

Abstract
Cancer progression begins when malignant cells colonize adjacent sites, and it is characterized by increasing tumor heterogeneity, invasion and dissemination of cancer cells. Clinically, progression is the most relevant stage in the natural history of cancers. A given virus is usually regarded as oncogenic because of its ability to induce malignant transformation of cells. Nonetheless, oncogenic viruses may also be important for the progression of infection-associated cancers. Recently this hypothesis has been addressed because of studies on the contribution of the Epstein–Barr virus (EBV) to the aggressiveness of nasopharyngeal carcinoma (NPC). Several EBV products modulate cancer progression phenomena, such as the epithelial–mesenchymal transition, cell motility, invasiveness, angiogenesis, and metastasis. In this regard, there are compelling data about the effects of EBV latent membrane proteins (LMPs) and EBV nuclear antigens (EBNAs), as well as nontranslated viral RNAs, such as the EBV-encoded small nonpolyadenylated RNAs (EBERs) and viral microRNAs, notably EBV miR-BARTs. The available data on the mechanisms and players involved in the contribution of EBV infection to the aggressiveness of NPC are discussed in this review. Overall, this conceptual framework may be valuable for the understanding of the contribution of some infectious agents in the progression of cancers.
Oncogenic Viruses
Worldwide, more than 50% of cancer cases are associated with preventable causes, including infections [1]. The population-attributable fraction for malignant neoplasms associated with infectious agents globally in 2008 was 16.1%, ranging from 3.3% in New Zealand to 32.7% in sub-Saharan Africa. About 2 million of all new malignant neoplasms reported in humans were associated with infections, and 1.6 million occurred in less developed regions. More than two-thirds of cancer cases (1.37 million) in 2008 were linked to well-known oncogenic viruses, namely, human T lymphotropic virus type 1 (HTLV-1), human hepatitis B and C viruses (HBV and HBC, respectively), human papillomavirus (HPV), Kaposi sarcoma-associated herpesvirus (KSHV), and EBV [2].
The oncogenic properties of a given virus are usually defined based on its ability to induce malignant cell transformation, and carcinogenic viruses typically interfere with multiple homeo-static cellular processes. For instance, viruses associated with malignant tumors hijack intracellular and extracellular signaling, induce genomic instability, increase the life span of the infected cell (by inhibiting apoptosis), and subvert cell senescence, resulting in unrestricted cell proliferation. These are some of the biological phenomena previously categorized as cancer hallmarks [3], and they can be actively induced during the infection by known human DNA oncoviruses [4,5].
The knowledge generated under this classical approach to viral carcinogenesis was pivotal to defining the etiopathogenesis of several neoplasms, as well as clarifying cancer biology itself. Nonetheless, an intriguing question has emerged in the past two decades: do cancers caused by oncogenic viruses evolve during progression like cancers that originate from other carcinogenic insults? Accumulating data indicate that viruses may be instrumental in phenomena related to cancer progression, and these findings provides new insights on the impact of infection on the natural history of cancers. This review consolidates the available information on putative effects of EBV on the aggressive behavior of nasopharyngeal carcinoma, aiming to delineate a conceptual framework for the impact of viral infection in cancer progression.
General Properties of EBV
Formerly designated human herpesvirus type 4 (HHV-4), EBV is a γ-herpesvirus associated with human proliferative diseases involving mostly lymphoid or epithelial cells. The former group of diseases predominantly encompasses Burkitt lymphoma (BL) and classical Hodgkin lymphoma (HL). EBV is also causative in immunodeficiency-associated lymphoproliferative disorders, such as post-transplant lymphoproliferative disease (PTLD), and non-Hodgkin lymphomas (NHL) in HIV-infected patients, such as primary central nervous system lymphoma (PCNSL), primary effusion lymphoma (PEL), and the plasmablastic lymphoma of the oral cavity [6]. Conversely, epithelial cancers associated with EBV infection include NPC and a subset of gastric and lymphoepithelioma-like carcinomas. In all cases of endemic BL and NPC, early onset PTLD, and PCNSL, the virus is consistently found within the neoplastic cells; conversely, only subsets of classical HL, NHL, gastric and the lymphoepithelioma-like carcinomas show evidence of EBV infection.
The EBV genome is composed of double-stranded DNA of approximately 180 kb, encoding more than 80 viral products. It is enclosed in an icosahedral nucleocapsid, surrounded by the viral tegument and a nuclear membrane-derived lipid envelope. Major targets for EBV infection are B lymphocytes and epithelial cells, though a few other cell types are rarely reported to be infected as well [7]. Box 1 summarizes the main features of EBV infection in humans and the viral life cycle.
Box 1. Epstein–Barr virus (EBV) Infection and Life Cycle.
The first contact with EBV usually occurs early in childhood, and infection lasts for life. The virus replicates initially in epithelial cells of the oropharynx and infects naïve B cells in the tonsils. Infection of B cells by EBV requires interaction of the viral gp350/220, the most abundant viral glycoprotein in the EBV envelope, with complement receptor 2 (CR2, also known as CD21) in target cells [86]. CD21-negative B cells can also be infected via EBV binding to the complement receptor 1 (CR1, or CD35), which is highly expressed in B lymphocytes (earlier than CD21 in B cell ontogeny), as well as follicular dendritic cells [87]. EBV entry in B cells relies on gp42 in complex with viral glycoproteins gH and gL. By contrast, infection of epithelial cells does not require gp350/220 and CD21. Since epithelial cells do not express significant levels of CD21, other cell receptors (yet to be characterized) are necessary for EBV infection. Moreover, EBV entry into epithelial cells is inhibited by an excess of viral gp42, but it is greatly dependent on gH alone or gHgL [88], and it can be enhanced with antibodies against gp350/gp220 [89]. These data indicate that the viral tropism for infection of B lymphocytes versus epithelial cells is finely controlled by a biological switch requiring the expression of proper viral and cellular surface proteins. Of note, carcinomas are much more common cancers overall, notably in adults (http://training.seer.cancer.gov/ disease/categories/classification.html). Thus, the biological basis of EBV cell tropism can be relevant to address the issue of whether viral strains with different cell tropisms might have distinct biological features regarding cell transformation and cancer progression.
The EBV life cycle is conventionally divided into latent and lytic (productive) phases. EBV's latency state is an evolutionary advantage that allows the virus to suspend lytic replication until a more favorable environment is sensed by the infectious agent. Viral products expressed during latency are either poorly immunogenic or subvert the immune responses, allowing viral persistence in latently infected cells [90]. During latency, the viral genome is maintained as a nuclear episome in chromatin, and viral expression is limited to few genes [91]. According to the set of viral products detected during the latent phase, three major latency programs are commonly described (see Table 1 in main text). They range from latency type III (found in EBV-infected naïve B cells and activated B lymphoblasts), with expression of all latent viral genes, to latency type I (observed in post-germinal center memory or dividing B cells), in which EBV nuclear antigen 1 (EBNA1) – which is essential for the maintenance of the viral episome – is the only viral protein expressed. Most neoplastic cells in EBV-associated malignancies show a gene expression profile resembling that found in their non-neoplastic counterparts infected by EBV [92]. By contrast, the viral lytic phase takes place when infected B lymphocytes differentiate towards plasma cells. During this phase, the viral genome become linear, is replicated, and new viral particles are assembled and released, ultimately causing the death of the infected cell due to cytopathic effects.
Cancer Biology: A Brief Overview
The assessment of possible effects of infection by a given agent in cancer progression is only possible with appropriate endpoints for analysis. Based on studies of chemically-induced malignant tumors, the prevailing model for the natural history of cancer encompasses three consecutive stages: initiation, promotion, and progression [8]. Initiation is characterized by the earlier, nonlethal, genomic insult occurring in a given cell population. During promotion, initiated cells are chronically stimulated to proliferate, at the same time that they accumulate new genetic and epigenetic lesions. In this stochastic model, the accumulation of DNA lesions in a given cell lineage eventually gives rise to a transformed cell clone, which shows properties of malignant behavior. Cancer progression takes place when transformed cells colonize their original tissue site in vivo, and it is characterized by augmented tumor heterogeneity and biological aggressiveness, as evidenced by local invasion and distant dissemination [9,10].
The tissue microenvironment has a key role in all stages of carcinogenesis, and it is crucial for cancer progression because malignant colonization relies on trophic signals for the neoplastic cells to survive and proliferate – even though these cells generate some signaling themselves.
Malignant cells within a tumor are heterogeneous, and only a rare subset of cells of a given cancer can produce de novo tumors in vivo when inoculated into immunocompromised animals, such as NOD/SCID mice. Those cancer-initiating cells (CICs) – referred to as cancer stem cells (CSCs) in some circumstances – are well documented in hematologic malignancies, but remain to be conclusively characterized in spontaneous solid tumors [11–13].
