERα/MED1 target genes including TFF-1, c-Myc, and cyclin D1 was significantly down-regulated after pRNA–HER2apt–siMED1 treatment38 (Figure 3F–H). Taken together, these data supported that pRNA–HER2apt–siMED1 nanoparticles could inhibit cell growth and metastatic capabilities of HER2-overexpressing breast cancer cells through silencing MED1 and its downstream gene expression.
Sensitizing HER2-Overexpressing Breast Cancer Cells to Tamoxifen Treatment Both in Vitro and in Vivo by the Biosafe-pRNA–HER2apt–siMED1 Nanoparticles
Since MED1 has recently been reported to play key roles in tamoxifen resistance of human breast cancer, we next examined the combinational effects of pRNA–HER2apt–siMED1 nanoparticles with tamoxifen on the growth and metastatic potential of BT474 cells. The results showed that the pRNA–HER2apt– siMED1 nanoparticles significantly enhanced the inhibitory effects of tamoxifen not only on the growth but also the migration and invasion capabilities of BT474 cells (Figure 4A–C and Figure S5). We also confirmed the combinational effect of pRNA–HER2apt–siMED1 nanoparticles with tamoxifen treatment on two other HER2-overexpressing breast cancer cells MCF-7/HER2 and MCF-7/TAM (Figure S4C,D). Moreover, we examined the effect of pRNA–HER2apt–siMED1 and its combination with tamoxifen on breast cancer stem cells using an in vitro mammosphere culture assay.39 We found that pRNA–HER2apt–siMED1 alone and in combination with tamoxifen not only greatly reduced the number of mammosphere formation but also the size of these mammospheres (Figure 4D–F).
Figure 4.
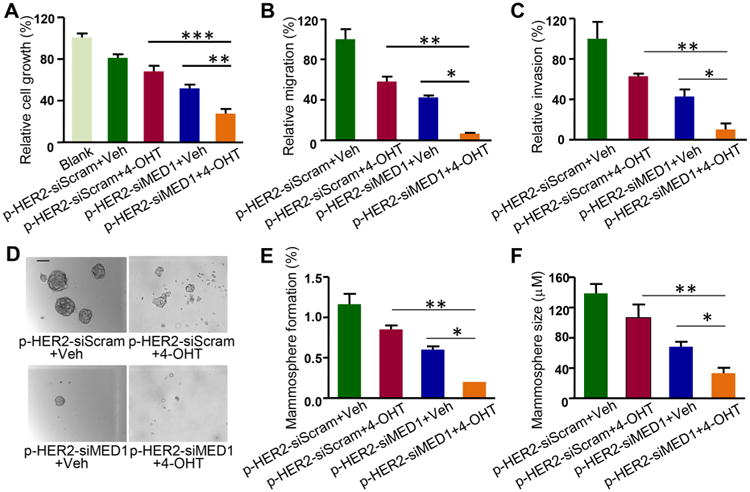
pRNA–HER2apt–siMED1 nanoparticles sensitized HER2-overexpressing BT474 cells to tamoxifen treatment. (A–C) BT474 cells were treated with pRNA nanoparticles in combination with vehicle or 1 μM 4-hydroxytamoxifen (4-OHT) as indicated for 48 h and assayed for cell viability by MTT assay (A). Cells were treated as above, and the migration (B) and invasion (C) capabilities were examined. (D–F) Cells were treated as above and assayed for in vitro mammosphere formation (D). The number of mammospheres was counted (E), and the mammosphere size was calculated (F). Scale bar: 10 μm.
To further examine the potential therapeutic effects of pRNA–HER2apt–siMED1 nanoparticles in vivo, we again employed an orthotopic xenograft mouse model generated using luciferase-overexpressing BT474 cells. After the tumor reached a size of ∼100 mm3, the mice were randomly divided into four groups and systemically administrated with pRNA nanoparticles alone or in combination with tamoxifen. As shown in Figure 5A–D, pRNA–HER2apt–siMED1 nanoparticles alone significantly inhibited tumor growth and reduced tumor burden, whereas tamoxifen treatment only weakly inhibited the growth of these tamoxifen resistant breast tumors as expected. More importantly, pRNA–HER2apt–siMED1 in combination with tamoxifen treatment further enhanced the inhibitory effect and almost completely suppressed tumor growth (Figure 5A–D). Immunochemistry staining and Western blot analyses further confirmed that the expression of MED1 was significantly depleted in the pRNA–HER2apt–siMED1-treated groups (Figure 5E–G). Consistent with the suppressive effect on tumor growth, we found a dramatic reduction of Ki-67 expression in the tumor sections from pRNA–HER2apt–siMED1-treated groups (Figure 5H,I). Importantly, the pRNA nanoparticles exhibited great biosafety after systemic administration, as indicated by no significant reduction of mouse body weight during the entire treatment period (Figure S6A). Moreover, we analyzed the histopathology of major organs including heart, liver, spleen, lung, and kidney by H&E staining and did not detect any apparent organ injury or disturbance in these tissues, further indicating excellent biosafety of pRNA–HER2apt–siMED1 nanoparticles (Figure S6B).
Figure 5.
