

Abstract
Virus–host cell interactions are most commonly analyzed in cells maintained in vitro as two-dimensional tissue cultures. However, these in vitro conditions vary quite drastically from the tissues that are commonly infected in vivo. Over the years, a number of systems have been developed that allow the establishment of three-dimensional (3D) tissue structures that have properties similar to their in vivo 3D counterparts. These 3D systems have numerous applications including drug testing, maintenance of large tissue explants, monitoring migration of human lymphocytes in tissues, analysis of human organ tissue development and investigation of virus–host interactions including viral latency. Here, we describe the establishment of tissue-like assemblies for human lung and neuronal tissue that we infected with a variety of viruses including the respiratory pathogens human parainfluenza virus type 3 (PIV3), respiratory syncytial virus (RSV) and SARS corona virus (SARS-CoV) as well as the human neurotropic herpesvirus, varicella-zoster virus (VZV).
Keywords: 3D tissue culture systems, Tissue-like assemblies (TLA), Rotating-wall vessel bioreactor, Varicella-zoster virus, Latency
1. Introduction
Three-dimensional (3D) tissue culture systems are used in cell biology and physiology, immunology, cancer research, microbiology (bacteriology and virology) and tissue engineering to facilitate investigations in a more physiological setting than is obtained with 2D-tissue culture systems. Many bioreactor systems and other experimental devices have been used to establish 3D tissues that emulate properties similar to human and mammalian tissues in vivo [1], [2], [3]. Here we describe our approach to establish an optimized 3D tissue culture system using a rotating wall vessel (RWV) reactor to study viral infections and virus/host interactions.
1.1. Early attempts at 3D tissue culture systems
Tissue culture explant models were established in the 1920s, which allowed maintenance and expansion of organ tissues in culture or on filters [4], [5]. For certain tissues (e.g. skin), it was critical to keep the cultures at the air–liquid interface, thereby providing conditions similar to the in vivo environment [6]. To maintain native tissue architecture, 1–5 mm2 tissue explants were grown on collagen-coated cellulose sponges surrounded by plasma clots in roller tubes [2], [7]. A number of these systems are well established and widely used in contemporary research. However, the disadvantages of explant systems are that ex vivo tissues from humans are limited and often difficult to obtain. Explant cultures are also short lived and prone to necrosis due to insufficient nutrient and oxygen transfer within the tissue. Furthermore, it remains almost impossible to genetically modify primary tissues, while cultured cells can be readily modified using various targeted genome-editing techniques such as transcription activator-like effector nucleases (TALENS) and clustered regularly interspaced short palindromic repeats (CRISPR/Cas9) [8], [9], [10]. To circumvent these limitations, culture systems have been established that allow de novo assembly of analog 3D tissues for a variety of applications.
The establishment of high-density 3D tissues in vitro using mammalian cells is challenging due to shear forces, turbulence, inadequate oxygenation and restricted nutrient transfer that can disrupt or damage the cultures. Peripheral cells commonly grow readily while cells in the center of the tissue undergo necrosis due to nutrient and oxygen deprivation. To provide a constant source of nutrients, chambers were developed that allowed metabolic exchange between a pool of medium and the culture chamber by diffusion across a membrane. This histo-physiological gradient mimics to some degree, the diffusion of essential nutrients in tissues [7], [11]. To enhance the establishment of 3D tissues in vitro, Nandi and colleagues embedded the cultures in collagenous gels providing a scaffold for tissue growth [12]. The collagen gels allow establishment of mammary-like tissue with ductile structures from mammary cells. These cultures show sustained growth for several weeks and closely resemble the corresponding tissue in vivo [12]. Taken together, the early advances allowed the maintenance of tissue explants and de novo assembly of 3D tissues, while retaining their normal shape and special associations [2].
1.2. Establishment of 3D tissues in bioreactors
To improve the 3D tissue culture systems, bioreactors were introduced. Bioreactors are commonly used to produce pharmaceutical proteins such as interferon, growth hormones, and insulin molecules and to allow the maintenance of stable growth conditions over a long time period. In addition, bioreactors can provide defined culture conditions at exact time points, which is often required for the assembly and differentiation of 3D structures. Basic steps involved in construction of 3D tissue structures include tissue assembly, growth, formation of extracellular matrix (ECM) and basement membranes. Cell differentiation is associated with cellular specialization and vascular (or pseudo-vasculature) formation and is required for the establishment of an ex vivo tissue comparable to their in vivo counterparts (Fig. 1 ). To achieve this, each 3D tissue structure must be optimized for the following parameters (i) co-location of particles (tissues) of different sedimentation rates, (ii) three-dimensional spacial freedom that maximizes cell-to-cell and cell-to-microcarrier adherence required for assembly of the complex tissues, (iii) extremely low fluid shear stress and turbulence and (iv) oxygenation by active or passive diffusion with the exclusion of all air bubbles and permitting only dissolved gasses to enter/exit the bioreactor chamber thereby yielding a vessel devoid gas/fluid interface (zero headspace) [13], [14]. Standard vertically oriented bioreactors allow minimal assembly of complex functional mammalian tissues; however, the normal fluid mechanical effects in these systems often leads to excessive shear forces, turbulence and in many cases, inadequate oxygenation and nutrient transfer that cause cell death and pose critical barriers to the establishment of the functional 3D culture systems (Fig. 1).
Fig. 1.

Stages of tissue assembly and development. The five stages of the assembly and development of tissue-like structures are shown. Each of these stages has been achieved in the RWV systems. The pseudo-vascularization of the TLAs is very hard to achieve [26].
To overcome these problems, NASA engineers and scientists developed the integrated rotating-wall vessel (RWV; US patent 5,026,650; Fig. 2 A and B) that allows establishment of 3D tissues on microcarriers or other matrices under near neutral buoyancy, controlled oxygenation, low shear stress and minimal turbulence [13], [15], [16]. The microcarriers serve as structural support for assembly and growth of 3D tissues [17]. Cells efficiently form bead-to-bead or matrix-to-matrix bridges in the RWV bioreactor at low agitation rates [18], typically resulting in aggregates of approximately 12–15 microcarrier beads. The RWV system eliminates excessive agitation from stirring (vertical bioreactors) and minimizes other shear forces that damage the microcarrier 3D tissues [19], [20]. RVW systems are available in various commercially available volumes and can be scaled up to 1 liter.