The interface between neoplastic cells and the stromal components in carcinomas is a favorable niche for the epithelial–mesenchymal transition (EMT) phenomena, in which epithelial cells switch the expression of several surface proteins (e.g., E-cadherin to N-cadherin), downregulate junctional complex components, and modify their cytoskeleton composition (e.g., vimentin becomes more abundant than cytokeratins). Cells under EMT are less attached to other cells and they have increased motility. Furthermore, signaling pathways that promote longer life span and extracellular matrix remodeling are activated during EMT, effects typically associated with induction of specific transcriptional factors, such as Snail, ZEB1, ZEB2, Slug, and Twist. Worth noting, the EMT and CSC phenotypes overlap in several key features [14].
Clinically, tumor progression is the most relevant phase in the natural history of cancer. Even when a cancer is limited to its primary site, its malignant cells already have a variable degree of genotypic and phenotypic heterogeneity, and local invasion might have occurred to some extent (except for in situ carcinomas, which are epithelial cancers in their very early stage of progression). Eventually, cancer cells seeded in the body are detected as distant metastasis, which arise as result of the successive events, as shown in Box 2.
Box 2. The Cancer Metastasis Cascade.
Cancer metastasis is a complex multistep process, comprehensively reviewed elsewhere [93]. Metastasis initially requires an initial phase of tumor growth at the primary site. Occasionally biological features developed during malignant transformation also enable the neoplastic cells to acquire metastatic behavior; thus, in some cases the cancer metastasizes very early, without significant growth. Nevertheless, in general, the risk for metastasis correlates directly with tumor size and the level of tumor angiogenesis. The properties of malignant cells and their microenvironment (e.g., tumor hypoxia and other signals provided by stromal cells) synergize to stimulate cell motility and extracellular matrix remodeling in tumors, both essential for tissue invasion. For carcinomas, these features are often found at the interface between neoplastic cells and the stroma (the invasive front), where malignant cells lose cell-to-cell adhesion and apical–basal polarity, along with other mesenchymal traits related to the EMT program. Once migrating malignant cells reach blood or lymphatic vessels they enter the circulation (intravasation) and travel to distant sites. Neoplastic emboli may form in the bloodstream, and the interaction with platelets and activation of hemostatic responses increase the odds for malignant cells to survive during transport within the bloodstream [94]. The neoplastic emboli can be trapped in the vasculature, or individual malignant cells may reach the endothelial wall at some point (for instance, due to low flow and/or chemoattractant stimuli). Upon interaction with the endothelium, malignant cells can exit the vessels and move into the interstitium (extravasation). This culminates in the last and rate-limiting step in cancer metastasis: the colonization of the foreign environment and formation of micrometastasis, initially, and the clinically detectable macrometastases, afterwards. Based on experimental models, it is estimated that only 1 out of 5 million intravenously implanted tumor cells will accomplish the invasion–metastasis cascade to form micrometastases at distant sites [93]. Although extremely inefficient biologically, metastasis is the most devastating pathological process in hosts harboring cancers. Figure I illustrates the invasion–metastasis cascade and indicates key molecules and phenomena in different steps of the process.
EBV in Cancer Progression
NPCs are malignant tumors of the head and neck that are rare in most parts of the world, but are notoriously more common in the east, south, and south-central parts of Asia, as well as in northern Africa, though to a lesser degree. Overall, patients with NPC are mostly males, with a peak around 40–60 years of age. Risk factors for this disease include genetics (e.g., susceptibility loci related to HLA and few non-HLA genes), and chronic exposure to environmental carcinogens, often associated with dietary and cultural practices (consumption of Chinese-style salted fish, for instance) [15]. NPC cases in endemic areas are invariably associated with EBV infection, which is clonal and is also found in premalignant and preinvasive nasopharyngeal lesions [16]. Even though NPC tumors typically show marked sensitivity to radiotherapy, and the disease's management has improved significantly lately, in biological terms this is a highly aggressive cancer, showing extensive local infiltration and early metastasis, especially to lymph nodes [17].
The current classification of head and neck tumors recognized by the World Health Organization [15] discriminates keratinized, nonkeratinized, and basaloid NPCs, the last being a quite unusual form. Keratinized tumors are equivalent to conventional squamous cell carcinomas arising in other sites, and nonkeratinized tumors are subdivided into differentiated and undifferentiated subtypes. NPC tumors usually show islands, trabeculae, or solid sheets of neoplastic cells within a stroma, with a variable degree of lymphoplasmacytic infiltrate. The commonest NPC presentations are undifferentiated tumors (WHO type 2b), which typically show large-sized malignant cells arranged in syncytium-like structures. These are high-grade carcinomas that may be remarkably rich in lymphoid cells (hence their alternative designations ‘lymphoepithelial carcinomas’ or ‘lymphoepitheliomas’) and are invariably EBV-positive [15], showing a viral latency type II infection profile (Table 1) [18].
Table 1. Viral Products Expressed and Disease Associated with Distinct Programs during Epstein–Barr Virus (EBV) Latent Infection.
| Latency | Main Viral Genes Expresseda | EBV Infection Context |
|---|---|---|
| Type I (EBNA1-only) | EBERs, BARF0, EBNA1, (LMP2A) |
|
| Type II (Default program) | EBERs, BARFs, EBERs, EBNA1, LMPs 1, 2A, and 2B |
|
| Type III (Growth program) | EBERs, BARFs, EBNAs (1, 2, 3A, 3B, 3C, and LP), LMPs (1, 2A, and 2B) |
|
In all programs, expression of a variable set of viral microRNAs is also observed.
Some cases of EBV-associated gastric carcinoma show also LMP2A expression.
Abbreviations: EBER, small nonpolyadenylated RNAs (EBERs 1 and 2); BARF, transcripts from the BamHI A region (BARF0 and/or BARF1); EBNA, EBV nuclear antigen; LMP, latent membrane protein; BL, Burkitt lymphoma; HL, Hodgkin lymphoma; NHL, non-Hodgkin lymphoma; NPC, nasopharyngeal carcinoma; PTLD, post-transplant lymphoproliferative disease.
Studies on NPC pathogenesis and EBV biology frequently take advantage of cell lines established from nasopharyngeal tissues. Regrettably, authenticity/contamination issues were reported for some of these cells, including CNE1, CNE2, HONE1, Ad/AH, and NPC-KT [19]. Results generated from these cells must therefore be interpreted with much caution; nonetheless, in some instances the published data are not entirely compromised because even though those cells cannot be regarded as reliable models for non-neoplastic or neoplastic cells of nasopharyngeal origin, some of their biological features might be considered, such as the epithelial phenotype and general behavior in vitro. Based on this premise, whenever a known compromised cell line and its derivatives are mentioned in this review, they will be marked (e.g., Ad/AH*, CNE1*, CNE2*, and HONE1*) in order to call the reader's attention to the reported issues.
The metastasis-prone behavior of EBV-positive NPC has been particularly associated with the expression of the EBV latent membrane protein 1 (LMP1). LMP1 is an integral viral protein that functionally resembles a constitutively active, ligand-independent, tumor necrosis factor (TNF) receptor (TNFR), such as the surface CD40 molecule. LMP1 is formed by a short N-terminal chain (24aa), six transmembrane domains, and three cytoplasmic domains in the C-terminal region, the C-terminal activation regions (CTAR) 1, 3, and 2. Recruitment of TNFR-associated factors (TRAFs) to CTAR1 and CTAR2 triggers multiple signaling pathways, notably those regulated by nuclear factor-κB (NF-κB), mitogen-activated protein kinases (MAPKs), janus kinase (JAK), and phosphatidylinositol 3-kinase (PI3K) [20]. Due to its interaction with Ucb9, a SUMO-conjugating enzyme [21], CTAR3 allows LMP1 to regulate sumoylation. Sumoylated proteins have their subcellular accumulation, turnover, and ability to interact with DNA or other proteins modified. For instance, sumoylation of the interferon-regulating factor 7 (IRF7) limits its transcriptional activity, with putative effects on the modulation of innate immune responses during latent EBV infection [22].
In several human cell lines (HeLa, SCC12F, and Rhek-1), and in the MDCK canine epithelial cell line, the expression of EBV LMP1 causes activation of the MAPK/ERK pathway independently of Ras activity. LMP1-expressing MDCK cells exhibit increased haptotaxis and invasiveness, effects consistently associated with activation of MAPK/ERK and Akt signaling pathways [23]. Although it is reported that all CTARs play a role in LMP1-induced migration, only CTAR3 seems to do so via Ubc9 interaction; accordingly, inhibition of LMP1-Ubc9 impairs the migration of MDA-MB231-EBV+ and U20S cells in vitro [21]. Conversely, the increase in cell migration and invasiveness of B lymphocytes in vitro associated with LMP1 expression is mediated by CTAR2-activated NF-kB signaling, and it culminates with upregulation of fascin (encoded by the FSCN-1 gene in humans; NCBI's gene ID: 6624), an actin-bundling protein essential for cytoskeleton changes required for cell motility [24].