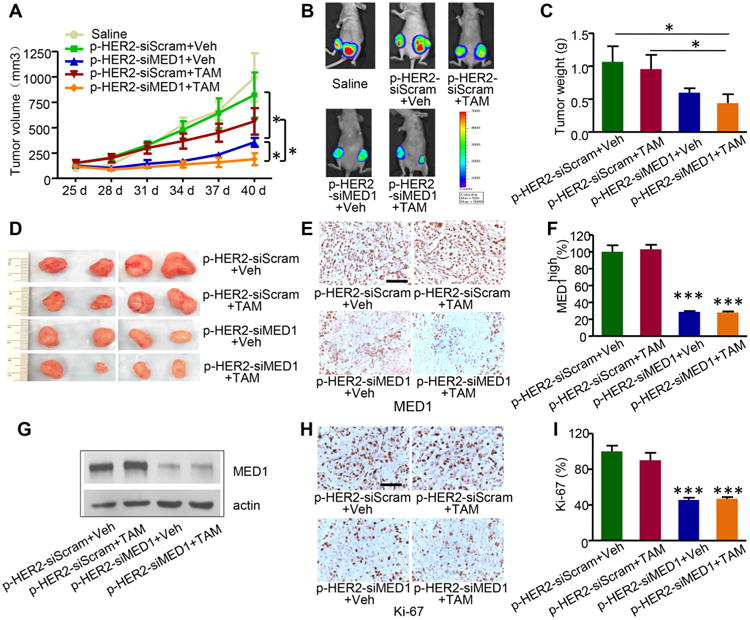
pRNA–HER2apt–siMED1 nanoparticles inhibited HER2-overexpressing breast tumor growth in vivo. (A) BT474 orthotopic xenograft mouse models were treated with control pRNA–HER2apt–siScram or pRNA–HER2apt–siMED1 (4 mg/kg) once a week, in combination with vehicle or tamoxifen (TAM, 0.5 mg/mice/day) 5 days per week. Tumor sizes were measured every 3 days. (B) After 3 weeks, mice were i.p. injected with d-luciferin, and representative in vivo images of BT474 tumors were recorded using IVIS Lumina imaging system. (C and D) Average weight of the tumors excised at the end of the treatment (C) and the representative photos of tumors (D). (E-G) MED1 expression in the BT474 tumors was examined using IHC staining (E and F) and immunoblotting (G). (H and I) The expression of Ki-67 in tumor tissues was analyzed using IHC staining (H), and the percentage of Ki-67 positive cells was counted (I). Scale bar: 100 μm.
Inhibition of Breast Cancer Lung Metastasis, Stem Cell Formation, and Associated Target Gene Expression by pRNA–HER2apt–siMED1 Nanoparticles in Vivo
Since enhanced tumor metastasis and cancer stem cell formation are the major hallmarks of endocrine therapy resistance, we next examined the tumor lung metastasis and cancer stem cell population in the tumor tissues after the therapeutic treatments. H&E staining results indicated that there were multiple tumor lung metastasis foci in the scramble control or tamoxifen-treated groups (Figure 6A,B). However, treatments with pRNA–HER2apt–siMED1 nanoparticles alone or in combination with tamoxifen totally eradicated tumor lung metastasis. Further flow cytometric results revealed that pRNA–HER2-apt–siMED1 nanoparticles significantly reduced the CD44+CD24−/low cancer stem cells in the breast tumors. Impressively, when combined with tamoxifen, pRNA–HER2apt–siMED1 nanoparticles led to almost complete depletion of the CD44+CD24−/low cancer stem cells in breast tumors (Figure 6C,D). To further understand the molecular mechanism underlying the anticancer effect of pRNA–HER2apt–siMED1 nanoparticles, we analyzed expression levels of key ERα-associated genes involved in metastasis and cancer stem cell formation. Consistent with the above results, we found that ERα target genes TFF-1, c-Myc, and cyclin D1 as well as MMP-9 were significantly down-regulated by pRNA–HER2apt–siMED1 treatment (Figure 6E–H). Importantly, the expression of these genes was further inhibited when we combined the treatment of pRNA–HER2apt–siMED1 nanoparticles with tamoxifen. Together, our data indicated that pRNA–HER2apt–siMED1 nanoparticles could overcome tamoxifen resistance of HER2-overexpressing breast cancer by inhibiting tumor growth, lung metastasis, cancer stem cells, and associated gene expression.
Figure 6.
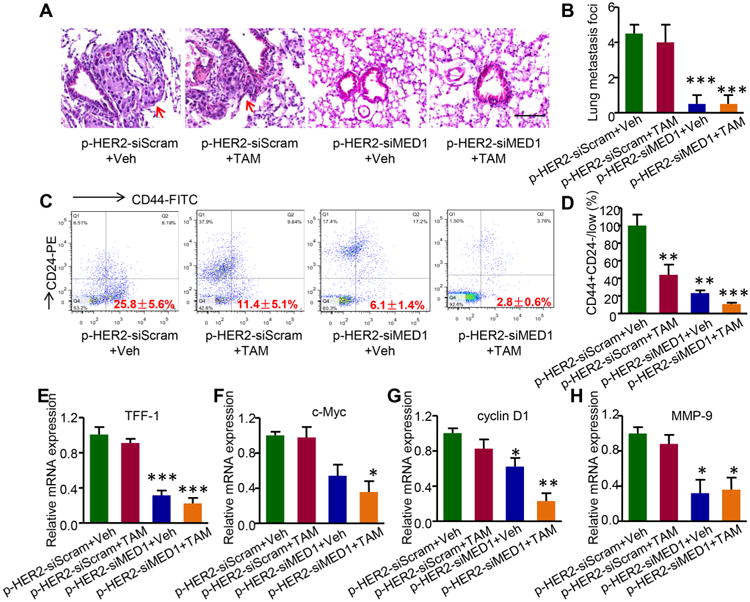
pRNA–HER2apt–siMED1 in combination with tamoxifen greatly impaired breast cancer lung metastasis, stem cell formation, and associated gene expression in vivo. (A and B) Whole lung tissues were fixed, embedded, and sectioned for H&E staining (A), and metastasis foci in the whole lung tissues were then counted (B). Red arrow indicated the metastasis foci. Scale bar: 100 μm. (C and D) After digesting tumor tissues with trypsin and 0.1% collagenase, tumor cells were stained for CD44 and CD24 and analyzed for CD44+CD24−/low stem cell population using flow cytometry. (E−H) Total RNA in the tumor tissues was extracted using TRIZOL reagent, and the expression of TFF-1 (E), c-Myc (F), cyclin D1 (G), and MMP-9 (H) was determined by real-time PCR.