Fig. 2.

RWV bioreactor system. Panel (A) schematic representation of a commercial RWV bioreactor is shown. A 24-V DC motor (A) drives a belt that rotates the culture vessel (B) around its horizontal axis. An air pump (C) draws incubator air through a 0.22 μm filter (D) and discharges it through a rotating coupling on the shaft that carries the vessel. The air pump moves about 1 l/min of incubator air. The oxygenator membrane (E) is wrapped around the center post. The vessel end caps and wall are made of Lexan. NASA Rotating-Wall Bioreactor. (Courtesy of NASA). Panel (B) Image of a RWV bioreactor prepared for a TLA experiment.
One advantage of the RVW system is the high reproducibility and efficiency of establishing 3D tissue-like assemblies (TLAs) with characteristics of human tissues. Numerous publications confirm the similarity of structural organization and biomarker expression on the surface of the TLAs with their in vivo counterparts [21], [22], [23], [24], [25]. For example, we selected both DNA (VZV) and RNA (RSV, PIV3 and SARS-CoV) viruses to infect neuronal or respiratory TLAs, respectively, that were shown by transcript and protein analysis to reflect their physiological counterparts [26], [27], [28], [29]. The unique feature of the RWV system maintains cell viability of the virus-infected TLA tissues for longer time periods than their 2D-tissue culture counterparts. Importantly, high-titer, cell free virus (1 × 105–107/ml; Hepatitis C, 1 × 105–107; RSV; and 1 × 107/ml, PIV3 5 × 106) [25], [28], [30], [31] can be obtained from the supernatants of infected TLAs. Cytopathic effects (CPE) were detected in TLAs infected with SARS and RSV; however, this CPE did not impair the ability of the RWV cultures to replicate. In the case of VZV we succeeded in our objective to construct and infect neuronal TLAs with the virus that remained viable for months during which time the virus/host interaction was monitored [27]. Here we describe the establishment of neuronal and lung (TLAs) in the RWV system that can be infected with various viruses.
2. Materials and equipment
2.1. Rotating-wall vessel (RWV) bioreactor assembly
RWV bioreactors are available as batch fed or continuous flow, and allow tissue growth under 3D conditions in an optimized fluid suspension environment. The system is based on a rotating cylinder completely filled with fluid. Shortly after the cylinder begins to rotate, the fluid in the cylinder becomes coupled to the wall and rotates at the same speed as the vessel and TLAs rotate at the same speed (angular velocity) as their supporting fluid. Since TLAs have a slightly higher density than the fluid, they become suspended without stirring and with extremely low shear (0.15 dynes/cm2). Oxygenation is achieved by passive diffusion to avoid gas bubbles and associated shear forces [14]. 3D cultures in the RWV system (Fig. 2) require the following equipment, materials, media and supplies.
2.2. Equipment
2.2.1. Required
Inverted microscope, hemocytometer Reichert, tissue culture incubator with sufficient air flow to dissipate heat (e.g. ThermoForma Model 3950), 1 laminar flow hood, fume hood; autoclave; bench top 4° refrigerated centrifuge, iStat handheld blood gas analyzer, 2 RWV bioreactor and power supply, model number RCCS1, Synthecon, 3 Milli-Q water system, 4 50-micron polyester mesh, No. 7-51/36 5 BEEM capsules, No. 2310, 6 Optional equipment: Scanning Electron Microscope Jeol 330T and JEOL-JEM 1010 transmission electron microscope 7 or Environmental Scanning electron microscope, FEI Quanta 250 Environmental Scanning Electron Microscope 8 (SEM, ESEM).
2.3. Materials (*, materials found to provide optimal results)
T-75 (Corning 430641U)∗, T-150 (Corning, 430824)∗ and T-225 (Nunc-159933)∗ culture flasks1; 50-ml (14-375-150)∗ and 15-ml (S50712)∗, polypropylene, sterile, Fisherbrand conical tubes1, Fisherbrand 1, 2, 5, 10, 25 and 50 ml, sterile, single pack serological pipettes1; Corning 9 1L disposable Filter units, (431096)1 ∗; Fisherbrand disposable borosilicate Glass 9-in. disposable pipettes, (1367820C)1 ∗; Pyrex Glass 6-l beaker, (4980-6L)1; 1 (309628), 3 (309657), 5 (309646), and 10 ml (NC9590365), single, sterile, luer lock syringes (Becton–Dickinson)1; #10 Feather Disposable Scalpel (NC9999403, Feather)1; Pyrex 50 ml reusable glass jars with polypropylene caps, autoclavable (139550, Corning)1 ∗, alcohol prep pads No. 0110 10, Cytodex-3 (C3275) or Cultispher G standard porosity (M9418) microcarriers 11 ∗.