In the majority of NPC tissues evaluated by Ho and colleagues [25], the expression of decoy receptor 3 (DcR3, a soluble protein from the TNFR superfamily) was detected at higher levels in metastatic NPC compared with primary tumors. DcR3 is induced by LMP1 in both lymphoid and epithelial cells via NF-κB and PI3K signaling, resulting in increased cell migration and invasiveness in vitro [25]. In another series of NPC cases, tumors of later stage at diagnosis and metastatic cases were more frequently associated with elevated levels of special AT-rich sequence-binding protein 1, encoded by the SATB1 gene (NCBI's gene ID: 6304), and LMP1-positive NPC cases in this series had significantly higher levels of SATB1 compared with LMP1-negative controls. Ectopic expression of LMP1 in NP-69 cells induced a robust increase in SATB1 levels; moreover, cell lines that overexpress SATB1 showed increased proliferation and migration rates compared to cells with low SATB1 expression [26].
The metastatic behavior of different human cancers is modulated by the intracellular signaling triggered by the interaction between the chemokine C–X–C motif receptor 4 (CXCR4) and its ligand CXCL12, also known as SDF-1α [27]. CXCR4 is sulfated by the TPST-1 enzyme, which can be induced by viral LMP1 in metastatic EBV-positive NPC 5–8F cells. In HNE2 cells (supposedly from NPC, but lacking EBV genome and proteins), the LMP1 induction of TPST-1 has increased cell motility towards CXCL12 and cell invasiveness. Nonmetastatic NPC 6-10B cells had low expression of EBV LMP1 and TPST-1, along with a low level of CXCR4 sulfation. It is worth noting that the immunodetection of TPSP-1 and LMP1 in 46 NPC tissues showed direct mutual association, as well as association with lymph node metastasis [28]. Overall, these results indicate that the metastatic behavior of these epithelial cells is directly linked to the expression of EBV LMP1, which upregulates endogenous proteins with distinct activity that converge to a similar phenotype of increased cell motility and invasion.
EBV products other than LMP1 may also enhance the metastatic behavior of neoplastic cells. BALF1, for instance, is an EBV homolog for the antiapoptotic cellular bcl-2 protein. It is required for transformation of resting B cells [29], and BALF1 is frequently expressed in EBV-positive NPC tissues and BL cell lines [30]. NIH3T3 and 293 cells stably expressing BALF1 showed increased motility in vitro, they were more tumorigenic, and they produced more numerous and larger distant tumors in BALB/c nude mice compared with mock-transfected control cells [31].
EBNA1 is the only viral protein expressed in all EBV latency types (Table 1). It enables the replication and segregation of EBV genomes in proliferating infected cells, resulting in viral persistence. The tumorigenic and metastatic potential of EBV EBNAs 1 and 3C were evaluated in an orthotopic model of murine breast carcinoma using MDA-MB-231T cells, with and without coexpression of nonmetastatic protein 23 (nm23-H1, a nucleoside diphosphate kinase and putative metastasis suppressor) [32]. It should be noted that the use of breast cancer cell lines does not address the disputed hypothesis on the role EBV infection in the etiopathogenesis of human breast cancers; as a matter of fact, some studies use cells that are not recognized targets for EBV infection due to their cell migration/invasion capabilities in vitro and/or metastatic potential in mouse models. That said, both EBV EBNAs 1 and 3C interact with nm23-H1 and accumulate in the cell nucleus [33,34]. Furthermore, they increase cell migration in vitro, reversing the negative effect of nm23-H1 and its effect in metastasis suppression. Cancer cells expressing these EBNAs show enhanced tumorigenesis in vivo and accelerated tumor development, irrespectively of coexpression of nm23-H1. Of note, expression of EBNA3C and EBNA1 was associated with 14- and 2-fold more micrometastasis in lung than in the control group, respectively, and the EBNA3C/nm23-H1 complexes upregulate matrix metalloproteinase-9 (MMP-9) via recruitment of the transcription factors Ap1, Sp1, and NF-κB [35].
A possible concern on results implicating EBNA3C expression in epithelial cells is that it is significantly expressed only in lymphoid cells with EBV latency type III; this makes its contribution to the aggressiveness of EBV-associated carcinomas questionable. Nevertheless, non-neo-plastic EBV-positive EBNA3C-expressing lymphoid cells within the tumor may affect the tumor microenvironment (for instance, inducing extracellular matrix remodeling due to increased synthesis of MMPs [36]). Thus, EBNA3C expression by EBV-positive tumor infiltrating lymphoid cells perhaps modulates the invasiveness and metastatic potential of the much more abundant EBV-positive carcinoma cells. As far as we could verify, this hypothesis has not been evaluated so far, but it deserves careful investigation.
In time, the metastatic behavior of cells expressing EBV products can be further enhanced by inhibition of antigen presentation in the context of MHC-I, as in the case of inhibition of tapasin by EBNA1 [37]. Furthermore, strategies of immune subversion, implicating viral proteins such as EBNA1 [38], can also have a role in modifying the progression of EBV-associated cancers. Nonetheless, this matter also remains to be adequately elucidated.
EBV Infection Sets up a Permissive Microenvironment for Invasion and Metastasis
Epithelial cells transformed by EBV may degrade extracellular collagens and become more able to invade the basement membrane due to increased expression of MMP-1, -3, or -9, directly induced by viral LMPs [39–41], EBNA-3C [35], and the viral lytic transactivator Zta [42]. The expression of MMP-1 is enhanced by an insertional polymorphism characterized by two guanines at positions -1607/-1608 (rs1799750, 1G->2G) in the MMP-1 gene promoter, which creates a binding site for Ets transcription factors. LMP1 induces MMP-1 via increased activity of the transcription factor AP1, and cells showing the 2G/2G genotype also have MMP-1 induction due to further Ets-mediated intracellular signaling. Not only do tumor cells in NPC patients have a higher frequency of the 2G/2G genotype for rs1799750 [39], Ets activation by LMP1 is also a putative mechanism for the expression of the hepatocyte growth factor receptor (HGFR; c-Met), which has been positively correlated with the presence of lymph node metastasis in NPC patients [43].
Genes encoding MMP-9, -10, and -14 were among the top hits of TNFα-induced genes in NPC tissues compared with adjacent normal tissues [44]. Moreover, overexpression of the gene for TNFα-induced protein 2 (TNFαIP2, also called B94 or M-sec) was also identified, among others. Although the biological properties of the TNF/IP2 protein are largely unknown, higher levels of the TNFAIP2 gene (NCBI's gene ID: 7127) transcripts were detected in 39% of NPC tissues evaluated, and it was associated with increased tumor angiogenesis and reduced distant metastasis-free survival. Nasopharyngeal HK1 cells expressing TNFAIP2 show increased migration and invasiveness that was disrupted by siRNA-based knockdown [44]. TNFAIP2 is induced by EBV LMP1 in several NPC cell lines via NF-κB activation, even more than the TNFα treatment. The role of TNFαIP2 in cell migration and invasion was related to modulation of actin-based filaments organization within cellular protrusions due to actin binding, and it can be disrupted upon depletion of Cdc42, a Rho-GTPase modeling protein [45]. These results are also in line with the increased migration reported for epithelial cells in vitro due to the activation of Cdc2 by LMP1 [46].
A cDNA microarray analysis of RHEK-1 cells (a nonmalignant cell line from normal human foreskin keratinocytes) transfected with a LMP1-encoding Tet-On vector (RHEK/Tet-LMP1) revealed 60 genes differentially expressed by fourfold or more, notably, in doxycycline-treated LMP1-expressing cells, the genes encoding endocan-1 (ESM1; NCBI's Gene ID: 11082) and MMP-9. By RT-PCR, ESM1 overexpression driven by LMP1 was verified in RHEK/Tet-LMP1 and two other human cell lines of epithelial cancers, NPC-TW04 (NPC) and H1299 (large-cell lung carcinoma). Endocan-1 is implicated in the angiogenic switch of cancers, and its expression was found to be mediated by LMP1 via activation of the NF-κB, MEK–ERK, and JNK signaling pathways. Expression of LMP1 and endocan-1 was directly correlated in 42 consecutive human NPC tissues by immunohistochemistry (endocan-1 was mostly detected in neoplastic cells and endothelial cells adjacent to tumor); moreover, endocan-positive patients have a shorter survival compared with endocan-negative cases [47].
LMP1 also provides angiogenic stimuli by inducing the secretion of proangiogenic cytokines, such as the vascular endothelial growth factor (VEGF) [48] and the 18 kDa isoform of the fibroblast growth factor 2 (FGF2, also known as bFGF) [49]. Remarkably, FGF2 is released with LMP1 in exosomes [49], membrane-limited nanovesicles (usually 40–100 nm in diameter) of endosomal origin. Exosomes participate in intercellular communication, in addition to communication via close cell contact (juxtacrine signaling) or by soluble mediators (autocrine, paracrine, and endocrine signaling). Malignant cells constitutively release high numbers of cancer-derived exosomes, also known as tumor-derived microvesicles (TMV), which play key roles in cancer biology (reviewed in [50]). For instance, exosomal products boost the subversion of immunosurveillance and contribute to shape a suitable microenvironment for tumor development and progression due to cancer-associated inflammation, tumoral angiogenesis, and formation of premetastatic niches (reviewed in [51]).