To date, a variety of nanocarriers have been exploited for siRNA delivery, such as liposomes, polymers, dendrimers, and RNA nanoparticles.23,24,40–43 In particular, the Phi29-derived 3-WJ pRNA nanoparticles show great promise as a highly desirable delivery system. In addition to the uniform nanoscale size, precise stoichiometry, and excellent stability and biocompatibility, the extended arms of the 3-WJ motif could be easily replaced with siRNAs or RNA aptamers without impairment of the ultrastability and conformation. Indeed, our pRNA–HER2apt–siMED1 nanoparticles are ultracompact (8.68 ± 1.87 nm) and have a very high Tm value (78 ± 2 °C), which greatly favored not only their serum stability but also their accumulation to tumors after systemic administration due to enhanced permeability and retention (EPR) effect.44 Importantly, we found these pRNA nanoparticles could be further utilized to combat endocrine therapy resistance, a major obstacle in current breast cancer treatment. Aside from its notable antibreast cancer activity, we found that pRNA–HER2apt–siMED1 nanoparticle greatly sensitized HER2-overexpressing breast cancer to tamoxifen treatment while eliciting no apparent toxicity to the normal organs after systemic administration, demonstrating a very promising siRNA delivery platform for cancer therapy.
HER2 is a well-known driver and biomarker for tamoxifen-resistant human breast cancer.8,9,11 HER2 monoclonal antibodies including trastuzumab and pertuzumab have been widely used to treat breast cancer patients.45,46 They have also been used for conjugation with chemotherapeutic drugs, such as emtansine, or nanocarriers for targeted drug delivery.47,48 Recently, RNA aptamers have emerged as promising targeting moieties for cancer diagnosis and therapy, and a number of HER2 RNA aptamers have been isolated through SELEX methods and tested in vitro.31,32 Nucleotide modification, such as 2′-fluoro, 2′-O-methyl, and 2′-amine modification, has now significantly improved the RNA stability under physiological conditions. In addition, RNA aptamers have several advantages for targeting, including small size, lower cost, convenient optimization, and conjugation. However, the potential of these HER2 RNA aptamers for in vivo targeting and drug delivery has not been examined. In the present study, we have tested and successfully identified one such aptamer capable of targeting HER2-overexpressing breast cancer cells and delivering MED1 siRNAs both in vitro and in vivo. Importantly, this HER2 aptamer not only specifically targeted the orthotopic xenograft tumors but also delivered siRNAs to dramatically silence MED1 expression in tumor tissues. Our observation of the greatly enhanced penetration and accumulation of pRNA–HER2apt–siMED1 nanoparticles into tumor cells is likely attributable to the specific targeting since the pRNA nanoparticles with mutant HER2 aptamers largely remain in the tumor blood vessels. Hence, our results highlighted the potential application of HER2 RNA aptamers in the development of future targeted drug delivery systems for HER2-overexpressing human breast cancer.
Cancer metastasis and cancer stem cells are the key drivers for therapy resistance and tumor recurrence.49,50 Significantly, our experimental evidence here indicated that the pRNA–HER2apt–siMED1 nanoparticles could reduce both breast cancer metastasis and the cancer stem cells. We found that the pRNA–HER2apt–siMED1 nanoparticles not only eliminated tumor lung metastasis in orthotopic xenograft mouse models but also dramatically reduced CD44+CD24−/low cancer stem cells in combination with tamoxifen. Consistent with these findings, we found their migration and invasion capabilities, and the expression of a number of key ERα-associated genes involved in metastasis and cancer stem cell formation are greatly inhibited by pRNA–HER2apt–siMED1 nanoparticles.51,52 These data suggested that the pRNA–HER2apt–siMED1 nanoparticles could be more advantageous by targeting both of these processes than most other currently available regimens only capable of targeting one such process. Finally, although we have only tested its combinational use with tamoxifen, our future studies can be further expanded to include other antiestrogens (e.g., fulvestrant) and possibly even anti-HER2 therapies. Such studies will undoubtedly provide further insights into pRNA–HER2apt–siMED1 functions and potentially broaden its applications.
Conclusions
The crosstalk between MED1 and HER2 plays an important role in the tamoxifen resistance of human breast cancer, rendering targeted MED1 silencing in HER2-overexpressing ERα-positive human breast cancer a promising strategy to overcome tamoxifen resistance. In the current study, we constructed pRNA–HER2apt–siMED1 nanoparticles for HER2-targeted MED1 siRNA delivery. Our results demonstrated that pRNA–HER2apt–siMED1 nanoparticles were specifically delivered to HER2-overexpressing ERα-positive breast cancer cells both in vitro and in vivo. These RNA nanoparticles successfully silenced MED1 expression and attenuated ERα function, consequently suppressing cancer cell proliferation and tumor growth. In combination with tamoxifen treatment, these nanoparticles notably decreased tumor lung metastasis and cancer stem cells content, thereby further reducing tumor burden in orthotopic xenograft mouse models. Overall, this study describes the delivery of MED1 siRNAs via the pRNA nanoparticle specifically to HER2-overexpressing ERα-positive human breast cancer cells to overcome tamoxifen resistance, and our findings provide a highly promising perspective for potential clinical treatment of advanced metastatic and tamoxifen resistant human breast cancer.