2.4. Culture media and supplements
We developed a unique media for all cultures and tissue engineering described in this article termed GTSF-2 [32] (patented) that is commercially available at GE Healthcare LifeScience 12. It consists of 40% MEM (Alpha Mod), 60% L-15 Leibovitz Medium, with HEPES (SH3A09912) supplemented with NaHCO3 1.35 g/l Sigma/S-576111, HEPES 3.0 g/l Research Organics/6003H-2, 13 Folic acid 6.667 μg/l Sigma/F-875813 and 0.5% Nicotinic acid 0.667 ml/l Sigma/N-412611, Bactopeptone 0.6 g/l Difco/0118-011, i-lnositol 0.024 g/l Sigma/l-512511, Fructose 0.1 3 g/l Sigma/F-351011, Galactose 0.25 g/l Sigma/G-538813, d-Glucose 0.25 g/l Sigma11, 300 mM l-Glutamine 10 ml/l Sigma/G-576311, 10 ml/l penicillin–streptomycin, No. 15140-122 Gibco/life technologies, 14 ThermoFisher Scientific, Waltham, MA1), Fungizone 2 ml/l 15290-018 Gibco/Life Technologies, ThermoFisher Scientific, Waltham, MA,1 Insulin–transferrin-sodium-selenite 5 ml/l Sigma/l-188411 and 7–10% fetal bovine serum Hyclone12. Serum concentration is dependent on cell line requirements. 10x Calcium/Magnesium-free phosphate buffered saline (PBS) containing 2 g/l KCl, 2 g/l KH2PO4, 11 g/l Na2HPO4 and 80 g/l NaCl; 0.2% trypsin/0.1% EDTA solution; Trypan blue stain, No. 630-5250.14
2.5. Optional reagents for electron microscopy
0.1 M cacodylic buffer, 0.2 M cacodylic buffer, No. 19180, EMS 15 diluted 1:1 with deionized water; Scanning electron microscopy (SEM) fixative, 12 ml 50% glutaraldehyde, No. 18428, Polysciences 16; 9.9 ml 2.0 M cacodylic buffer, 39.6 ml 10% paraformaldehyde, cat. No. 4018, Polysciences16; 136.5 ml deionized water; solution pH 7.3; 2.0 M cacodylic buffer, made from cacodylic acid sodium salt, No. E258 17; 1% Osmium tetroxide in 0.1 M cacodylic buffer, No. 19180, EMS15; 1.0% thiocarbohydrazide (TCH), No. 2190026; 20.0%, 50.0%, 70.0%, 75.0%, 95.0% and 100% ethyl alcohol; Liquinox detergent, Alconox, Inc., No. 501411,1 HMDS, no 1670015; Isoton, No. 8546719.1
2.6. Cells and viruses
Primary cells and stable cell lines were used to establish various 3D tissues as shown in Table 1 [15], [17], [21], [22], [27]. Human lung TLAs were established using primary human bronchio-tracheal cells (HBTCs) 18 (Cambrex BioWittaker, San Diego, CA) and BEAS-2B cells, an SV-40 transformed human bronchial epithelial cell line from ATCC 19 (Rockville, MD). Cells were propagated in GTSF-2 media as described previously [15]. For the establishment of human neuronal TLAs, we used normal human neural progenitor (NHNP) cells (Lonza 20; Walkersville, MD, USA). NHNPs from multiple donors were initially propagated in GTSF-2 on human fibronectin-coated flasks 21 (BD Biosciences, San Jose, CA) and pooled from at least five donors as described previously [32] (US patent 5,846,807 and 5,858,783). Stock cultures were prepared, tested for viral contaminants as described by the manufacturer and stored in liquid nitrogen. All cells and viruses used to infect RWV 3D tissues were either generated by us (VZV) [27], [33], [34] or obtained through ATCC Rockville MD19 (SARS-CoV, RSV and PIV3).
Table 1.
Overview of established TLAs.
| References | |
|---|---|
| Normal tissue models (virus infection) | |
| Cartilage | Baker and Goodwin [38] |
| Liver (hepatitis viruses) | Sainz et al. [30] |
| Human embryonic kidney | Hammond et al. [39] |
| Human liver (extracorporeal support) | Barzegari et al. [40] |
| Human lymphoid tissue (HIV) | Margolis et al. [31] |
| Thyroid | Becker et al. [41] |
| Skin | Lei et al. [42] |
| Pancreatic islet cells | Qureshi et al. [43] |
| Neuroendocrine cells | Lelkes et al. [44] |
| Human intestinal epithelium (Norwalk virus) | Goodwin et al. [15] |
| Human Cardiac muscle | Lu et al. [48] |
| Cancer models | |
| Colon | Jessup et al. [45] |
| Goodwin [17] | |
| Breast | Vertrees et al. [23] |
| Ovary | Goodwin et al. [46] |
| Prostate | Wang et al. [47] |
| Endocrine | Unsworth and Lelkes [24] |
3. Preparation of material and RWV bioreactor
3.1. Microcarrier preparation
Microcarriers are prepared two days before initiation of an RWV experiment. Prepare four 50-ml autoclavable corning tubes with a 20 mg/ml stock suspension of Cytodex-311 or 3 mg/ml Cultispher11 microcarriers for each bioreactor. Steps 3–7 are performed in a laminar flow hood to avoid contamination.
-
(1)
Add 1 g of dry microcarriers into each of the four tubes.
-
(2)
Sterilize 500 ml of calcium- and magnesium-free 1× PBS by filtration (0.22 μm filter) or purchase sterile PBS.
-
(3)
Add 50 ml of PBS to each microcarrier tubes and incubate at room temperature (RT) overnight.
-
(4)
Autoclave microcarriers in PBS in corning tube with caps loosened for 15 min at 110 °C (liquid cycle) with slow cool down.
-
(5)
Aspirate PBS and resuspend microcarriers in 50 ml of growth media.
3.2. RWV preparation
The RWV bioreactor should be disassembled and properly cleaned prior to use as described below.
-
(1)
Remove center bolt at top of the RWV bioreactor using an Allen wrench.
-
(2)
Gently twist outer wall while holding one end cap to disassemble vessel. Remove O-rings from each end cap.
-
(3)
Place all pieces of vessel in a 4-l Pyrex beaker filled with warm 20% solution of Alconox or Liquinox. Allow to soak for 1 h.
-
(4)
Gently clean oxygenator membrane with your fingers. Harsh scrubbing may damage the membrane.
-
(5)
Discard detergent solution from the 4-l beaker and rinse vessel parts with continuous flow of Milli-Q ultrapure H2O for 20 min.
-
(6)
Let the bioreactor soak in ultrapure Milli-Q H20 overnight.
-
(7)
Remove vessel parts from H2O and dry on absorbent pads. Follow manufacturer’s’ instructions for the assembly.
-
(8)
Place the vessel and cap for the fill port in two separate sterilization pouches and seal.
-
(9)
Autoclave on dry cycle (vacuum cycle) at 121 °C, for 30 min.
-
(10)
Remove the equipment from the autoclave, open the autoclave pouch in a laminar flow hood and allow to dry for 2 h.
3.3. Filling vessel with growth medium and experiment initiation
-
(1)
In the sterile laminar flow hood, aseptically unwrap sterilized vessel and tighten center bolt with a sterile Allen wrench.
-
(2)
Fill vessel with antibiotic free media containing 2% fetal bovine serum using a 25-ml pipette.
-
(3)
Wipe all ports with sterile alcohol pad and aseptically open pouch containing fill port cap.