Exosomes produced by neoplastic cells infected by EBV provide a variety of cellular and viral molecules that can manipulate the tumor microenvironment. LMP1 accumulated in exosomes plays a role in immune evasion of cells latently infected by EBV, including malignant ones [52]. By contrast, expression of LMP1 in Ad/AH* epithelial cells redistributes the usually diffuse FGF2 and causes colocalization of both proteins in perinuclear and peripheral late endosomal structures. Moreover, isolated LMP1-positive exosomes induce proliferation of human umbilical vein endothelial cells (HUVECs), an effect abrogated by the anti-FGF2 antibody [49]. These data suggest a role for exosomes produced by EBV in latently infected cancer cells in the induction of angiogenesis in the tumor microenvironment.
Nasopharyngeal NP69 cells expressing LMP1 secrete exosomes with higher levels of HIF-1-α – a key transcription factor for angiogenesis response – compared with the LMP1-negative counterparts; furthermore, transient transfection of Ad/AH* cells also increases the exosomal levels of HIF-1- α. After exposure to LMP1-positive exosomes, recipient LMP1-negative cells exhibit a dose–response increase in functionally active HIF-1-α along with increased expression of N-cadherin, suppression of E-cadherin, and morphological changes suggestive of EMT change, and motility and invasiveness in vitro were substantially increased in NP69 and Ad/AH* cells exposed to LMP1-positive exosomes. Therefore, cell-to-cell exosomal transfer of active HIF-1-α was correlated with EMT-associated features in the recipient cells [53]. Indeed, as discussed in the next section, EMT is a key cancer progression phenomenon also targeted by EBV proteins, notably LMPs.
EBV Induces EMT and the Cancer Cell State
The undifferentiated nature of EBV-associated NPC has been attributable to EMT, and NPC cells can be successfully induced to a mesenchymal-like state in vitro when they express the EBV oncoproteins LMP1 [54,55] or LMP2A [56], as well as several EBV microRNAs, as will be discussed later. The maintenance of the EMT state in NPC cells was associated with the activity of the polycomb group protein Bmi [57]. Furthermore, knocking-down expression of the SATB1 protein – which was previously mentioned to be induced by LMP1 in NPC cells – increases the expression of E-cadherin and suppresses vimentin, suggesting the reversion of the EMT phenotype [26]. In NPC tissues, immunodetection of LMP-1 and Twist-related protein 1 (encoded by TWIST1 gene, a positive regulator of EMT; NCBI's gene ID: 7291) were higher in cases with lymph node metastasis (n = 26) compared with negative cases (n = 11) [54].
One of the major functions of LMP1 is the constitutive activation of the NF-κB, which has an important role in activating the EMT program in epithelial cells (reviewed in [58]). Indeed, the NF-κB signaling pathway is efficiently hijacked both by EBV and KSHV (reviewed in [59]). LMP1 induces key transcription factors associated with the EMT signature [54,55]. In this regard, ectopic expression of LMP-1 in Ad/AH* (EBV-negative) and the immortalized nonmalignant nasopharyngeal epithelial cells NP69SV40T upregulated the EMT-associated transcription factor Snail in a dose-dependent manner. Not only did NP69SV40T cells expressing LMP1 show EMT morphological hallmarks, they also acquired increased mobility and invasiveness capabilities in vitro. The EMT-like features acquired by NP69SV40T expressing LMP1 rely on Snail, as Twist is not upregulated [55]. Additionally, LMP1-negative epithelial cells exposed to LMP1-positive exosomes showed E-cadherin downregulation, increase in N-cadherin, and increased motility and invasiveness. Therefore, LMP1 can modulate the EMT features and the metastatic behavior of malignant cells of EBV-associated cancers via prometastatic factors present in exosomes released in the tumor microenvironment [53].
Self-renewal capabilities and CD44high/CD24low expression were found in LMP1-expressing epithelial cells [60], consistent with the overlap between the EMT and CICs phenotypes [14]. The Hedgehog (HH) signaling pathway, which is implicated in the maintenance of stemness, was found to be activated in epithelial cells by EBNA1, and LMPs 1 and 2A via an autocrine loop involving the SHH ligand. HH activation was required for tumor-sphere formation by the NPC cell line C666.1, constitutively EBV-positive, as well as the EBV-negative A549 (derived from lung adenocarcinoma) and CNE2* cell lines that were infected in vitro by EBV. Furthermore, the same study showed that the LMPs evaluated, but not EBNA1, induced the expression of CD44v6 and CD271/NGFR (induced by LMP1), or CD133 and CXCR4 (induced by LMP2A), all stemness-associated markers [61]. Also, immortalized or malignant lung epithelial cells (HPL1D and A549, respectively) are more sensitive to TGF-β1-induced EMT when they express EBV LMP1. Compared with exposure to TGF-β1 alone, cells exposed to both TGF-β1 and LMP1 had a reduced transcriptional level of E-cadherin and a significant increase of N-cadherin, vimentin, and MMP9. Remarkably, while cell migration in vitro was increased by 2- and 3-fold by TGF-β1 and LMP1, respectively, it was increased by 9-fold when cells were exposed to both proteins combined [62].
EBV LMP2A interferes with multiple intracellular signaling pathways in both B cells and epithelial cells, including PI3K/AKT, NF-κB, Wnt/beta-catenin, and Jak/STAT. In B cells, LMP2A simulates the active B cell receptor, providing constitutive cell survival signals. Nontransformed, immortalized mouse embryonic NIH3T3 fibroblasts expressing LMP2A are tumorigenic when inoculated in nude mice. In vitro, they grow in soft agar more abundantly than do cells carrying the vector control, an effect linked to activation of Stat3, PI3K/AKT, and MEK/ERK signaling. It is worth noting that cells expressing LMP2A are enriched for cells excreting the Hoechst 33342 dye, a feature associated with cell stemness [63]. LMP2A contributes to the migration of latently infected B cells to lymphoid tissues [64]; furthermore, it was reported that LMP2A enhanced the migration and invasion of TW03 EBV-negative cells due to Syk hijacking, which causes displacement and surface redistribution of the ITAM-like motif of integrin β4 (ITGβ4), which result in changes in cell adhesion and cytoskeleton reorganization [65].
Based on these data, it would not be unexpected that LMP2A might also be relevant for the invasiveness of malignant cells when expressed in EBV-associated cancers. In fact, LMP2A immunodetection was exclusively found in neoplastic cells in 19/33 (57.6%) NPC cases evaluated, notably at the invasive edges of the tumor. In vitro, ectopic expression of LMP2A in SUNE1 NPC cells and CNE2* downregulated E-cadherin and α-catenin, simultaneously inducing fibronectin and Snail. In EBV-positive NPC cells C666, suppression of LMP2A caused E-cadherin upregulation and downregulation of vimentin; maintenance of EMT-like features requires LMP2A in constitutively EBV-infected cells. Both in vitro and in NPC tissues the detection of the stem cell markers ABCG2 and Bmi-1 positively correlated with the expression of LMP2A [56]. Furthermore, LMP2A increases the number of tumor-initiating cells and enhances tumorigenesis in nude mice [56]. Remarkably, primary tonsil epithelial cells showed increased cell migration and invasiveness in vitro upon expression of LMP2A, an effect mediated by upregulation of the integrin-α6 gene (ITGAG; NCBI's gene ID: 3655) [64]. ITGAG encodes a component of the laminin receptor, and it is consistently implicated in the metastatic behavior of human carcinomas (reviewed in [66]). Finally, LMP2 expression in NPC does seem to have a prognostic value, as supported by findings that the expression of this EBV oncoprotein was inversely correlated with survival for 15 NPC patients evaluated [64].
An increase in the mRNA levels for the key EMT regulators Snail, ZEB1, and Slug was reported in human breast carcinoma MDA-MB-231T cells transfected with vectors encoding EBV EBNA1 or EBNA3C. Accordingly, these cells showed an upregulation of vimentin transcripts, along with suppression of mRNAs for E-cadherin and the zona occludens protein 1 (ZO-1), typically expressed in normal epithelial cells; overall, these results were also confirmed at the protein level. It is worth noting that nude mice had an increase in metastatic foci in lungs when inoculated with cancer cells expressing EBNAs 1 or 3C compared with controls (3- and 12-fold, respectively); besides, primary tumors and lung metastasis derived from EBNAs-expressing cancer cells showed EMT features more often, compared with cells lacking these viral proteins [67].
In summary, EMT can be effectively induced in epithelial cells due to the expression of several EBV proteins. This can be critical to the aggressiveness of EBV-associated NPC because EMT encompasses many biological features pivotal for cancer progression, such as cell proliferation, survival, and cell motility, as well as invasiveness and ultimately increased metastatic behavior. Furthermore, besides the most studied EBV oncoproteins, other viral products may have an important role modulating the aggressiveness of NPC, as discussed in the following section.