Experimental Section
Cell Culture
Human breast cancer luciferase-overexpressing BT474 and MDA-MB-231 cells were maintained in DMEM medium (Hyclone, Thermo Scientific) supplemented with 10% fetal bovine serum (FBS, Sigma) and 1% penicillin/streptomycin (Corning) at 37 °C in a humidified atmosphere with 5% CO2.53
Generation of pRNA Nanoparticles
The two strands of pRNA were synthesized with the DNA templates, which were amplified with overlapping primers (Sigma) by PCR methods, using a T7 promoter-controlled in vitro RNA transcription system as previously described33 (Table S1). All of the CTP and UTP used for synthesis of the pRNA nanoparticles were 2′-fluoro modified, except the control unmodified pRNA nanoparticles used in the stability assays. Following an overnight transcription at 37 °C, RNA strands were purified using RNA clean and concentrator kits (ZYMO research) and examined by 8 M urea denatured 8% PAGE gel. pRNA nanoparticles were assembled by mixing the equal molar amounts of p1 and p2 strands and gradually annealing from 90 to 20 °C in 1× RNA annealing buffer containing 50 mM Tris–HCl (pH 7.5), 50 mM NaCl, and 1 mM EDTA on a PCR machine. The generated pRNA nanoparticles were examined by 8% native PAGE gels as previously described. To generate AF647-conjugated pRNA nanoparticles, a 5′-termial AF647-conjugated RNA strand was synthesized (TriLink) for annealing.
Characterization of pRNA Nanoparticles
The hydrodynamic diameter and Tm value of pRNA nanoparticles were determined using dynamic light scattering and TGGE assay, respectively, as previously described.54 The AFM imaging of the pRNA nanoparticles was performed at the University of Nebraska Medical Center imaging facility as previously reported.55 Briefly, the pRNA nanoparticles (5 μL) were deposited on APS modified mica56 for a total of min of incubation time. Excess samples were washed with DI water and dried under a flow of Argon gas. AFM images in air were acquired using a MultiMode AFM NanoScope IV system (Bruker Instruments) operating in tapping mode with 1.5 Hz scanning rate. TESPA probes from Bruker were used for tapping mode imaging. The probe had a spring constant of about 40 N/m and a resonance frequency between 320 kHz. For stability analyses, the pRNA nanoparticles were treated with RNase A, 8 M urea, or 10% FBS supplemented DMEM medium at 37 °C, respectively. The integrity of the pRNA nanoparticles was examined through native PAGE gel electrophoresis.
pRNA Nanoparticle Cellular Uptake Assay
BT474 and MDA-MB-231 cells were seeded into 4-well Lab-Tek chamber slides (Nalge Nunc international, Thermo scientific) and incubated with 10 μg/mL AF647-conjugated pRNAs at 4 °C for 30 min. After washing with PBS three times, cells were incubated at 37 °C for another 1 h. Cells were then fixed with 4% formaldehyde, permeated with 0.25% Trinton X-100, and stained with FITC-conjugated β-actin antibody (Sigma) and DAPI (Sigma). pRNA nanoparticle uptake was examined using a laser scanning confocal microscope (Zeiss). For flow cytometry analyses, BT474 cells were incubated with AF647-labeled pRNA nanoparticles at 37 °C for 3 h. After washing three times with PBS, the cellular uptake of pRNAs was determined using a BD LSRFortessa flow cytometer.
Real-Time PCR
Total RNA was extracted from cultured cells using an RNeasy mini kit (Qiagen) or from tumor tissues by Trizol reagent (Invitrogen), according to the manufacturer's instructions, followed by reverse-transcription using Superscript IV reverse transcriptase (Invitrogen). Real-time PCR was performed using fast start SYBR master mix (Roche) in a 7900HT Fast Real-time system (Applied Biosystem). The expression of MED1, TFF1, c-Myc, cyclinD1, and MMP-9 was analyzed using GAPDH as the internal reference.
Western Blot analyses
Cells were lysed in RIPA buffer supplemented with cocktail protease inhibitors (Roche) and PMSF, and subjected to SDS-PAGE gel electrophoresis. After transferring to nitrocellulose membranes, immunoblotting was performed by incubation with anti-MED1 and anti-β actin primary antibodies, followed by horseradish peroxidase-coupled goat antirabbit secondary antibody (Jackson ImmunoResearch) treatment and visualization with an enhanced chemiluminescence system (Pierce).
MTT Assay
BT474 cells were treated with pRNAs alone or together with 4-hydroxytamoxifen (4-OHT, Sigma) for 48 h. After the treatment, 10 μL of MTT reagent ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Sigma) was added to the cell culture medium and incubated at 37 °C for 4 h. The formazan crystals were dissolved in DMSO, and the absorbance of the solution at 570 nm (A570 nm) was determined using a multifunctional microplate reader (BioTek).