-
(4)
Attach 10 ml sterile syringe to each syringe port. One 75% filled syringe with sterile growth media to compensate for evaporation through the oxygenator and one empty syringe for the removal of air bubbles.
-
(5)
Fill vessel to 50% of total volume with growth medium, allowing space for the microcarriers and cells for the establishment of the TLA culture.
4. General protocol for establishment and maintenance of TLAs
-
(1)
Remove cells from T-flasks by enzymatic digestion with 0.2% trypsin–0.1% EDTA solution. Count cells with hemocytometer and check for viability by Trypan blue dye exclusion (details for each TLA culture system are provided in Sections 4.1 and 4.2).
-
(2)
Add microcarriers to RWV (Section 3.3 (5)).
-
(3)
Add appropriate cell density to RWV with microcarriers (see Sections 4.1, 4.2).
-
(4)
Fill RWV completely with growth media.
-
(5)
Remove air bubbles in the RWV by gentle tilting to push bubbles beneath syringe ports and use empty syringe to pull bubbles out.
-
(6)
Once bubbles are removed, apply slight head pressure to RWV using syringe with media to restore the culture volume.
-
(7)
Once slight head pressure achieved, close both syringe ports and thoroughly examine if RWV assembly has any leaks. Transfer vessel from laminar flow hood to humidified CO2 incubator and screw onto base. Set initial rotation speed to 18–20 rpm. Check again after 1 h for appropriate rotation speed, potential leaks and lack of bubbles.
-
(8)
Monitor vessel metabolism every 24 h using the iStat handheld blood gas analyzer2 according to the manufacturer’s instructions. The following 13 basic physiological parameters were analyzed: glucose concentration, Cl/Na/K ion concentrations, pH, partial pressure of CO2 (PCO2), bicarbonate concentration (HCO3), total CO2 (TCO2) anion gap, base excess, hematocrit (Hct), hemoglobin (Hgb), blood urea nitrogen (BUN). These metabolic levels should be stably maintained at physiologic levels.
4.1. Human lung TLAs
HBTCs were injected at a density of 2.5 × 105/ml in 1.0 ml along with 3 mg/ml Cultispher G microcarriers, into a 55.0 ml NASA RWV bioreactor and allowed to attach and assemble for 48 h at 18–20 rpm. The Cultispher G beads functioned as support matrix for the attachment of the HBTCs, which provide a mesenchymal and endothelial cell (basal cell layer). At 48–72 h post HBTCs inoculation, 3.5 × 105 BEAS-2B cells were added to the RWVs. The BEAS-2B cells adhered to the existing bead-cell matrix and the co-culture continuously develops until the TLAs reach a size of 3–5 mm. Cultures were then ready for infection with either SARS-CoV [29], human parainfluenza virus type 3 (PIV3), respiratory syncytial virus (RSV) [21], [22], [26], [28].
4.2. Normal human neural progenitor TLAs
3D NHNP TLAs were generated by seeding 3.5 × 105 NHNP cells/ml and 3 mg/ml Cultispher11 beads into a 55 ml RWV and grown at 37 °C under a 5% CO2. Cells were allowed to attach to the beads for 48 h in the bioreactor before re-feeding with GTSF-2 containing 10% FBS. To maintain the TLA cultures within normal human physiological blood chemistry parameters (80–120 mg/dL glucose and pH 7.2), 20–90% of the media was replaced as required with fresh GTSF-2 media every 24–48 h, thus facilitating efficient TLA tissue growth. All metabolic determinations were made using an iStat analyzer as described above.
4.3. Media change in RWV bioreactor
-
(1)
Turn power off. Remove vessel from base and place in laminar flow hood. Stand vessel on the threaded end to allow all cell/bead aggregates to settle at the bottom.
-
(2)
Remove empty syringe, and discard. Wipe ports with sterile alcohol pads.
-
(3)
Remove fill cap and place in a sterile dish. Aspirate medium through fill port with sterile Pasteur pipette without disturbing the settled microcarriers/cells. Aspirate air bubbles from syringe ports without breaking the tip of the pipette.
-
(4)
Fill vessel with growth medium using sterile pipette. It is important to fill vessel slowly that the cell/bead aggregates are not disturbed. This is accomplished by allowing the medium to flow down wall of vessel.
-
(5)
Wipe fill ports with sterile alcohol pads and replace fill cap.
-
(6)
Fill 20-ml sterile syringe with growth medium and attach to one syringe port. Attach 5- or 10-ml sterile syringe to other syringe port.
-
(7)
Gently rock RWV to expel air bubbles that are often present at the sample ports. Maneuver air bubbles under empty syringe. With valves open, gently press on syringes to replace air bubbles with medium.
-
(8)
When all bubbles are expelled, apply slight head pressure, close syringe valves. Discard one syringe, wipe port with alcohol pad and replace protective cap. The second syringe filled with growth media remains in the other port to compensate for evaporation through the oxygenator.
-
(9)
Screw vessel onto rotating base and turn power on. Rotation speed must be increased for increasing cell/bead aggregate sizes. Check rotation speed and adjust as necessary.
4.4. Removing samples from the RWV culture
-
(1)
Turn off the power to stop vessel rotation. Remove syringe port cap and place in sterile petri plate. Attach either a 10-ml sterile syringe to the Luer lock sample valve and open syringe port valve. The vessel can remain in the incubator for this procedure.
-
(2)
Turn power on and allow vessel to rotate until cell/bead aggregates are evenly distributed (approximately 2 min). Gently press on compliant syringe originally placed on vessel at time of inoculation and pull on empty sample syringe to remove sample (two syringes). This step must be done while vessel is rotating to prevent sedimentation of the TLAs during sampling; therefore, care must be taken to prevent syringes from detaching from the vessel.
-
(3)
When desired volume of sample has been removed and replaced with equal volume of fresh medium from compliance syringe, turn off power. Close valve on sampling syringe port and remove sampling syringe. Repeat this step if multiple samples are required.
-
(4)
Replace Lure lock cap and turn on rotator power. Note: we routinely take samples every 24–48 h and analyze them by light microscopy, RT-PCR, proteomics (Luminex), SEM or ESEM. In addition, cells can be dissociated from microcarriers to determine viability and concentration.
-
(5)
TLA Culture Monitoring procedure 3D TLA cultures are monitored daily using the iStat as described above.