EBV in Cancer Progression: LMPs and EBNAs Are Not the Whole Story
EBV latently infected cells and tissues copiously express EBV-encoded small nonpolyadenylated RNAs (EBERs), irrespective of the viral latency program. Besides their activity on the regulation of cytokines and growth factor synthesis in infected cells, EBV noncoding RNAs putatively contribute to several features of malignant transformation. For instance, Akata cells (from BL) transfected with a vector allowing low expression levels of EBERs (EK plasmid) had scant numbers of colonies in soft agar compared with cells transfected with a vector allowing a high expression of EBERs (EKS10 plasmid) or EBV-reinfected cell clones. In addition, in sharp contrast with EK-transfected cells, EKS10-transfected cells and EBV-reinfected cells were tumorigenic [68]. AGS gastric carcinoma cells stably expressing EBERs have increased migration and invasion capabilities, effects associated with FAK and PAK1 phosphorylation [69]. Remarkably, EBERs were identified in exosomes from EBV-infected malignant cell lines [70]. Therefore, the contribution of these small viral transcripts in EBV-induced carcinogenesis, and even cancer progression, might be much more relevant than initially suspected.
EBV also produces miRNAs, which are encoded within two regions of the viral genome: in the BHRF (at the 3′UTR of BamHI fragment H rightward ORF1) and BART (BamHI A region rightward transcript, respectively) clusters [71]. BHRF and BART encode 4 and 40 viral miRNAs, respectively. The genome of EBV prototype strain B95-8 shows a 12 kb deletion in BARTs, compromising 17 of their pre-miRNAs. Recently, Kanda and colleagues generated vectors encoding the wild-type EBV B95 (wt-EBV-BAC) and one derivative in which the deleted BART segment was orthotopically restored (BART(+)-EBV-BAC) [72]. Downregulation of the NDRG1 gene (NCBI's gene ID: 10397), which encodes a well-studied metastasis suppressor protein, was reported in HEK293 and Ad/AH* epithelial cells stably transfected with BART(+)-EBV-BAC compared to cells transfected with wild-type, BART-deleted vector, and this effect was mostly associated with the activity of EBV miR-BART22. Considering that the NDRG1 protein was reported to be consistently expressed in epithelium-derived cell lines (Ad/AH*, HBEC1, Caco-2, PC3, PrEC, and C666-1), but barely detected in the B cells from BL (Akata, P3HR-1, and Daudi) or lymphoblastoid cell lines, a role for NDRG1 in epithelial differentiation was postulated [72], in line with the effect of this gene in inhibiting the dissemination of carcinoma cells.
Different studies reported upregulation of several EBV miR-BARTs in NPC [73–77]. Strikingly, among 24 upregulated miRNAs differently expressed between NPC and nonmalignant nasopharyngeal samples, Cai and coworkers found that EBV miR-BARTs were more than twice the number of endogenous human miRNAs (62.5% vs 29.2%, respectively) [76]. The upregulation of BART miRNAs was also reported in mice xenograft tumors produced from EBV-positive carcinoma cells C666-1 (nasopharyngeal) and AGS-BX1 (gastric), as well as the EBV-positive BL36 (Burkitt Lymphoma) [77]. In this study, expression of BART miRNAs in EBV-negative AGSBX1 cells resulted in increased tumor growth in vivo and poorer survival when inoculated heterotopically in the nasopharynx of NOD scid gamma (NSG) mice. Interestingly, the expression of BART miRNAs along with EBNA1 in AGS cells showed no significant effect in the rates of in vitro cell invasion and in vivo metastasis, and the EBV BART miRNAs levels were not significantly different in metastatic versus matched primary tumors. Unfortunately, no comparative data regarding nasopharyngeal carcinoma cells (C666-1, for instance) were provided in this study [77]; that would have been useful in addressing the hypothesis of histogenetic specificity of BART miRNAs in carcinoma progression.
Combined detection of EBV miRs-BART 7 and 13 in the blood achieved 90% predictive value for NPC, and a higher expression of these viral miRs was predominantly found in advanced disease [78]. Accordingly, in a series of 102 radiotherapy-refractory NPC cases, the expression of EBV miR-BART7 in surgical margins of resected tumors was inversely associated with recurrence, even though no difference in cancer progression or free survival was verified [79]. In another series, elevated levels of EBV miR-BART1 were associated with advanced clinical stages of NPC. Nude mice with xenograft tumors overexpressing EBV-miR-BART1 generated from the EBV-negative 5-8F NPC cell line had an increase in the frequency of metastasis in the liver and lymph node (86%) or lung (43%), compared to controls (14% for each site), as well as a higher number of lesions (45 versus 5 nodules, respectively). The enhanced aggressiveness of EBVmiR-BART1 in this model was convincingly associated with post-transcriptional inhibition of the PTEN tumor suppressor gene (NCBI's gene ID: 5728) [76].
In both EBV-positive NPC tissues and cultured cells, miR-BART9 is expressed at levels higher than miR-21, an endogenous microRNA with known oncogenic activities (oncomiR). The depletion of miR-BART9 suppresses migration and invasion of NPC cells in vitro, an effect not associated with changes in the levels of LMP1, LMP2A, or EBNA1. The ectopic expression of mature miR-BART9 in EBV-negative BM1, TW04, and HK1 NPC cells significantly enhanced cell migration and invasion capabilities in vitro, and both effects are inhibited when miR-BART9 is suppressed. BM1 cells expressing miR-BART9 inoculated into nude mice produced tumors with an increased rate of metastasis to lymph nodes, lung, and liver. Putative targets for miR-BART9 include several transcripts implicated in motility-related pathways, including E-cadherin, which have a binding site for miR-BART9 within its 3′ UTR. Further experiments showed that the increase in cell motility and invasiveness by EBV miR-BART9 was indeed associated with several mesenchymal EMT properties, namely, E-cadherin downregulation, induction of β-catenin signaling, expression of vimentin and MMPs, and spindle-cell morphology [80]. Essentially the same properties reported for EBV miR-BART9 were found for viral miR-BART10-3p, which expression was directly associated with patient poor survival in a series of 106 NPCs [81].
In conclusion, the expression of several EBV oncoproteins or noncoding RNAs contributes to the aggressive behavior of NPC. Key reported effects of these viral products in tumor growth and metastasis are summarized in Figure 1. These data strengthen the concept that cancers associated with infection by oncogenic viruses might have particular biological features in terms of their progression capabilities. Moreover, some EBV products are quite promising in terms of their putative value as biomarkers for either NPC diagnosis or disease monitoring, such as viral miR-BARTs, whose expression can be readily assessed using current molecular biology techniques.
Figure 1. Reported Effects of Epstein–Barr Virus (EBV) Products That May Contribute to the Progression of EBV-Associated Cancers.
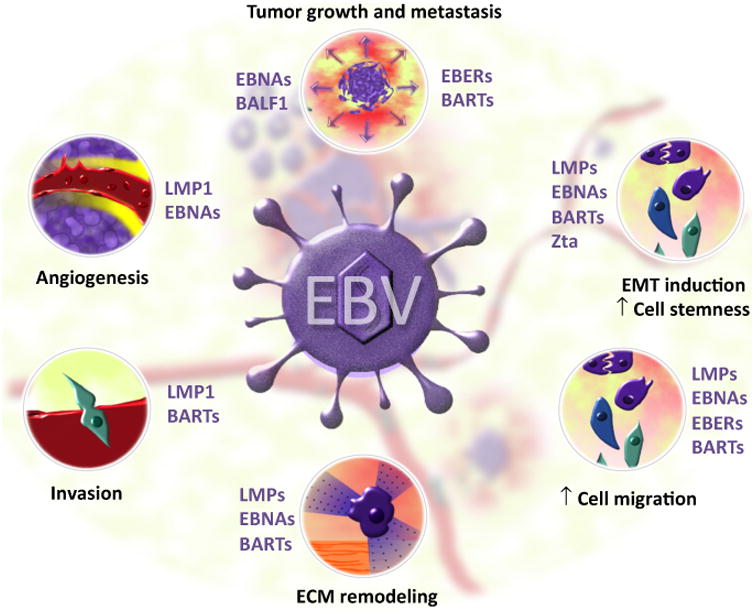
This matter has been mostly studied by considering the natural history of the EBV-associated nasopharyngeal carcinoma. Both EBV proteins – such as latent viral proteins (LMPs), EBV nuclear antigens (EBNAs), and the apoptosis regulator BALF1 – and nontranslated viral RNAs, notably EBV-encoded small nonpolyadenylated RNAs (EBERs) and BamHI A region rightward transcripts (BARTs), interfere with different phenomena that may account for increased tumor aggressiveness, favoring the dissemination of malignant cells and ultimately causing cancer metastasis. Tumors expressing some EBV viral products have accelerated growth, as accessed in different animal models. Mechanistically, the EBV products indicated in the figure may induce an epithelial–mesenchymal transition, which is associated with a higher rate of metastasis for epithelial cancers due to increased cell motility and degradation of the extracellular matrix (e.g., due to the synthesis of metalloproteinases), and consequently higher invasive behavior of malignant cells. Furthermore, tumor angiogenesis is induced by cells expressing viral products, notably LMP1 and EBNAs. This can be due to either upregulation of some cytokines in EBV-infected neoplastic cells (e.g., VEGF) or stimulation of adjacent cells by proangiogenic factors (e.g., HIF-1-α and FGF2) inside LMP1-positive exosomes released by EBV-infected cells. Abbreviation: Zta, the viral lytic transactivator.