Migration and metastasis assay
The 8-μm-pore-size polycarbonate membrane, which separates the two chambers of a 6.5 mm Transwell (Costar), was coated with or without 1:8 DMEM-diluted Matrigel (Corning). Transwell assay was performed as previously described.57 Briefly, BT474 cells were treated with pRNAs alone or together with 4-OHT for 48 h and starved with serum-free DMEM containing 0.2% BSA overnight. On the next day, cells were collected and 2 × 105 cells were seeded to the upper chamber of Transwell. The lower chamber was filled with 600 μL DMEM medium containing 10% FBS. After incubation at 37 °C for 24 h, cells on the membranes were fixed and stained with 0.1% crystal violet in 20% ethanol solution. Cells at top side of membranes were removed completely, and cells at the bottom side were examined with an Olympus SZX12 microscope and counted.
Mammosphere Culture and Breast Cancer Stem Cell Analyses
BT474 cells were treated with pRNAs for 48 h. Then, cells were suspended in DMEM/F12 medium supplemented with 1×B27 (Thermo Fisher), 20 ng/mL bFGF and 20 ng/mL EGF (R&D), and added into 24-well ultralow attachment plates. After treatment with or without 1 μM 4-OHT for 7 days, mammosphere formation was recorded using an Axiovert S100 TV inverted microscope (Zeiss). Xenograft tumor tissues were cut into small pieces and digested with trypsin and collagenase at 37 °C for 1 h, and tumor cells were collected and rinsed with precold PBS. All cells were stained with FITC-conjugated anti-CD44 antibody and PE-conjugated anti-CD24 antibody (BD Pharmingen) in the dark on ice for 30 min. After resuspension with 0.5 mL of PBS buffer, CD44+/CD24−/low cell population was analyzed by flow cytometry.
Orthotopic Xenograft Breast Tumor Mouse Model, in Vivo Imaging and Antitumor Therapy
Six-week-old female athymic nude mice were purchased from Charles River Laboratories, and all animal procedures were performed under IACUC-approved protocols at the University of Cincinnati. Athymic nude mice were orthotopically injected with 1 × 107 BT474-luc cells mixed with matrigel into the fat pads of the fourth pair of mammary glands. When the tumor sizes reached 100–150 mm3, the mice were randomly divided into four groups (four/group) and intravenously (i.v.) injected with pRNAs (4 mg/kg) once a week for 3 weeks. For tamoxifen treatments, 0.5 mgof tamoxifen (Sigma) dissolved in sesame oil was intraperitoneally (i.p.) injected into mice 5 days a week for 3 weeks. The tumor volume was monitored every 3 days, and tumor size was calculated using the formula: volume = 0.5 × (width)2 × (length). For in vivo pRNA nanoparticle targeting and tumor imaging, mice were i.v. injected with AF647-conjugated pRNA nanoparticles or i.p. injected with d-luciferin and imaged using the IVIS Lumina imaging system with Living Images 3.0 software (Caliper Life Sciences). At the end of the treatment, mice were sacrificed and the organs including heart, liver, spleen, lung, kidneys, and tumors were also excised for imaging. After the tumor weights were recorded, part of the tumor tissues were digested with trypsin and collagenase, lysed for extraction of total proteins and RNAs, or fixed in 10% neutral buffered formalin along with organ tissues, respectively, for further Western blot, real-time PCR and IHC studies, etc. To examine the biodistribution of pRNA nanoparticles within the tumor issues, O.C.T.-embedded frozen sections (5 μm) were examined by confocal microscopy. The same frozen sections were also stained with anti-CD31 primary antibody (BD Biosciences) and Alexa488-conjugated secondary antibody for blood vessels.
H&E and Immunohistochemical (IHC) Staining
The mice organs (heart, liver, spleen, lung, and kidney) and tumors were collected and fixed in 10% neutral buffered formalin, followed by dehydration by gradient series of ethanol (75%, 85%, 95%, and 100%). The dehydrated tissues were embedded in paraffin and cut into 5 μm sections. For H&E staining, the tissue sections were sequentially stained with hematoxylin and eosin (Sigma) and assessed for histology. To examine the MED1 and Ki-67 expression in tumor tissues, IHC staining was performed as previously described.14 Briefly, tumor tissue sections were first deparaffinized and heat-induced antigen retrieval was performed using citrate buffer before incubation with anti-MED1 or anti-Ki-67 primary antibodies (Thermo scientific) at 4 °C overnight. Then, the sections were incubated with biotin-SP-conjugated secondary antibody (Jackson ImmunoResearch) and developed using Vectastain ABC kit with DAB substrates (Vector Lab). The nuclei were counterstained with hematoxylin. Finally, the sections were dehydrated and mounted with neutral balsam. The images were analyzed via an Axioplan Imaging 2e microscope (Zeiss).
Statistics
Data were represented as mean ± SEM from at least three independent experiments. The differences among groups were calculated using Student's t test or one-way ANOVA analysis followed by Tukey's post test (GraphPad Prism, GraphPad Software).
Supplementary Material
Acknowledgments
We thank the members of the laboratories of Drs. X. Zhang, S. Khan (University of Cincinnati), and C. Chen (Colorado State University for help and discussions. We thank Drs. A. Lushnikov and A. Krasnoslobodtsev of the Nanoimaging Core Facility at the University of Nebraska Medical Center for assisting us with atomic force microscopy imaging and B. Ehmer and G. Doerman (University of Cincinnati) for technical and editorial support, respectively. This study was supported by a University of Cincinnati Cancer Institute Drake Pilot Award, Ride Cincinnati Award, Cincinnati Cancer Center Pilot Grant, Komen for the Cure Foundation Career Catalyst Grant (KG110028), NIH Grant Nos. R01CA197865 (to X.Z.) and R01 EB019036 (to P.G.), and a DOD Idea Award W81XWH-15-1-0052 (to D.S.).
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.6b05910.