5. Infection of RWV cultures
5.1. Preparation of cell-free viruses
All virus cultures in this article were prepared by a similar protocol. Cell-free virus SARS-CoV, RSV, PIV3 and VZV [27], [35], [36] was used for TLA infections to avoid cellular contamination of the TLA cultures. In the case of each virus, infected cells were harvested at 96 h post-infection (p.i.) of infected tissue cultures and resuspended in reticulocyte standard buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris–HCl, pH 7.4). The cells were disrupted by 20–25 strokes of a Dounce (type A) homogenization, debris was centrifuged at 9000×g for 15 min and supernatant containing virus filtered through a 1.0 mm Millex [25] filter unit.
5.2. Viral inoculation
TLA constructs in the RWV were infected with cell-free virus. Multiplicities of infection (MOI) varied for each virus from 0.01 (RSV, PIV3 and SARS) to 0.1 (VZV) determined by plaque assay and cell counts [26], [27], [28], [29], [34].
-
(1)
For the inoculation, follow the protocol outlined in Section 4.3. At step Section 4.3(4) the inoculum is injected into the bioreactor before replacing the removed volume with fresh media.
-
(2)
Inoculate the RWV with the desired virus at the appropriate MOI in 20 ml GTSF-2 in the vessel and allow to absorb at 35 °C (lung TLAs) and 37 °C (NHNP TLAs) for 30 min in a CO2 incubator.
-
(3)
Then fill the RWVs with fresh GTSF-2/8% FBS and transfer to a humidified incubator with a 5% CO2 at 37 °C following steps Section 4.3(4–9).
-
(4)
Every 24 h.p.i., replace 80% of the culture media with fresh GTSF-2 or media of choice containing 8–10% FBS.
-
(5)
Samples can be collected every desired intervals for viral titration, biochemical analysis or TLAs analyses.
6. Analysis of virus infected TLAs
6.1. Sample preparation for analysis of VZV, SARS-CoV, RSV or PIV3 infected TLAs
-
(1)
Samples are taken from the cultures as described in Section 4.4 at the desired intervals.
-
(2)
TLAs and culture supernatant are gently deposited into 15 ml conical tubes (as needed) and allowed to stand until the aggregates are sedimented.
-
(3)
Remove the culture supernatant and centrifuge at 9000 g to remove any cell debris and transfer to 2 ml aliquots in cryovials and freeze at −80 °C for later analyses (proteomics/Luminex, metabolomics and viral assays).
-
(4)
Wash TLAs with calcium and magnesium free PBS (2×) and place in either the appropriate SEM or immunohistochemistry fixative. Some unfixed samples may be placed in 1 ml cryovials for flash freezing at −196 °C or at −80 °C for DNA or RNA analyses (RT-PCR, Transcriptome).
6.2. Electron microscopy protocols (Optional)
6.2.1. Scanning electron microscopy (SEM, Fig. 3)
Fig. 3.

Scanning electron microscopy (SEM) of human lung TLA. (A) SEM image showing the surface of a complex human lung TLA at 1000× magnification. Distinct cell are no longer discernible as the cells have formed an integrated tissue. (B–D) Images of the surface of the complex lung TLA at 3500, 5000, and 7500× respectively after infection with RSV. Budding virus (BV) can be clearly seen covering the surface of the infected cells. Images were taken using a Jeol 330T scanning electron microscope.7
-
(1)
Make a solution of 0.1 M cacodylic buffer prior to analyses and store at 4 °C; prepare 1.0% saturated Thiocarbohydrazide (TCH) by stirring for 24 h before sample preparation. Filter solution with a Corning disposable filter unit just before using to remove any crystals. Solution is light sensitive keep in dark bottle and protected by wrapping in aluminum foil (shelf life 5 days).
-
(2)
Prepare 1.0% OsO4 in 0.1 M cacodylic buffer. This solution is also light sensitive and has a 5 day shelf life.
-
(3)
Prepare modified BEEM capsules (one capsule for each sample).
-
(4)
Remove the caps from two standard capsules and discard extra body section. Cut off conical end of each capsule with a razor blade. Make a hole in each cap using a hole punch. Cut 50-micron polyester mesh in 4 cm2 squares (two squares per capsule). Attach a punched cap on one end and cover the other with a piece of the mesh.
-
(5)
To prepare for scanning place the SEM-fixed sample at room temperature in 0.1 M cacodylic buffer and put sample inside modified BEEM capsule.
-
(6)
Cover the end of capsule and attach hole punched cap. Repeat for all samples. Place capsules in 50-ml tube. Add 10 ml of OsO4 solution to each tube and allow to fix for 45–60 min. Swirling tube intermittently to move the solution. Cover with foil to protect from light. Rinse sample very well with deionized water.
-
(7)
Next add 10-ml filtered 1.0% TCH solution to sample tubes and as before swirl tube and allow to fix for 10 min. Again, protect from light. Then rinse 5× times for 5 min each with deionized H2O.
-
(8)
Fix samples again for 10 min by adding 10 ml of OsO4 solution while protecting from light and rinse 4× for 5 min each with deionized H2O
-
(9)
Perform sequential dehydration using increasing concentrations of ethanol as follows: 20% for 5 min, 50% for 5 min, 75% for 5 min, 95% for 5 min, 100% 2 × 5 min. Samples are ready for dehydration with hexamethyldisilazane (HMDS) or critical-point drying.
6.2.2. Environmental scanning electron microscopy (ESEM, Fig. 4)
Fig. 4.