Concluding Remarks
Although there are convincing data suggesting that EBV contributes to the aggressiveness of NPC, the effects of viral infection in cancer progression is conceivably context-dependent. A glimpse of this matter can be taken when considering the EBV-associated gastric carcinoma, which represents about 10% of all epithelial cancers in the stomach worldwide [82,83]. Analogous to NPC, EBV-associated gastric carcinoma is a peculiar clinicopathological entity. For instance, patients affected are predominantly male and Caucasian, and overall they are younger than patients with the more common EBV-negative gastric carcinomas. The EBV-associated gastric cancers more often arise in the cardia or body of the stomach, and they are more often either diffuse or lymphoepithelioma type. Despite these features, EBV-associated gastric carcinomas do not seem to be more aggressive: for instance, they are not more likely to be associated with deep tissue invasion, higher clinical stage, or metastasis, and data are conflicting regarding the differences in patient survival compared with EBV-negative cases [83]. For Hodgkin lymphoma, another EBV-associated human cancer, a recent meta-analysis encompassing 13 045 subjects from 119 published studies indicated that EBV infection does not have an impact on the lymphoma's prognosis [84].
It seems that not much can be expected in terms of EBV influencing the aggressiveness of a group of the cancers recognized as associated with viral infection. Moreover, even considering that EBV products may enhance the migratory and invasive capabilities of infected malignant cells for some cancers, the overall impact of these effects on prognosis cannot be anticipated. For instance, it is generally accepted that patients with nonkeratinized EBV-associated NPC have a better prognosis compared with keratinized EBV-negative NPC cases [85]. Though this might be taken as contradictory considering the results discussed in this review, we must recall that cancer survival data do not reflect exclusively the natural history of the disease, since it has some bias owing to responses to the treatment protocols available to date. This is not different for NPC, in the sense that patients' outcomes result from an interplay among the disease's biological, clinical, and therapeutic parameters.
In conclusion, EBV infection not only has an impact on malignant cell transformation, it may also contribute to the biological aggressiveness of some of the cancers associated with the viral infection, at least for NPC. EBV products modulate critical cancer progression phenomena (e.g., cell motility, cell invasion, and metastasis) due to either direct or indirect effects. Only recently has this topic received more attention, so that there are plenty of intriguing questions remaining to be addressed (see Outstanding Questions). As the knowledge on how EBV participates in cancer progression becomes clearer, we anticipate that new exciting data on the role of other oncogenic viruses in the behavior of cancers associated with viral infection will emerge. Indeed, it is plausible that, perhaps once again, the field of viral carcinogenesis will become key for understanding the biology of cancers.
Outstanding Questions.
Does EBV contribute to the progression of epithelial cancers only, or does this virus also have a role in other malignant neoplasms, such as EBV-associated lymphomas?
Could the activity of products of oncogenic viruses within the infected cell and in the tumor microenvironment inhibit the progression of cancers? If they do, what are the molecular mechanisms?
Could we extend this new viral carcinogenesis paradigm to other infective agents consistently linked to human cancers, such as KSHV, the hepatitis viruses HBV and HCV, HPVs, or even nonviral oncogenic microorganisms, such as Helicobacter pylori?
How can we use knowledge about the modulation of cancer progression by oncoviruses to improve outcomes for cancer patients?
Figure I. Overview of the Metastatic Cascade.
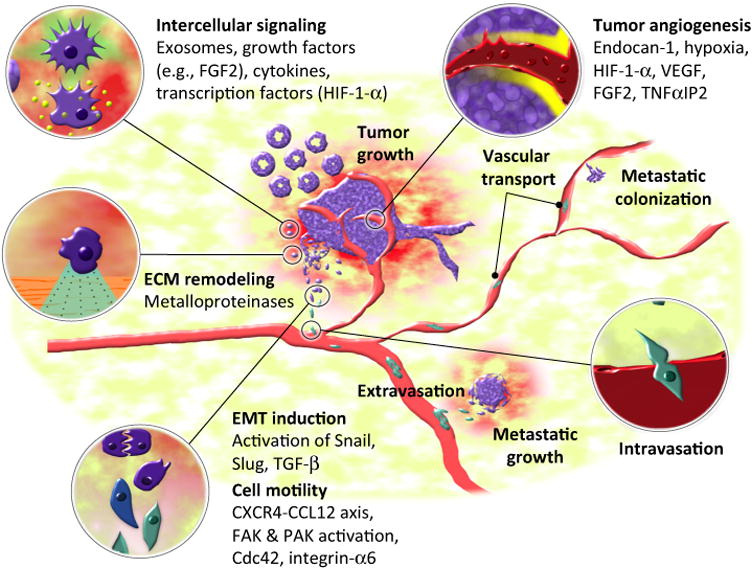
Cancer metastasis is a complex and multifactorial process that relies on successful interplay among a large repertoire of molecules that contribute to a propitious phenotype of the cancer cells, along with a permissible tissue microenvironment for tumor development, both locally and at distant sites. The term ‘metastatic cascade’ refers to a theoretical attempt to organize chronologically recognized key phenomena for successful establishment of malignant cells discontinuous to the primary tumor. Molecules indicated in the figure were selected among those that are known players in the metastatic cascade and can be modulated during EBV infection or are induced by one or more viral products (see Figure 1 in main text).
Trends.
The Epstein–Barr virus (EBV) is implicated in many neoplastic diseases, notably lymphomas and epithelial cancers.
Nasopharyngeal carcinoma (NPC) is strongly associated with EBV infection, and EBV products partially contribute to the aggressiveness of this cancer.
Several EBV products enhance cell motility and invasiveness, and they can also modulate the epithelial–mesenchymal transition.
Challenges in the manipulation of EBV genomes hampered the assessment of the extent of cancer addiction to viral products. New genetic editing tools (e.g., CRISPR/Cas9) will be valuable to create new informative models to address this issue.
Accumulated data on the role of EBV in the biological features of NPC makes it conceivable that some oncoviruses contribute to malignant transformation and have a role in the aggressiveness of the associated cancers due to effects on tumor progression phenomena.
Acknowledgments
ViriCan's studies on EBV and cancer progression at the State University of Sao Paulo, SP, Brazil, are sponsored by Sao Paulo Research Foundation (FAPESP), which awarded a scholarship to BGMC (Proc. MS 2014/14678-5), as well as research funds to DEO (Proc. AP 2014/17326-9). The authors sincerely apologize for papers relevant to the topic that were not mentioned in this review due to space and citations constraints.
Footnotes
Disclaimer statement The authors declare that they have no conflict of interest to disclose.