Table of sequences used for construction of pRNA nanoparticles; Figures S1–S6 as described in the text (PDF)
ORCID: Peixuan Guo: 0000-0001-5706-2833; Xiaoting Zhang: 0000-0002-1328-4255
Notes: The authors declare the following competing financial interest(s): P.G. is the consultant of Oxford Nanopore and Nanobio RNA Technology Co, Ltd. He is the cofounder of P&Z Biology Medical Co. Ltd.
References
- 1.Jensen EV, Jacobson HI. Basic Guides to the Mechanism ofEstrogen Action. Recent Prog Horm Res. 1962;18:387–414. [Google Scholar]
- 2.Deroo BJ, Korach KS. Estrogen Receptors and Human Disease. J Clin Invest. 2006;116:561–570. doi: 10.1172/JCI27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang B, Warner M, Gustafsson JA. Estrogen Receptors in Breast Carcinogenesis and Endocrine Therapy. Mol Cell Endocrinol. 2015;418:240–244. doi: 10.1016/j.mce.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Jordan VC. Tamoxifen: A Most Unlikely Pioneering Medicine. Nat Rev Drug Discovery. 2003;2:205–213. doi: 10.1038/nrd1031. [DOI] [PubMed] [Google Scholar]
- 5.Osborne CK, Schiff R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali S, Coombes RC. Endocrine-Responsive Breast Cancer and Strategies for Combating Resistance. Nat Rev Cancer. 2002;2:101–112. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 7.Ross JS, Fletcher JA. The Her-2/Neu Oncogene in Breast Cancer: Prognostic Factor, Predictive Factor, and Target for Therapy. Stem Cells. 1998;16:413–428. doi: 10.1002/stem.160413. [DOI] [PubMed] [Google Scholar]
- 8.Osborne CK, Schiff R. Mechanisms of Endocrine Resistance in Breast Cancer. Annu Rev Med. 2011;62:233–247. doi: 10.1146/annurev-med-070909-182917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurokawa H, Lenferink AEG, Simpson JF, Pisacane PI, Sliwkowski MX, Forbes JT, Arteaga CL. Inhibition of Her2/Neu (Erbb-2) and Mitogen-Activated Protein Kinases Enhances Tamoxifen Action against Her2-Ovorexpresessed Tamoxifen-Resistant Breast Cancer Cells. Cancer Res. 2000;60:5887–5894. [PubMed] [Google Scholar]
- 10.Luoh SW. Amplification and Expression of Genes from the 17q11 Similar to Q12 Amplicon in Breast Cancer Cells. Cancer Genet Cytogenet. 2002;136:43–47. doi: 10.1016/s0165-4608(01)00657-4. [DOI] [PubMed] [Google Scholar]
- 11.Cui J, Germer K, Wu T, Wang J, Luo J, Wang SC, Wang Q, Zhang X. Cross-Talk between Her2 and MED1 Regulates Tamoxifen Resistance of Human Breast Cancer Cells. Cancer Res. 2012;72:5625–5634. doi: 10.1158/0008-5472.CAN-12-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang XT, Krutchinsky A, Fukuda A, Chen W, Yamamura S, Chait BT, Roeder RG. MED1/TRAP220 ExistsPredominantly in a Trap/Mediator Subpopulation Enriched in RNAPolymerase II and Is Required for ER-Mediated Transcription. Mol Cell. 2005;19:89–100. doi: 10.1016/j.molcel.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Kang YK, Guermah M, Yuan CX, Roeder RG. The TRAP/Mediator Coactivator Complex Interacts Directly with Estrogen Receptors Alpha and Beta through the TRAP220 Subunit and Directly Enhances Estrogen Receptor Function in Vitro. Proc Natl Acad Sci U S A. 2002;99:2642–2647. doi: 10.1073/pnas.261715899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang P, Hu Q, Ito M, Meyer S, Waltz S, Khan S, Roeder RG, Zhang X. Key Roles for Med1 LxxLL Motifs in Pubertal Mammary Gland Development and Luminal-Cell Differentiation. Proc Natl Acad Sci U S A. 2010;107:6765–6770. doi: 10.1073/pnas.1001814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu Y, Qi C, Jain S, Le Beau MM, Espinosa R, 3rd, Atkins GB, Lazar MA, Yeldandi AV, Rao MS, Reddy JK. Amplification and Overexpression of Peroxisome Proliferator-Activated Receptor Binding Protein (PBP/PPARBP) Gene in Breast Cancer. Proc Natl Acad Sci U S A. 1999;96:10848–10853. doi: 10.1073/pnas.96.19.10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS. Differential Oestrogen Receptor Binding Is Associated with Clinical Outcome in Breast Cancer. Nature. 2012;481:389–393. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagalingam A, Tighiouart M, Ryden L, Joseph L, Landberg G, Saxena NK, Sharma D. MED1 Plays a CriticalRole in the Development of Tamoxifen Resistance. Carcinogenesis. 2012;33:918–930. doi: 10.1093/carcin/bgs105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 19.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 20.Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi Therapies: Drugging the Undruggable. Sci Transl Med. 2014;6:240ps7. doi: 10.1126/scitranslmed.3008362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wittrup A, Lieberman J. Knocking Down Disease: A Progress Report on SiRNA Therapeutics. Nat Rev Genet. 2015;16:543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bobbin ML, Rossi JJ. RNA Interference (RNAi)-Based Therapeutics: Delivering on the Promise? Annu Rev Pharmacol Toxicol. 2016;56:103–122. doi: 10.1146/annurev-pharmtox-010715-103633. [DOI] [PubMed] [Google Scholar]