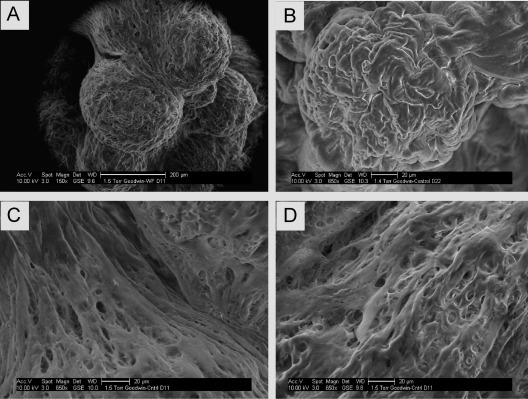
Environmental scanning electron microscopy (ESEM) of NHNP TLAs. (A)–(D) Images show human NHNP TLAs at 150× (A) and 650× (B–D) magnification. The integrated nature and alignment of the neural TLA is similar to the native neural tissues ex vivo (D). Images were taken using a FEI Quanta 250 environmental scanning electron microscope.8
-
(1)
Aliquot desired cell sample volume and add an equal volume of 2× ESEM fix for a final 1× concentration (2% glutaraldehyde/3% formaldehyde final concentration). 2× ESEM Fix 4% glutaraldehyde, 6% formaldehyde prepared in filter/sterilized PBS pH 7.4
-
(2)
Fix samples for a minimum of 30 min at room temperature or leave in fix at 4 °C overnight.
-
(3)
For large cell aggregates transfer several aggregates to a new tube and fill tube with filter/sterilized Milli-Q; allow cells to settle to the bottom and remove supernatant. For single cell/bead samples or very small aggregates skip to step 6.
-
(4)
Repeat step 3, 3–4 times to remove any fixative. Ensure that all fixative/buffer is removed to avoid crystal formation during sample drying as this will interfere with the imaging).
-
(5)
Load aggregates on to a T-stub with a silicon wafer chip sitting on double-sided carbon tape. Sample can immediately be loaded into the ESEM chamber for image analysis.
-
(6)
Load 50–75 μl cell suspension at a concentration of 2 × 106 cells/ml onto a 2 μm TSTP filter paper loaded into a filter unit and rinse with 1 ml of 1x PBS (at least the sample volume) to allow for even dispersal of sample.
-
(7)
Rinse with 1 ml of 0.2 μm filtered Milli-Q to remove 1x PBS crystals before dehydration.
-
(8)
Dehydrate samples with increasing dilutions of Ethanol (25%, 50%, 60%, 70%, 80%, 90% and 100%) by running 0.5–1 ml of each dilution into the filter housing unit and allowing it to filter by gravity at room temperature. Note: prepare fresh ethanol solutions using absolute ethyl alcohol and Milli-Q water, filter sterilized.
-
(9)
Dehydration: Steps: Dehydrate samples sequentially with 25% 2 times, 30 min each, then 50%, 60%, 70%, 80%, 90%, 100% ethanol, 2 times, 10 min each.
-
(10)
Place double-sided carbon tape on stub. Remove filter from housing and place sample side up on carbon tape.
-
(11)
Air dry (or place in ESEM chamber to dry).
-
(12)
Optional: Sputter coat with Platinum (recommend 2 nm coating) for best results in viewing small structures Fig. 4 .
6.2.3. Transmission and immuno-electron microscopy (TEM, Fig. 5)
Fig. 5.
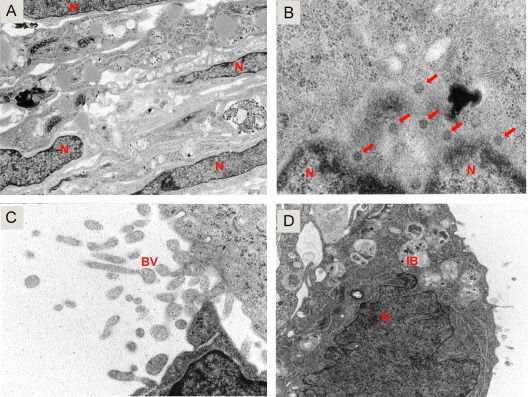
Transmission electron microscopy (TEM) of human lung TLA. TEM image of a human lung TLA at 50,000× (A–C) and 12,000× (D). (A) The highly integrated nature of the uninfected TLA with healthy cell layers from the microcarrier to the external cell surface. Cell nuclei (N) are indicated. (B) Infected lung TL with RSV nucleocapsids in the peri-nuclear region of the cells (VNC). (C and D) Budding RSV virions (BV) at the surface of lung TLA. Images were taken using a JEOL-JEM 1010 transmission electron microscope.
-
(1)
TLA samples were washed 3× with 0.1 M sodium cacodylate buffer pH 7.4, then fixed in 2.5% glutaraldehyde-paraformaldehyde in 0.1 M sodium cacodylate buffer with 0.3 M sucrose13 – 1% DMSO17 and allow to stand overnight at 4 °C.
-
(2)
The fixed tissue was washed 3× in buffer, and then post-fixed stained in 0.3 M tannic acid in 0.1 M sodium cacodylate pH 7.4 for 3 h at room temperature.
-
(3)
There after TLAs were washed 3× in buffer, and fixed again in 1.0 M OsO4 for 1.5 h at room temperature. Samples were dehydrated in a series of graded ETOH baths (as outlined previously), and then embedded in Spurs resin.
-
(4)
Samples were sectioned at silver-gray 6 (100–300 μm), mounted on Ni grids, and examined using a JEOL-JEM 1010 transmission electron microscope (JEOL, USA) at 80 kV Fig. 5 .
6.3. Plaque assay for viruses
-
(1)
Seed appropriate number of MeWo (VZV) or Vero cells (SARS, RSV, and PIV3) into 6-wells and allow them to adhere overnight.
-
(2)
Inoculate MeWo or Vero cells with 10-fold dilutions of supernatant from virus infected TLAs at various times post infection.
-
(3)
Allow the virus to adsorb for 1–2 h at 37 °C.
-
(4)
Aspirate the supernatant and add fresh culture media (GTSF-2 or 1x MEM supplemented with 10% FBS)
-
(5)
Incubate at 37 °C for 3–4 days until CPE is visible.
-
(6)
If fluorescent virus were use, analyze the plaques by fluorescent microscopy. Otherwise continue.
-
(7)
Fix cells with 2% paraformaldehyde in PBS and incubate for 15 min.
-
(8)
Wash cells 3× with PBS.
-
(9)
Add pathogen specific antibodies and incubate for 30–60 min.
-
(10)
Wash cells 3× with PBS.
-
(11)
Add fluorescently labeled secondary antibodies and incubate for 30–60 min.
-
(12)
Analyze the plaques by fluorescent microscopy.