References
- 1.Colditz GA, et al. Applying what we know to accelerate cancer prevention. Sci Transl Med. 2012;4:127rv4. doi: 10.1126/scitranslmed.3003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Martel C, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. 2012;13:607–615. doi: 10.1016/S1470-2045(12)70137-7. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Elgui de Oliveira D. DNA viruses in human cancer: an integrated overview on fundamental mechanisms of viral carcino-genesis. Cancer Lett. 2007;247:182–196. doi: 10.1016/j.canlet.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 5.Mesri EA, et al. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15:266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bibas M, Antinori A. EBV and HIV-related lymphoma. Mediterr J Hematol Infect Dis. 2009;1:e2009032. doi: 10.4084/MJHID.2009.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chandran B, Hutt-Fletcher L. Gammaherpesviruses entry and early events during infection. In: Arvin A, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press; 2007. [PubMed] [Google Scholar]
- 8.Pitot HC, et al. The natural history of carcinogenesis: implications of experimental carcinogenesis in the genesis of human cancer. J Supramol Struct Cell Biochem. 1981;17:133–146. doi: 10.1002/jsscb.380170204. [DOI] [PubMed] [Google Scholar]
- 9.Pitot HC. Progression: the terminal stage in carcinogenesis. Jpn J Cancer Res Gann. 1989;80:599–607. doi: 10.1111/j.1349-7006.1989.tb01683.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pitot HC. The molecular biology of carcinogenesis. Cancer. 1993;72:962–970. doi: 10.1002/1097-0142(19930801)72:3+<962::aid-cncr2820721303>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 11.Ishizawa K, et al. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell. 2010;7:279–282. doi: 10.1016/j.stem.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Driessens G, et al. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–530. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbertson RJ, Graham TA. Cancer: Resolving the stem-cell debate. Nature. 2012;488:462–463. doi: 10.1038/nature11480. [DOI] [PubMed] [Google Scholar]
- 14.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barnes L. Pathology and Genetics of Head and Neck Tumours. IARC Press; 2005. [Google Scholar]
- 16.Pathmanathan R, et al. Clonal proliferations of cells infected with Epstein–Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N Engl J Med. 1995;333:693–698. doi: 10.1056/NEJM199509143331103. [DOI] [PubMed] [Google Scholar]
- 17.Chua MLK, et al. Nasopharyngeal carcinoma. Lancet. 2016;387:1012–1024. doi: 10.1016/S0140-6736(15)00055-0. [DOI] [PubMed] [Google Scholar]
- 18.Raab-Traub N. Epstein–Barr virus in the pathogenesis of NPC. Semin Cancer Biol. 2002;12:431–441. doi: 10.1016/s1044579x0200086x. [DOI] [PubMed] [Google Scholar]
- 19.Strong MJ, et al. Comprehensive high-throughput RNA sequencing analysis reveals contamination of multiple nasopharyngeal carcinoma cell lines with HeLa cell genomes. J Virol. 2014;88:10696–10704. doi: 10.1128/JVI.01457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li HP, Chang YS. Epstein–Barr virus latent membrane protein 1: structure and functions. J Biomed Sci. 2003;10:490–504. doi: 10.1007/BF02256110. [DOI] [PubMed] [Google Scholar]
- 21.Bentz GL, et al. Epstein–Barr virus latent membrane protein 1 (LMP1) C-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J Virol. 2011;85:10144–10153. doi: 10.1128/JVI.05035-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bentz GL, et al. Epstein–Barr virus latent membrane protein 1 regulates the function of interferon regulatory factor 7 by inducing its sumoylation. J Virol. 2012;86:12251–12261. doi: 10.1128/JVI.01407-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dawson CW, et al. Epstein–Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J Virol. 2008;82:3654–3664. doi: 10.1128/JVI.01888-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohr CF, et al. The tumor marker Fascin is induced by the Epstein–Barr virus-encoded oncoprotein LMP1 via NF-kB in lymphocytes and contributes to their invasive migration. Cell Commun Signal CCS. 2014;12:46. doi: 10.1186/s12964-014-0046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ho CH, et al. Decoy receptor 3, upregulated by Epstein– Barr virus latent membrane protein 1, enhances nasopharyngeal carcinoma cell migration and invasion. Carcinogenesis. 2009;30:1443–1451. doi: 10.1093/carcin/bgp135. [DOI] [PubMed] [Google Scholar]
- 26.Shen Z, et al. Over-expression of the special AT rich sequence binding protein 1 (SATB1) promotes the progression of nasopharyngeal carcinoma: association with EBV LMP-1 expression. J Transl Med. 2013;11:217. doi: 10.1186/1479-5876-11-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domanska UM, et al. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, et al. Tyrosylprotein sulfotransferase-1 and tyrosine sulfation of chemokine receptor 4 are induced by Epstein–Barr virus encoded latent membrane protein 1 and associated with the metastatic potential of human nasopharyngeal carcinoma. PLoS ONE. 2013;8:e56114. doi: 10.1371/journal.pone.0056114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altmann M, Hammerschmidt W. Epstein-Barr Virus Provides a New Paradigm: A Requirement for the Immediate Inhibition of Apoptosis. PLoS Biol. 2005;3:e404. doi: 10.1371/journal.pbio.0030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cabras G, et al. Epstein–Barr virus encoded BALF1 gene is transcribed in Burkitt's lymphoma cell lines and in nasopharyngeal carcinoma's biopsies. J Clin Virol. 2005;34:26–34. doi: 10.1016/j.jcv.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 31.Hsu WL, et al. A role for Epstein–Barr viral BALF1 in facilitating tumor formation and metastasis potential. Virus Res. 2012;163:617–627. doi: 10.1016/j.virusres.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 32.Kaul R, et al. Epstein–Barr virus latent nuclear antigens can induce metastasis in a nude mouse model. J Virol. 2007;81:10352–10361. doi: 10.1128/JVI.00886-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murakami M, et al. Epstein–Barr virus nuclear antigen 1 interacts with Nm23-H1 in lymphoblastoid cell lines and inhibits its ability to suppress cell migration. J Virol. 2005;79:1559–1568. doi: 10.1128/JVI.79.3.1559-1568.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian C, et al. Epstein–Barr virus nuclear protein EBNA-3C interacts with the human metastatic suppressor Nm23-H1: a molecular link to cancer metastasis. Nat Med. 2001;7:350–355. doi: 10.1038/85499. [DOI] [PubMed] [Google Scholar]
- 35.Kuppers DA, et al. Regulation of matrix metalloproteinase 9 expression by Epstein–Barr Virus nuclear antigen 3C and the suppressor of metastasis Nm23-H1. J Virol. 2005;79:9714–9724. doi: 10.1128/JVI.79.15.9714-9724.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trocmé C, et al. Human B lymphocytes synthesize the 92-kDa gelatinase, matrix metalloproteinase-9. J Biol Chem. 1998;273:20677–20684. doi: 10.1074/jbc.273.32.20677. [DOI] [PubMed] [Google Scholar]
- 37.Cao JY, et al. Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA1 protein of Epstein–Barr virus reveal potential roles for EBNA1 in metastasis and oxidative stress responses. J Virol. 2011;86:382–394. doi: 10.1128/JVI.05648-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin Y, et al. Self-inhibition of synthesis and antigen presentation by Epstein–Barr virus-encoded EBNA1. Science. 2003;301:1371–1374. doi: 10.1126/science.1088902. [DOI] [PubMed] [Google Scholar]
- 39.Kondo S, et al. Epstein–Barr virus latent membrane protein 1 induces the matrix metalloproteinase-1 promoter via an Ets binding site formed by a single nucleotide polymorphism: Enhanced susceptibility to nasopharyngeal carcinoma. Int J Cancer. 2005;115:368–376. doi: 10.1002/ijc.20849. [DOI] [PubMed] [Google Scholar]
- 40.Chew MMS, et al. Interleukins, laminin and Epstein–Barr virus latent membrane protein 1 (EBV LMP1) promote metastatic phenotype in nasopharyngeal carcinoma. BMC Cancer. 2010;10:574. doi: 10.1186/1471-2407-10-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lan YY, et al. Epstein–Barr virus latent membrane protein 2A promotes invasion of nasopharyngeal carcinoma cells through ERK/Fra-1-mediated induction of matrix metalloproteinase 9. J Virol. 2012;86:6656–6667. doi: 10.1128/JVI.00174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lan YY, et al. Epstein–Barr virus Zta upregulates matrix metalloproteinases 3 and 9 that synergistically promote cell invasion in vitro. PLoS ONE. 2013;8:e56121. doi: 10.1371/journal.pone.0056121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horikawa T, et al. Induction of c-Met proto-oncogene by Epstein–Barr virus latent membrane protein-1 and the correlation with cervical lymph node metastasis of nasopharyngeal carcinoma. Am J Pathol. 2001;159:27–33. doi: 10.1016/S0002-9440(10)61669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen LC, et al. A novel role for TNFAIP2: its correlation with invasion and metastasis in nasopharyngeal carcinoma. Mod Pathol. 2011;24:175–184. doi: 10.1038/modpathol.2010.193. [DOI] [PubMed] [Google Scholar]
- 45.Chen CC, et al. NF-κB-mediated transcriptional upregulation of TNFAIP2 by the Epstein–Barr virus oncoprotein, LMP1, promotes cell motility in nasopharyngeal carcinoma. Oncogene. 2014;33:3648–3659. doi: 10.1038/onc.2013.345. [DOI] [PubMed] [Google Scholar]
- 46.Liu HP, et al. Epstein–Barr virus-encoded LMP1 interacts with FGD4 to activate Cdc42 and thereby promote migration of nasopharyngeal carcinoma cells. PLoS Pathog. 2012;8:e1002690. doi: 10.1371/journal.ppat.1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu PH, et al. Upregulation of endocan by Epstein–Barr virus latent membrane protein 1 and its clinical significance in nasopharyngeal carcinoma. PLoS ONE. 2013;8:e82254. doi: 10.1371/journal.pone.0082254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakisaka N, et al. Epstein–Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2004;24:5223–5234. doi: 10.1128/MCB.24.12.5223-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ceccarelli S, et al. Epstein–Barr virus latent membrane protein 1 promotes concentration in multivesicular bodies of fibro-blast growth factor 2 and its release through exosomes. Int J Cancer. 2007;121:1494–1506. doi: 10.1002/ijc.22844. [DOI] [PubMed] [Google Scholar]