- 23.Guo P. The Emerging Field of Rna Nanotechnology. Nat Nanotechnol. 2010;5:833–842. doi: 10.1038/nnano.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shu Y, Pi F, Sharma A, Rajabi M, Haque F, Shu D, Leggas M, Evers BM, Guo P. Stable RNA Nanoparticles as Potential New Generation Drugs for Cancer Therapy. Adv Drug Delivery Rev. 2014;66:74–89. doi: 10.1016/j.addr.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Guo S, Cinier M, Shlyakhtenko LS, Shu Y, Chen C, Shen G, Guo P. Fabrication of Stable and RNase-Resistant RNA Nanoparticles Active in Gearing the Nanomotors for Viral DNA Packaging. ACS Nano. 2011;5:237–246. doi: 10.1021/nn1024658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdelmawla S, Guo S, Zhang L, Pulukuri SM, Patankar P, Conley P, Trebley J, Guo P, Li QX. Pharmacological Characterization of Chemically Synthesized Monomeric Phi29 pRNA Nanoparticles for Systemic Delivery. Mol Ther. 2011;19:1312–1322. doi: 10.1038/mt.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo S, Huang F, Guo P. Construction of Folate-Conjugated Prna of Bacteriophage Phi29 DNA Packaging Motor for Delivery of Chimeric SiRNA to Nasopharyngeal Carcinoma Cells. Gene Ther. 2006;13:814–820. doi: 10.1038/sj.gt.3302716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shu D, Shu Y, Haque F, Abdelmawla S, Guo P. Thermodynamically Stable RNA Three-Way Junction for Constructing Multifunctional Nanoparticles for Delivery of Therapeutics. Nat Nanotechnol. 2011;6:658–667. doi: 10.1038/nnano.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui DX, Zhang CL, Liu B, Shu Y, Du T, Shu D, Wang K, Dai FP, Liu YL, Li C, Pan F, Yang YM, Ni J, Li H, Brand-Saberi B, Guo PX. Regression of Gastric Cancer by Systemic Injection of RNA Nanoparticles Carrying Both Ligand and SiRNA. Sci Rep. 2015;5:10726. doi: 10.1038/srep10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rychahou P, Haque F, Shu Y, Zaytseva Y, Weiss HL, Lee EY, Mustain W, Valentino J, Guo PX, Evers BM. Delivery of RNA Nanoparticles into Colorectal Cancer Metastases Following Systemic Administration. ACS Nano. 2015;9:1108–1116. doi: 10.1021/acsnano.5b00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thiel KW, Hernandez LI, Dassie JP, Thiel WH, Liu X, Stockdale KR, Rothman AM, Hernandez FJ, McNamara JO, 2nd, Giangrande PH. Delivery of Chemo-Sensitizing SiRNAs to Her2+-Breast Cancer Cells Using RNA Aptamers. Nucleic Acids Res. 2012;40:6319–6337. doi: 10.1093/nar/gks294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MY, Jeong S. In Vitro Selection of RNA Aptamer and Specific Targeting of Erbb2 in Breast Cancer Cells. Nucleic Acid Ther. 2011;21:173–178. doi: 10.1089/nat.2011.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shu Y, Shu D, Haque F, Guo P. Fabrication of pRNA Nanoparticles to Deliver Therapeutic RNAs and Bioactive Compounds into Tumor Cells. Nat Protoc. 2013;8:1635–1659. doi: 10.1038/nprot.2013.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo S, Tschammer N, Mohammed S, Guo P. Specific Delivery of Therapeutic RNAs to Cancer Cells Via the Dimerization Mechanism of Phi29 Motor pRNA. Hum Gene Ther. 2005;16:1097–1109. doi: 10.1089/hum.2005.16.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, Tang P. The Expression Patterns of ER, PR, Her2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer (Auckl) 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 36.Long X, Fan M, Bigsby RM, Nephew KP. Apigenin Inhibits Antiestrogen-Resistant Breast Cancer Cell Growth through Estrogen Receptor-Alpha-Dependent and Estrogen Receptor-Alpha-Independent Mechanisms. Mol Cancer Ther. 2008;7:2096–2108. doi: 10.1158/1535-7163.MCT-07-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin L, Zhang XT, Bian XW, Guo YM, Wang ZY. Disruption of the ER-Alpha36-EGFR/Her2 Positive Regulatory Loops Restores Tamoxifen Sensitivity in Tamoxifen Resistance Breast Cancer Cells. PLoS One. 2014;9:e107369. doi: 10.1371/journal.pone.0107369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM. Her-2 Amplification, Her-1 Expression, and Tamoxifen Response in Estrogen Receptor-Positive Metastatic Breast Cancer: A Southwest Oncology Group Study. Clin Cancer Res. 2004;10:5670–5676. doi: 10.1158/1078-0432.CCR-04-0110. [DOI] [PubMed] [Google Scholar]