7. Future directions
The TLA models for human lung and neuronal tissues using RWV bioreactors provide a very versatile tool. The tissues can be maintained for several months and allow the infection with a number of viruses. Both DNA (VZV) and RNA (SARS-CoV, PIV3 and RSV) viruses have been used to infect these systems to analyze infection in a more physiological setting [26], [27], [28], [29]. We recently demonstrated that VZV can establish a persistent infection in these NHNP TLAs, which can be maintained for at least 3 months [27]. This system will provide the basis for future studies to decipher the establishment of VZV latency and reactivation. The use of genetically modified NHNP cells would provide a suitable system to address the cellular factors that influence VZV latency. Similarly, neuronal TLAs could be used as a system to study other neurotropic viruses such as herpes simplex (HSV), Dengue and West Nile viruses. Similarly, lung TLAs can be infected with SARS for up to 10 days proving a system that more closely resembles the infection in vivo. Intriguingly, TLAs can be seeded with various immune cells [23], [37], allowing analyses of virus infection in the presence of defined components of the immune system. Furthermore, production of live attenuated VZV vaccines is difficult due to the highly cell-associated nature of the virus. However, the virus is cell-free in the skin lesions of chicken pox. Engineering skin TLA may provide the cell system required to generation of high-titer, cell free attenuated virus for vaccine production.
Acknowledgements
This work was supported in part by the NASA Human Research Program Grant/Rapid Operational Investigation (T.J.G.) and Public Health Service Grant AG032958 and NS082228 (R.J.C.) from the National Institutes of Health (NIH). We acknowledge Mayra A. Nelman, Laurie Graff, Igor Trktinskiy and Nathan Bos for excellent technical assistance, and Ms. Millie Young for editorial assistance.
Footnotes
ThermoFisher Scientific, Waltham, MA.
iStat Abbott Laboratories, Abbott Park, IL.
Synthecon, Friendswood, TX.
Millipore Corp., Milford, MA.
Tetko, Inc., Briarcliff, NY.
SPI, West Chester, NY.
Jeol, Peabody. MA.
FEI Quanta, Hillsboro, Oregon.
Corning, Corning, NY.
Clinipad Corp., Guilford, CN.
Sigma–Aldrich Chemical Co., St. Louis, MO.
GE Life Science/Hyclone Laboratories, Logan, UT.
Research Organics, Cleveland, OH.
Grand Island Biological Supply Co., Grand Island, NY.
Electron Microscopy Sciences. Ft. Washington, PA.
Polysciences, Inc., Warrington, PA.
J.T. Baker, Phillipsburg.
Cambrex BioWittaker, San Diego, CA.
ATCC (Rockville, MD).
Lonza, Walkersville, MD, USA.
BD Biosciences, San Jose.
References
- 1.Carrel A. J. Exp. Med. 1912;15:516–528. doi: 10.1084/jem.15.5.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman R.M. Cancer Cells. 1991;3:86–92. [PubMed] [Google Scholar]
- 3.Sherwin R.P., Richters A., Yellin A.E., Donovan A.J. J. Surg. Oncol. 1980;13:9–20. doi: 10.1002/jso.2930130103. [DOI] [PubMed] [Google Scholar]
- 4.Strangeways T. W. Heffer & Sons; Cambridge, England: 1924. Tissue Culture in Relation to Growth and Differentiation. [Google Scholar]
- 5.Fell H.B., Robison R. Biochem. J. 1929;23(767–784):765. doi: 10.1042/bj0230767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browning T.H., Trier J.S. J. Clin. Invest. 1969;48:1423–1432. doi: 10.1172/JCI106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoffman R.M. Cancer Metastasis Rev. 1994;13:169–173. doi: 10.1007/BF00689634. [DOI] [PubMed] [Google Scholar]
- 8.Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leighton J. In Vitro Cell. Dev. Biol. Anim. 1992;28A:482–492. doi: 10.1007/BF02634131. [DOI] [PubMed] [Google Scholar]
- 12.Yang J., Richards J., Bowman P., Guzman R., Enami J., McCormick K., Hamamoto S., Pitelka D., Nandi S. Proc. Natl. Acad. Sci. U.S.A. 1979;76:3401–3405. doi: 10.1073/pnas.76.7.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goodwin T.J., Prewett T.L., Wolf D.A., Spaulding G.F. J. Cell. Biochem. 1993;51:301–311. doi: 10.1002/jcb.240510309. [DOI] [PubMed] [Google Scholar]
- 14.Schwarz R.P., Goodwin T.J., Wolf D.A. J. Tissue Cult. Methods. 1992;14:51–57. doi: 10.1007/BF01404744. [DOI] [PubMed] [Google Scholar]
- 15.Goodwin T.J., Schroeder W.F., Wolf D.A., Moyer M.P. Proc. Soc. Exp. Biol. Med. 1993;202:181–192. doi: 10.3181/00379727-202-43525. [DOI] [PubMed] [Google Scholar]
- 16.Glacken M.W., Fleischaker R.J., Sinskey A.J. Ann. N. Y. Acad. Sci. 1983;413:355–372. doi: 10.1111/j.1749-6632.1983.tb47912.x. [DOI] [PubMed] [Google Scholar]
- 17.Goodwin T.J., Jessup J.M., Wolf D.A. Animal. 1992;28A:47–60. doi: 10.1007/BF02631079. [DOI] [PubMed] [Google Scholar]
- 18.Cherry R.S., Papoutsakis E.T. Biotechnol. Bioeng. 1988;32:1001–1014. doi: 10.1002/bit.260320808. [DOI] [PubMed] [Google Scholar]
- 19.Cherry R.S., Hulle C.T. Biotechnol. Prog. 1992;8:11–18. doi: 10.1021/bp00013a003. [DOI] [PubMed] [Google Scholar]
- 20.Croughan M.S., Wang D.I.C. Biotechnol. Bioeng. 1989;33:731–744. doi: 10.1002/bit.260330611. [DOI] [PubMed] [Google Scholar]
- 21.Vertrees R.A., Zwischenberger J.B., Boor P.J., Popov V., McCarthy M., Solley T.N., Goodwin T.J. Cancer Biol. Ther. 2008;7:404–412. doi: 10.4161/cbt.7.3.5368. [DOI] [PubMed] [Google Scholar]
- 22.Vertrees R.A., McCarthy M., Solley T., Popov V.L., Roaten J., Pauley M., Wen X., Goodwin T.J. Cancer Biol. Ther. 2009;8:356–365. doi: 10.4161/cbt.8.4.7432. [DOI] [PubMed] [Google Scholar]
- 23.Vertrees R.A., Jordan T.M., Solley T., Goodwin T.J. In: Basic Concepts of Molecular Pathology. Allen T.C., Cagel P.T., editors. Springer Science and Business Media; New York, NY: 2009. [Google Scholar]
- 24.Unsworth B.R., Lelkes P.I. Nat. Med. 1998;4:901–907. doi: 10.1038/nm0898-901. [DOI] [PubMed] [Google Scholar]
- 25.Barrila J., Radtke A.L., Crabbé A., Sarker S.F., Herbst-Kralovetz M.M., Ott C.M., Nickerson C.A. Nat. Rev. Microbiol. 2010;8:791–801. doi: 10.1038/nrmicro2423. [DOI] [PubMed] [Google Scholar]
- 26.T.J. Goodwin, M. McCarthy, Y.-H. Lin, A.M. Dealtly, NASA Tech Paper, TP-2008-214771 (2008).