- 50.Filipazzi P, et al. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Semin Cancer Biol. 2012;22:342–349. doi: 10.1016/j.semcancer.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 51.Martins VR, et al. Tumor-cell-derived microvesicles as carriers of molecular information in cancer. Curr Opin Oncol. 2013;25:66–75. doi: 10.1097/CCO.0b013e32835b7c81. [DOI] [PubMed] [Google Scholar]
- 52.Middeldorp JM, Pegtel DM. Multiple roles of LMP1 in Epstein–Barr virus induced immune escape. Semin Cancer Biol. 2008;18:388–396. doi: 10.1016/j.semcancer.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Aga M, et al. Exosomal HIF1α supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene. 2014;33:4613–4622. doi: 10.1038/onc.2014.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Horikawa T, et al. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007;67:1970–1978. doi: 10.1158/0008-5472.CAN-06-3933. [DOI] [PubMed] [Google Scholar]
- 55.Horikawa T, et al. Epstein–Barr virus latent membrane protein 1 induces Snail and epithelial–mesenchymal transition in metastatic nasopharyngeal carcinoma. Br J Cancer. 2011;104:1160–1167. doi: 10.1038/bjc.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kong QL, et al. Epstein–Barr virus-encoded LMP2A induces an epithelial–mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS Pathog. 2010;6:e1000940. doi: 10.1371/journal.ppat.1000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song LB, et al. The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelialmesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest. 2009;119:3626–3636. doi: 10.1172/JCI39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Min C, et al. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733–744. doi: 10.1002/jcb.21695. [DOI] [PubMed] [Google Scholar]
- 59.de Oliveira DE, et al. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 2010;18:248–257. doi: 10.1016/j.tim.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 60.Kondo S, et al. Epstein–Barr virus latent membrane protein 1 induces cancer stem/progenitor-like cells in nasopharyngeal epithelial cell lines. J Virol. 2011;85:11255–11264. doi: 10.1128/JVI.00188-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Port RJ, et al. Epstein–Barr virus induction of the Hedgehog signalling pathway imposes a stem cell phenotype on human epithelial cells. J Pathol. 2013;231:367–377. doi: 10.1002/path.4245. [DOI] [PubMed] [Google Scholar]
- 62.Sides MD, et al. The Epstein–Barr virus latent membrane protein 1 and transforming growth factor–β1 synergistically induce epithelial–mesenchymal transition in lung epithelial cells. Am J Respir Cell Mol Biol. 2011;44:852–862. doi: 10.1165/rcmb.2009-0232OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakaya T, et al. Enrichment of stem-like cell population comprises transformation ability of Epstein–Barr virus latent membrane protein 2A for non-transformed cells. Virus Res. 2013;174:108–115. doi: 10.1016/j.virusres.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 64.Pegtel DM, et al. Epstein–Barr-virus-encoded LMP2A induces primary epithelial cell migration and invasion: possible role in nasopharyngeal carcinoma metastasis. J Virol. 2005;79:15430–15442. doi: 10.1128/JVI.79.24.15430-15442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhou X, et al. SYK interaction with ITGb4 suppressed by Epstein–Barr virus LMP2A modulates migration and invasion of nasopharyngeal carcinoma cells. Oncogene. 2015;34:4491–4499. doi: 10.1038/onc.2014.380. [DOI] [PubMed] [Google Scholar]
- 66.Rathinam R, Alahari SK. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010;29:223–237. doi: 10.1007/s10555-010-9211-x. [DOI] [PubMed] [Google Scholar]
- 67.Gaur N, et al. Epstein–Barr virus latent antigens EBNA3C and EBNA1 modulate epithelial to mesenchymal transition of cancer cells associated with tumor metastasis. Tumour Biol. 2014;36:3051–3060. doi: 10.1007/s13277-014-2941-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Komano J, et al. Oncogenic role of Epstein–Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol. 1999;73:9827–9831. doi: 10.1128/jvi.73.12.9827-9831.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Banerjee AS, et al. Epstein–Barr virus-encoded small non-coding RNAs induce cancer cell chemoresistance and migration. Virology. 2013;443:294–305. doi: 10.1016/j.virol.2013.05.020. [DOI] [PubMed] [Google Scholar]
- 70.Ahmed W, et al. Epstein–Barr virus-encoded small RNAs (EBERs) are present in fractions related to exosomes released by EBV-transformed cells. PLoS ONE. 2014;9:e99163. doi: 10.1371/journal.pone.0099163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu JY, et al. Identification of novel Epstein–Barr virus microRNA genes from nasopharyngeal carcinomas. J Virol. 2009;83:3333–3341. doi: 10.1128/JVI.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanda T, et al. Clustered microRNAs of the Epstein–Barr virus cooperatively downregulate an epithelial cell-specific metastasis suppressor. J Virol. 2015;89:2684–2697. doi: 10.1128/JVI.03189-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen SJ, et al. Characterization of Epstein–Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS ONE. 2010;5:e12745. doi: 10.1371/journal.pone.0012745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chan JYW, et al. The role of Epstein–Barr virus-encoded microRNA BART7 status of resection margins in the prediction of local recurrence after salvage nasopharyngectomy for recurrent nasopharyngeal carcinoma. Cancer. 2015;121:2358–2366. doi: 10.1002/cncr.29380. [DOI] [PubMed] [Google Scholar]
- 75.Zhang G, et al. Circulating Epstein–Barr virus microRNAs miR-BART7 and miR-BART13 as biomarkers for nasopharyngeal carcinoma diagnosis and treatment. Int J Cancer. 2015;136:E301–E312. doi: 10.1002/ijc.29206. [DOI] [PubMed] [Google Scholar]
- 76.Cai L, et al. Epstein–Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat Commun. 2015;6:7353. doi: 10.1038/ncomms8353. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Qiu J, et al. The Epstein–Barr virus encoded BART miRNAs potentiate tumor growth in vivo. PLoS Pathog. 2015;11:e1004561. doi: 10.1371/journal.ppat.1004561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang G, et al. Circulating Epstein–Barr virus microRNAs miR-BART7 and miR-BART13 as biomarkers for nasopharyngeal carcinoma diagnosis and treatment: EBV BART miRNA as bio-marker for NPC diagnosis and treatment. Int J Cancer. 2015;136:E301–E312. doi: 10.1002/ijc.29206. [DOI] [PubMed] [Google Scholar]
- 79.Chan JYW, et al. The role of Epstein–Barr virus-encoded microRNA BART7 status of resection margins in the prediction of local recurrence after salvage nasopharyngectomy for recurrent nasopharyngeal carcinoma: Margin EBV miRNA BART7 in Recurrent NPC. Cancer. 2015;121:2358–2366. doi: 10.1002/cncr.29380. [DOI] [PubMed] [Google Scholar]
- 80.Hsu CY, et al. The Epstein–Barr virus-encoded microRNA MiR-BART9 promotes tumor metastasis by targeting E-cadherin in nasopharyngeal carcinoma. PLoS Pathog. 2014;10:e1003974. doi: 10.1371/journal.ppat.1003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yan Q, et al. EBV-miR-BART10-3p facilitates epithelialmesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget. 2015;6:41766–41782. doi: 10.18632/oncotarget.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen JN, et al. Epstein–Barr virus-associated gastric carcinoma: a newly defined entity. J Clin Gastroenterol. 2012;46:262–271. doi: 10.1097/MCG.0b013e318249c4b8. [DOI] [PubMed] [Google Scholar]
- 83.Lee JH, et al. Clinicopathological and molecular characteristics of Epstein–Barr virus-associated gastric carcinoma: A meta-analysis. J Gastroenterol Hepatol. 2009;24:354–365. doi: 10.1111/j.1440-1746.2009.05775.x. [DOI] [PubMed] [Google Scholar]
- 84.Lee JH, et al. Prevalence and prognostic significance of Epstein–Barr virus infection in classical Hodgkin's lymphoma: a meta-analysis. Arch Med Res. 2014;45:417–431. doi: 10.1016/j.arcmed.2014.06.001. [DOI] [PubMed] [Google Scholar]
- 85.Petersson F. Nasopharyngeal carcinoma: a review. Semin Diagn Pathol. 2015;32:54–73. doi: 10.1053/j.semdp.2015.02.021. [DOI] [PubMed] [Google Scholar]
- 86.Fingeroth JD, et al. Epstein–Barr virus receptor of human B lymphocytes is the C3d receptor CR2. Proc Natl Acad Sci USA. 1984;81:4510–4514. doi: 10.1073/pnas.81.14.4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ogembo JG, et al. Human complement receptor type 1/CD35 is an Epstein–Barr virus receptor. Cell Rep. 2013;3:371–385. doi: 10.1016/j.celrep.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borza CM, et al. Use of gHgL for attachment of Epstein– Barr virus to epithelial cells compromises infection. J Virol. 2004;78:5007–5014. doi: 10.1128/JVI.78.10.5007-5014.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Turk SM, et al. Antibodies to gp350/220 enhance the ability of Epstein–Barr virus to infect epithelial cells. J Virol. 2006;80:9628–9633. doi: 10.1128/JVI.00622-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taylor GS, Blackbourn DJ. Infectious agents in human cancers: Lessons in immunity and immunomodulation from gammaherpesviruses EBV and KSHV. Cancer Lett. 2011;305:263–278. doi: 10.1016/j.canlet.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 91.Young LS, et al. EBV gene expression and regulation. In: Arvin A, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press; 2007. [PubMed] [Google Scholar]
- 92.Thorley-Lawson DA. EBV the prototypical human tumor virus – just how bad is it? J Allergy Clin Immunol. 2005;116:251–261. doi: 10.1016/j.jaci.2005.05.038. quiz 262. [DOI] [PubMed] [Google Scholar]
- 93.Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi: 10.1016/j.cell.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11:123–134. doi: 10.1038/nrc3004. [DOI] [PMC free article] [PubMed] [Google Scholar]