- 39.Grimshaw MJ, Cooper L, Papazisis K, Coleman JA, Bohnenkamp HR, Chiapero-Stanke L, Taylor-Papadimitriou J, Burchell JM. Mammosphere Culture of Metastatic Breast Cancer Cells Enriches for Tumorigenic Breast Cancer Cells. Breast Cancer Res. 2008;10:R52. doi: 10.1186/bcr2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afonin KA, Bindewald E, Yaghoubian AJ, Voss N, Jacovetty E, Shapiro BA, Jaeger L. In Vitro Assembly of Cubic RNA-Based Scaffolds Designed in Silico. Nat Nanotechnol. 2010;5:676–682. doi: 10.1038/nnano.2010.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Afonin KA, Viard M, Martins AN, Lockett SJ, Maciag AE, Freed EO, Heldman E, Jaeger L, Blumenthal R, Shapiro BA. Activation of Different Split Functionalities on Re-Association of RNA-DNA Hybrids. Nat Nanotechnol. 2013;8:296–304. doi: 10.1038/nnano.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitehead KA, Langer R, Anderson DG. Knocking Down Barriers: Advances in SiRNA Delivery. Nat Rev Drug Discovery. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee JB, Hong J, Bonner DK, Poon Z, Hammond PT. Self-Assembled RNA Interference Microsponges for Efficient SiRNA Delivery. Nat Mater. 2012;11:316–322. doi: 10.1038/nmat3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K. Accumulation of Sub-100 nm Polymeric Micelles in Poorly Permeable Tumours Depends on Size. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 45.Rimawi MF, Schiff R, Osborne CK. Targeting Her2 for the Treatment of Breast Cancer. Annu Rev Med. 2015;66:111–128. doi: 10.1146/annurev-med-042513-015127. [DOI] [PubMed] [Google Scholar]
- 46.Amiri-Kordestani L, Wedam S, Zhang L, Tang S, Tilley A, Ibrahim A, Justice R, Pazdur R, Cortazar P. First FDA Approval of Neoadjuvant Therapy for Breast Cancer: Pertuzumab for the Treatment of Patients with Her2-Positive Breast Cancer. Clin Cancer Res. 2014;20:5359–5364. doi: 10.1158/1078-0432.CCR-14-1268. [DOI] [PubMed] [Google Scholar]
- 47.Hurvitz SA, Dirix L, Kocsis J, Bianchi GV, Lu J, Vinholes J, Guardino E, Song C, Tong B, Ng V, Chu YW, Perez EA. Phase II Randomized Study of Trastuzumab Emtansine Versus Trastuzumab Plus Docetaxel in Patients with Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. J Clin Oncol. 2013;31:1157–1163. doi: 10.1200/JCO.2012.44.9694. [DOI] [PubMed] [Google Scholar]
- 48.Calce E, Monfregola L, Saviano M, De Luca S. Her2-Mediated Anticancer Drug Delivery: Strategies to Prepare Targeting Ligands Highly Specific for the Receptor. Curr Med Chem. 2015;22:2525–2538. doi: 10.2174/0929867322666150521091103. [DOI] [PubMed] [Google Scholar]
- 49.Singh A, Settleman J. Emt, Cancer Stem Cells and DrugResistance: An Emerging Axis of Evil in the War on Cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, Leggett D, Li W, Pardee AB, Li CJ. Suppression of Cancer Relapse and Metastasis by Inhibiting Cancer Stemness. Proc Natl Acad Sci U S A. 2015;112:1839–1844. doi: 10.1073/pnas.1424171112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nair R, Roden DL, Teo WS, McFarland A, Junankar S, Ye S, Nguyen A, Yang J, Nikolic I, Hui M, Morey A, Shah J, Pfefferle AD, Usary J, Selinger C, Baker LA, Armstrong N, Cowley MJ, Naylor MJ, Ormandy CJ, Lakhani SR, Herschkowitz JI, Perou CM, Kaplan W, O'Toole SA, Swarbrick A. C-Myc and Her2 Cooperate to Drive a Stem-Like Phenotype with Poor Prognosis in Breast Cancer. Oncogene. 2014;33:3992–4002. doi: 10.1038/onc.2013.368. [DOI] [PubMed] [Google Scholar]
- 52.Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. Tumor Cell-Produced Matrix Metalloproteinase 9 (MMP-9) Drives Malignant Progression and Metastasis of Basal-Like Triple Negative Breast Cancer. Oncotarget. 2014;5:2736–2749. doi: 10.18632/oncotarget.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Cui J, Leonard M, Nephew K, Li Y, Zhang X. Silencing MED1 Sensitizes Breast Cancer Cells to Pure Anti-Estrogen Fulvestrant in vitro and in vivo. PLoS One. 2013;8:e70641. doi: 10.1371/journal.pone.0070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shu D, Li H, Shu Y, Xiong G, Carson WE, 3rd, Haque F, Xu R, Guo P. Systemic Delivery of Anti-MiRNA for Suppression of Triple Negative Breast Cancer Utilizing RNA Nanotechnology. ACS Nano. 2015;9:9731–9740. doi: 10.1021/acsnano.5b02471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haque F, Shu D, Shu Y, Shlyakhtenko LS, Rychahou PG, Evers BM, Guo P. Ultrastable Synergistic Tetravalent RNA Nanoparticles for Targeting to Cancers. Nano Today. 2012;7:245–257. doi: 10.1016/j.nantod.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shlyakhtenko LS, Gall AA, Filonov A, Cerovac Z, Lushnikov A, Lyubchenko YL. Silatrane-Based Surface Chemistry for Immobilization of DNA, Protein-DNA complexes and Other Biological Materials. Ultramicroscopy. 2003;97:279–287. doi: 10.1016/S0304-3991(03)00053-6. [DOI] [PubMed] [Google Scholar]
- 57.Zhang Y, Li Y, Ma Y, Liu S, She Y, Zhao P, Jing M, Han T, Yan C, Wu Z, Gao J, Ye L. Dual Effects of Interleukin-18: Inhibiting Hepatitis B Virus Replication in HepG2.2.15 Cells and Promoting Hepatoma Cells Metastasis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G565–573. doi: 10.1152/ajpgi.00058.2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.