- 27.Goodwin T.J., McCarthy M., Osterrieder N., Cohrs R.J., Kaufer B.B. PLoS Pathog. 2013;9:e1003512. doi: 10.1371/journal.ppat.1003512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.A.M. Deatly, Y.H. Lin, M. McCarthy, W. Chen, L.Z. Miller, J. Quiroz, R.A. Nowak, S.A. Lerch, S.A. Udem, T.J. Goodwin, NASA Tech Paper, TP-2012-217363 (2012).
- 29.M.T. Suderman, M. McCarthy, E. Mossell, D.M. Watts, C.J. Peters, R. Shope, T.J. Goodwin, NASA Tech Paper, (2006).
- 30.Sainz B., Jr., TenCate V., Uprichard S.L. Virol. J. 2009;6:103. doi: 10.1186/1743-422X-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margolis L.B., Fitzgerald W., Glushakova S., Hatfill S., Amichay N., Baibakov B., Zimmerberg J. AIDS Res. Hum. Retroviruses. 1997;13:1411–1420. doi: 10.1089/aid.1997.13.1411. [DOI] [PubMed] [Google Scholar]
- 32.Lelkes P.I., Ramos E., Nikolaychik V.V., Wankowski D.M., Unsworth B.R., Goodwin T.J. Animal. 1997;33:344–351. doi: 10.1007/s11626-997-0004-7. [DOI] [PubMed] [Google Scholar]
- 33.Tischer B.K., von Einem J., Kaufer B., Osterrieder N. Biotechniques. 2006;40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 34.Tischer B.K., Kaufer B.B., Sommer M., Wussow F., Arvin A.M., Osterrieder N. J. Virol. 2007;81:13200–13208. doi: 10.1128/JVI.01148-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grose C., Perrotta D.M., Brunell P.A., Smith G.C. J. Gen. Virol. 1979;43:15–27. doi: 10.1099/0022-1317-43-1-15. [DOI] [PubMed] [Google Scholar]
- 36.Grose C., Brunel P.A. Infect. Immun. 1978;19:199–203. doi: 10.1128/iai.19.1.199-203.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellis N.R., Goodwin T.J., Risin D., McIntyre B.W., Pizzini R.P., Cooper D., Baker T.L., Spaulding G.F. Animal. 1997;33:398–405. doi: 10.1007/s11626-997-0012-7. [DOI] [PubMed] [Google Scholar]
- 38.Baker T.L., Goodwin T.J. Animal. 1997;33:358–365. doi: 10.1007/s11626-997-0006-5. [DOI] [PubMed] [Google Scholar]
- 39.Hammond T.G., Benes E., O’Reilly K.C., Wolf D.A., Linnehan R.M., Taher A., Kaysen J.H., Allen P.L., Goodwin T.J. Physiol. Genomics. 2000;3:163–173. doi: 10.1152/physiolgenomics.2000.3.3.163. [DOI] [PubMed] [Google Scholar]
- 40.Barzegari A., Saei A.A. BioImpacts: BI. 2012;2:23–32. doi: 10.5681/bi.2012.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Becker J.L., Souza G.R. Nat. Rev. Cancer. 2013;13:315–327. doi: 10.1038/nrc3507. [DOI] [PubMed] [Google Scholar]
- 42.Lei X.H., Ning L.N., Cao Y.J., Liu S., Zhang S.B., Qiu Z.F., Hu H.M., Zhang H.S., Liu S., Duan E.K. PLoS One. 2011;6:e26603. doi: 10.1371/journal.pone.0026603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qureshi K.M., Lee J., Paget M.B., Bailey C.J., Curnow S.J., Murray H.E., Downing R. Clin. Transplant. 2015;29:90–98. doi: 10.1111/ctr.12488. [DOI] [PubMed] [Google Scholar]
- 44.Lelkes P.I., Galvan D.L., Hayman G.T., Goodwin T.J., Chatman D.Y., Cherian S., Garcia R.M., Unsworth B.R. Animal. 1998;34:316–325. doi: 10.1007/s11626-998-0008-y. [DOI] [PubMed] [Google Scholar]
- 45.Jessup J.M., Frantz M., Sonmez-Alpan E., Locker J., Skena K., Waller H., Battle P., Nachman A. Animal. 2000;36:367–373. doi: 10.1290/1071-2690(2000)036<0367:mcraai>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 46.Goodwin T.J., Prewett T.L., Spaulding G.F., Becker J.L. Animal. 1997;33:366–374. doi: 10.1007/s11626-997-0007-4. [DOI] [PubMed] [Google Scholar]
- 47.Wang R., Xu J., Juliette L., Castilleja A., Love J., Sung S.Y., Zhau H.E., Goodwin T.J., Chung L.W. Semin. Cancer Biol. 2005;15:353–364. doi: 10.1016/j.semcancer.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 48.Lu S., Liu S., He W., Duan C., Li Y., Liu Z., Zhang Y., Hao T., Wang Y., Li D., Wang C., Gao S. Cloning Stem Cells. 2008;10:363–370. doi: 10.1089/clo.2007.0093. [DOI] [PubMed] [Google Scholar]